
Comparative Study on the Performance and Mechanism of Adsorption–Oriented Phosphorus–Modified High–Efficiency and Durable Activated Biochar from Fast Pyrolysis



Abstract
1. Introduction
2. Materials and Methods
2.1. Feedstocks and Reagents
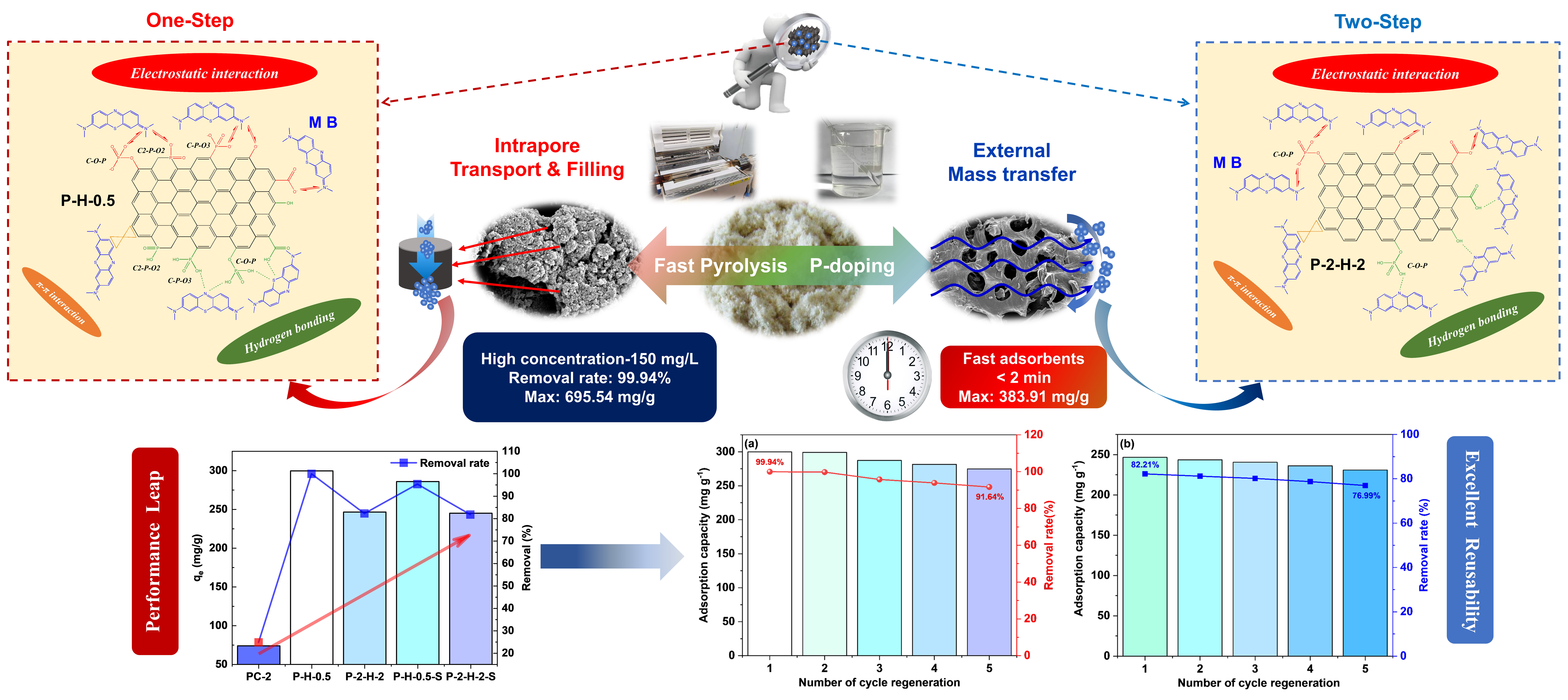
2.2. Synthesis of Modified Biochar (PS–ACs)
2.3. Characterization of the Samples
2.4. Batch Adsorption Experiments
3. Results and Discussion
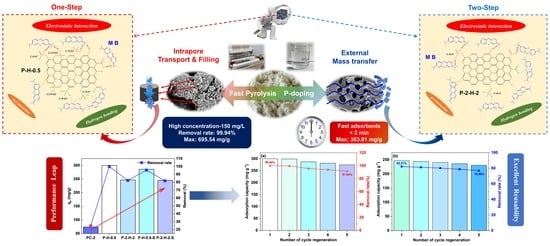
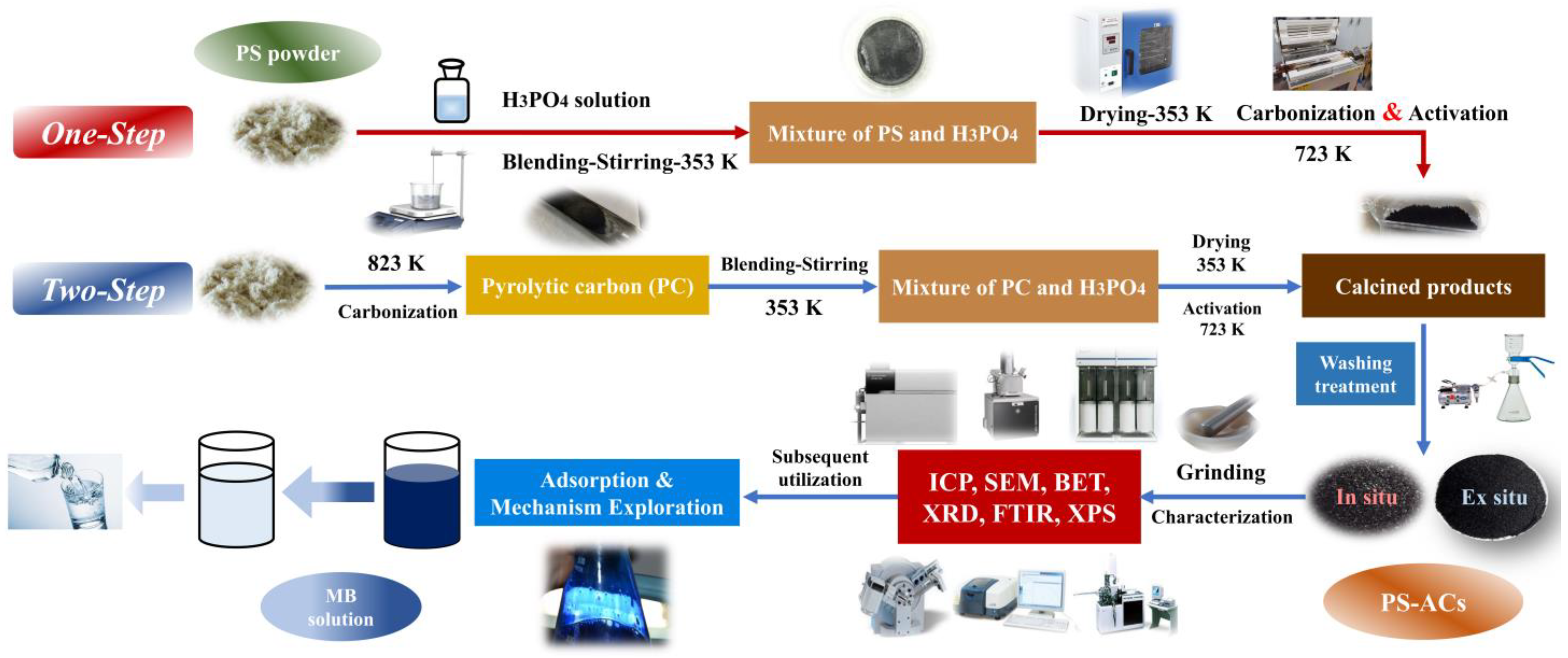
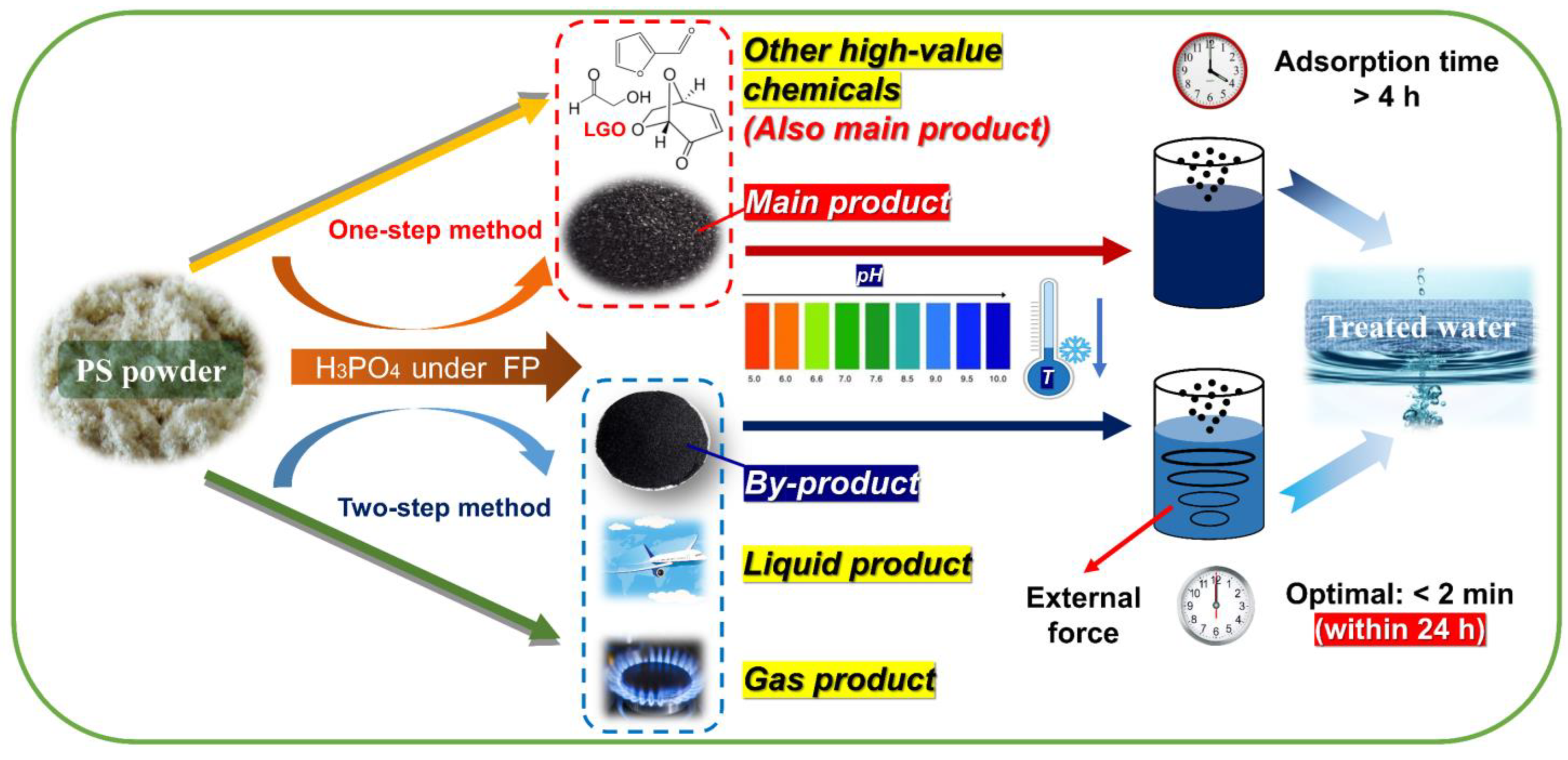
3.1. Yield Analysis and Initial Screening of PS–ACs
3.2. Comparative Characterization of the Samples
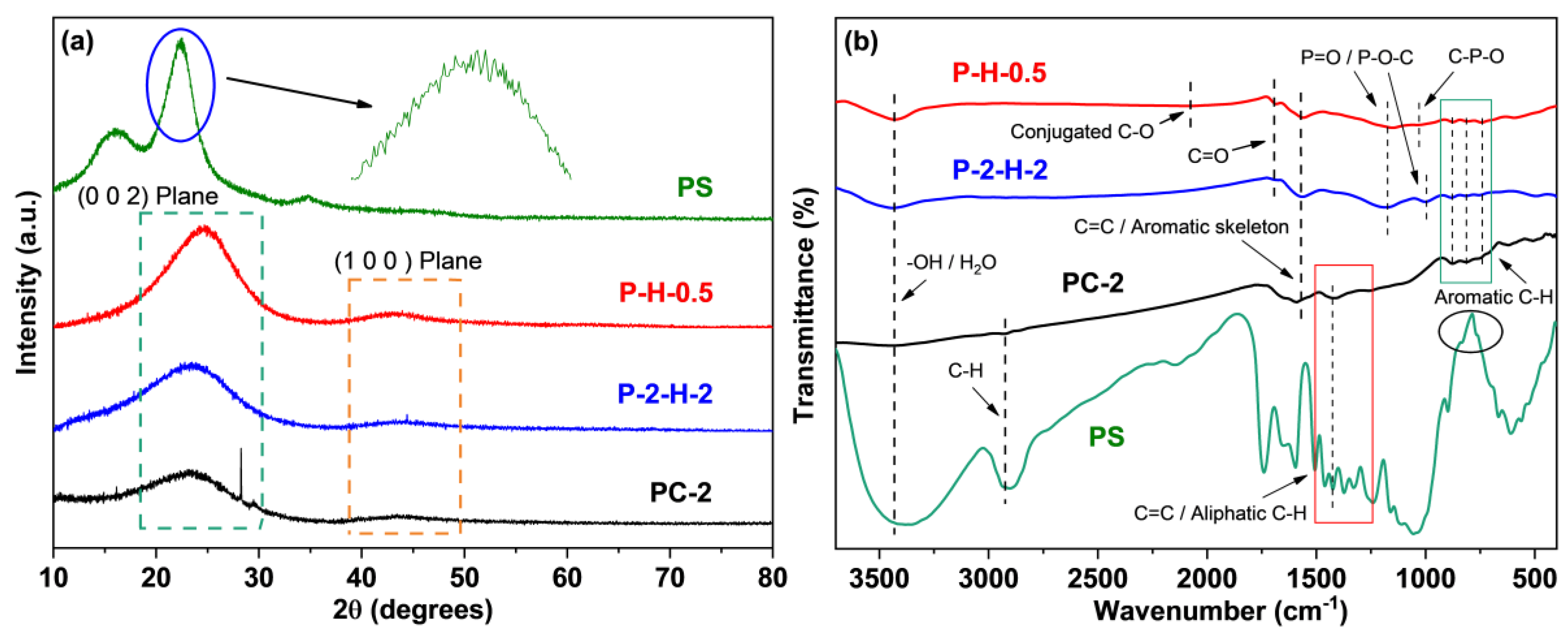
3.2.1. Analysis of Modified Biochar’s Chemical Composition and Stability
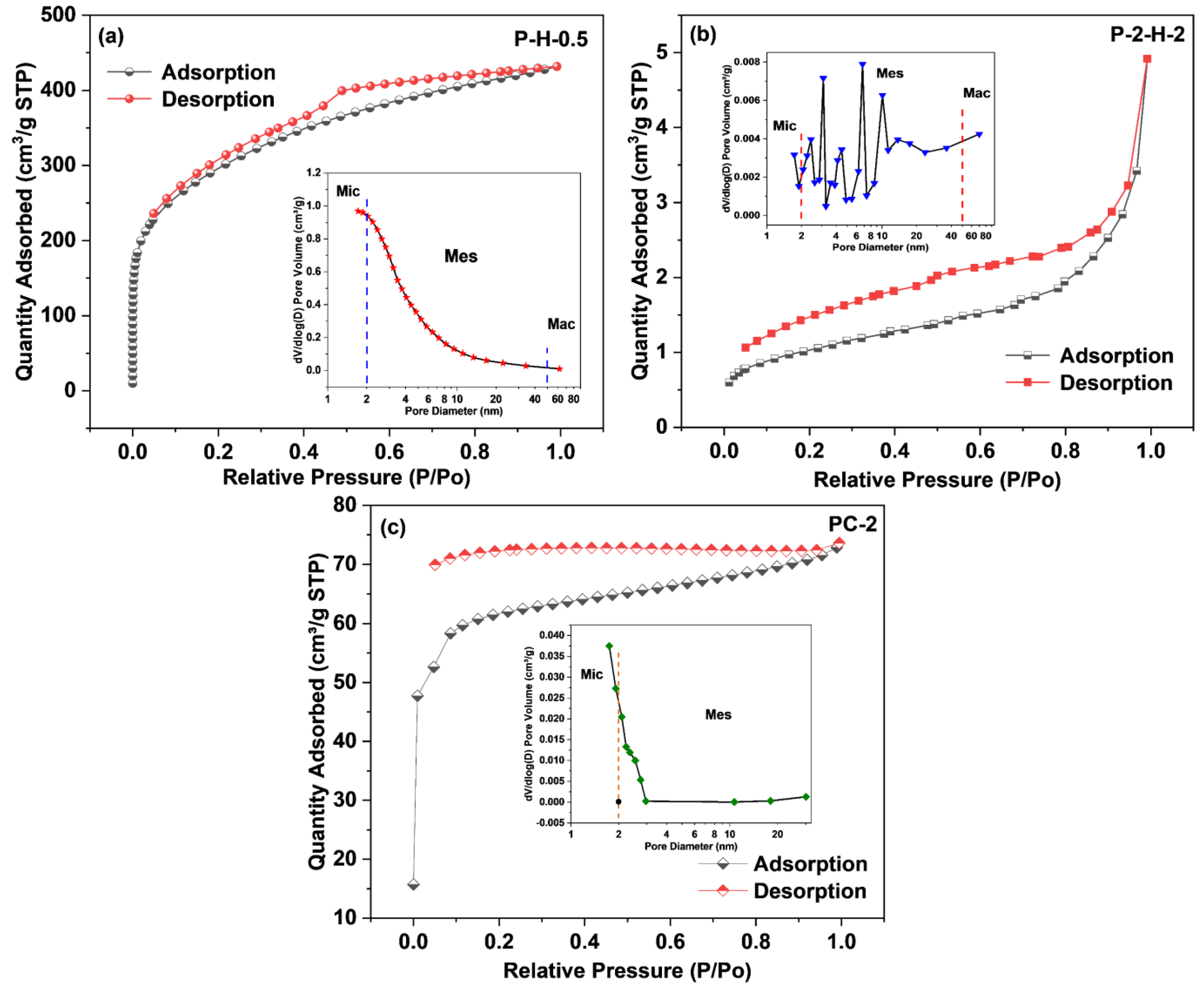
3.2.2. Analysis of Surface Morphology, Pore Structure, and Crystallinity
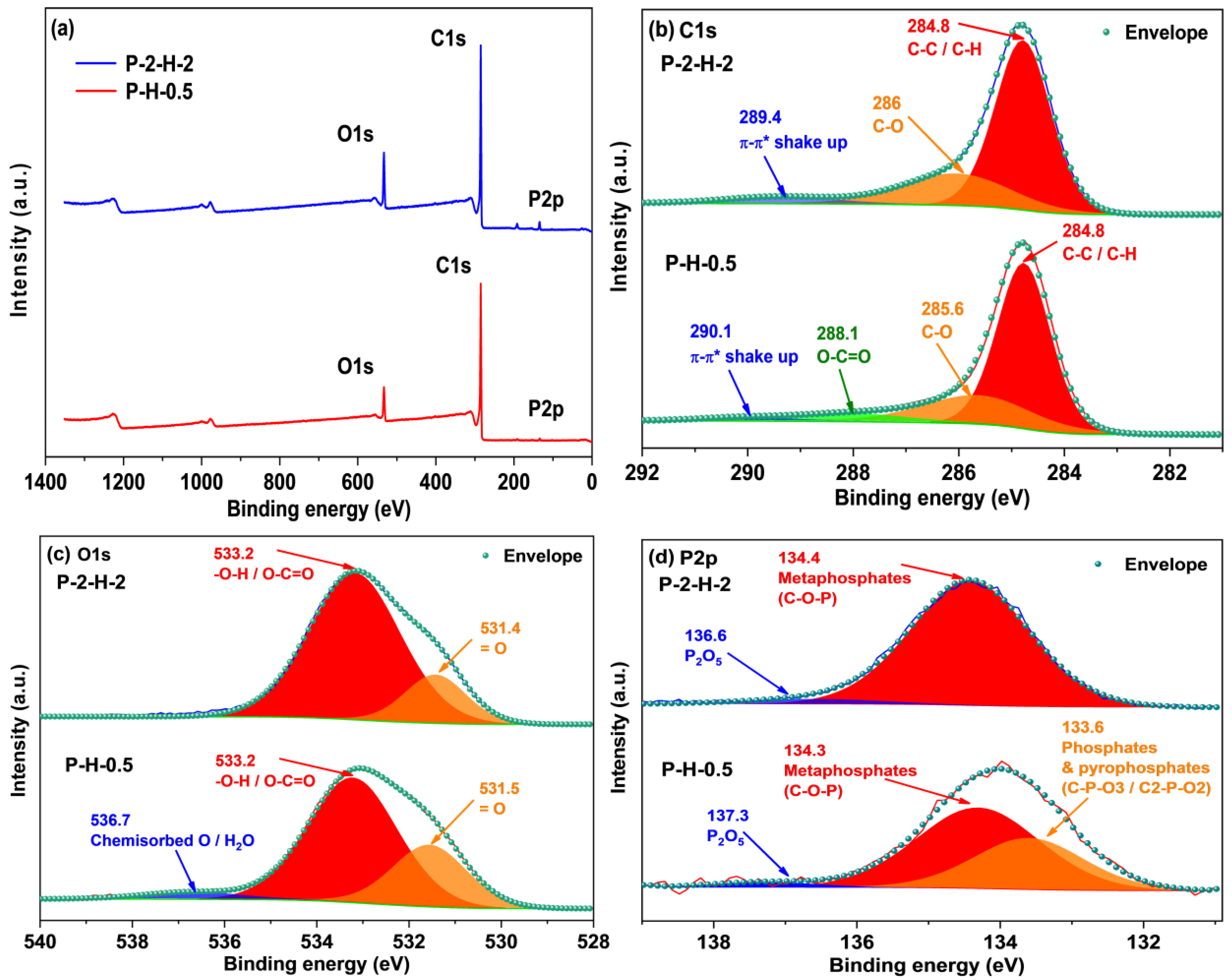
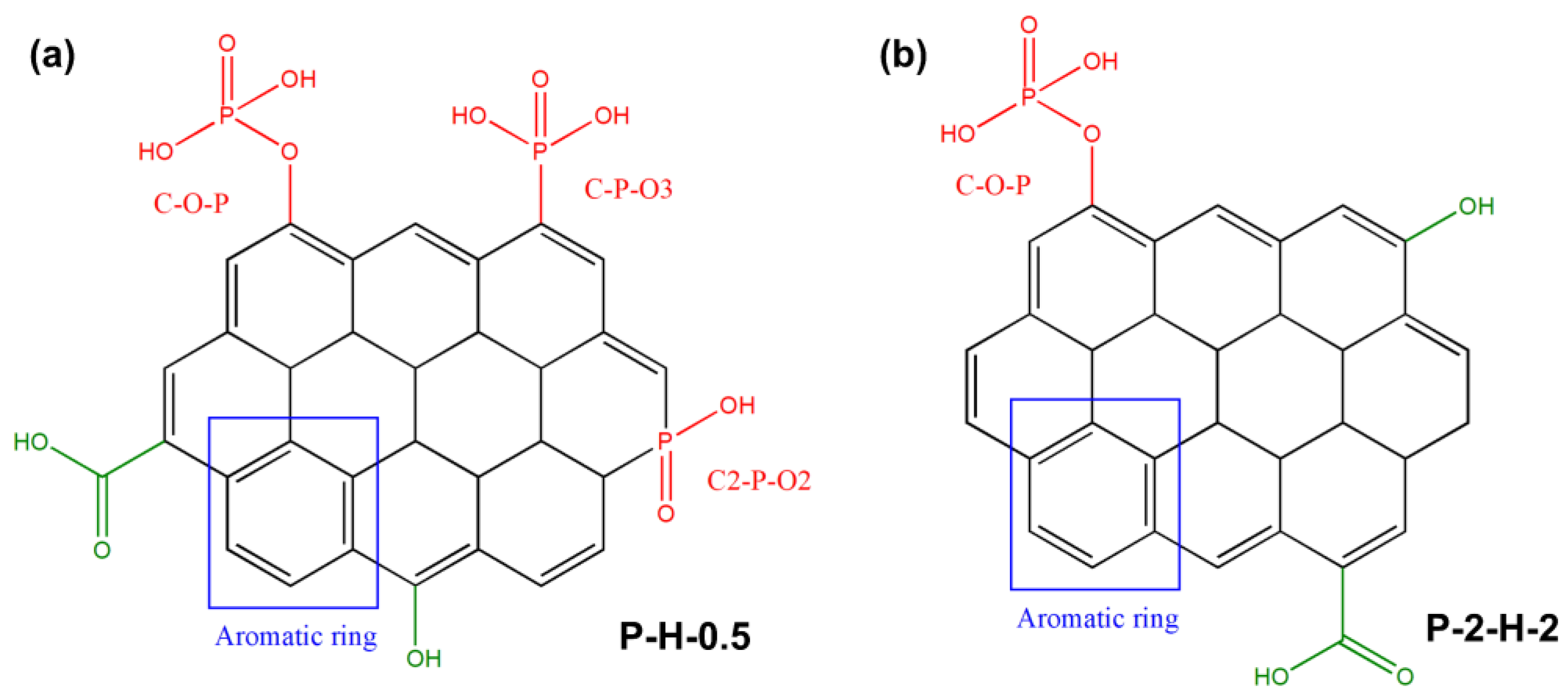
3.2.3. Analysis of Functional Groups and Surface Element
3.3. Adsorption Properties and Potential Application Strategies
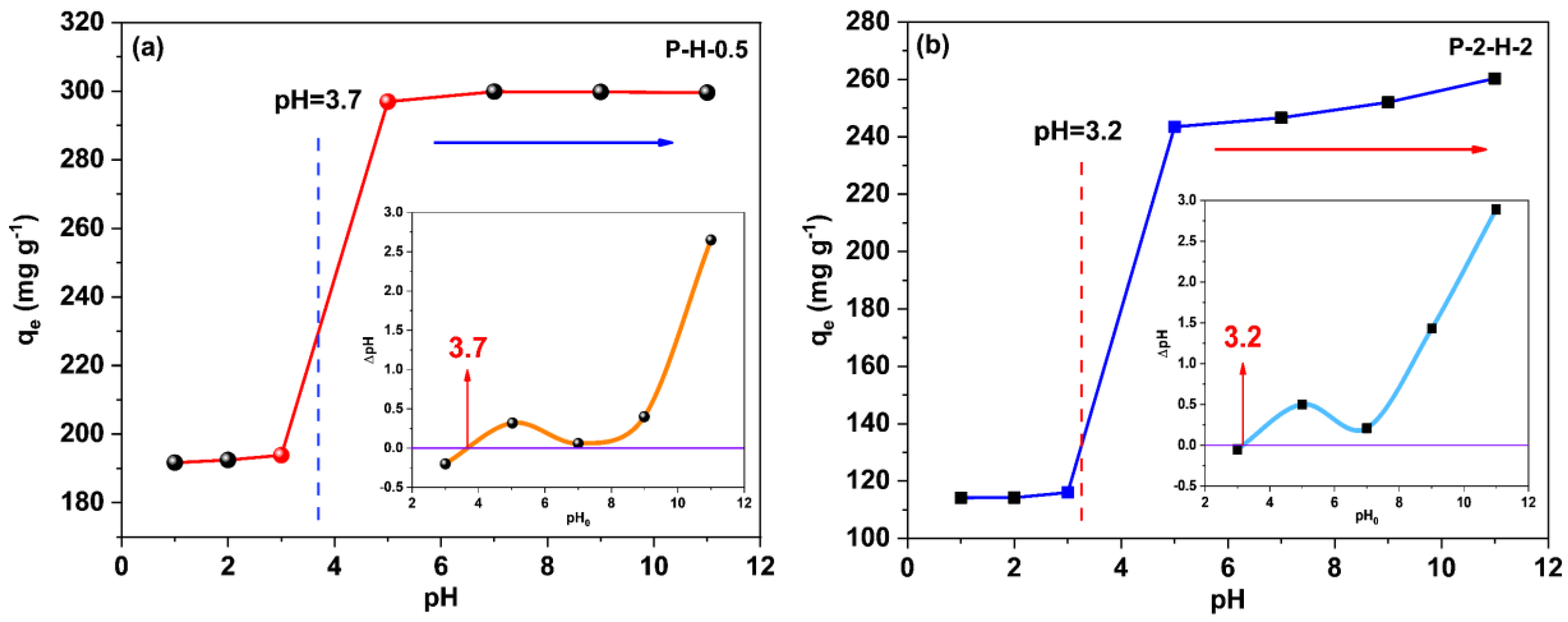
3.3.1. System pH Effect on Adsorption
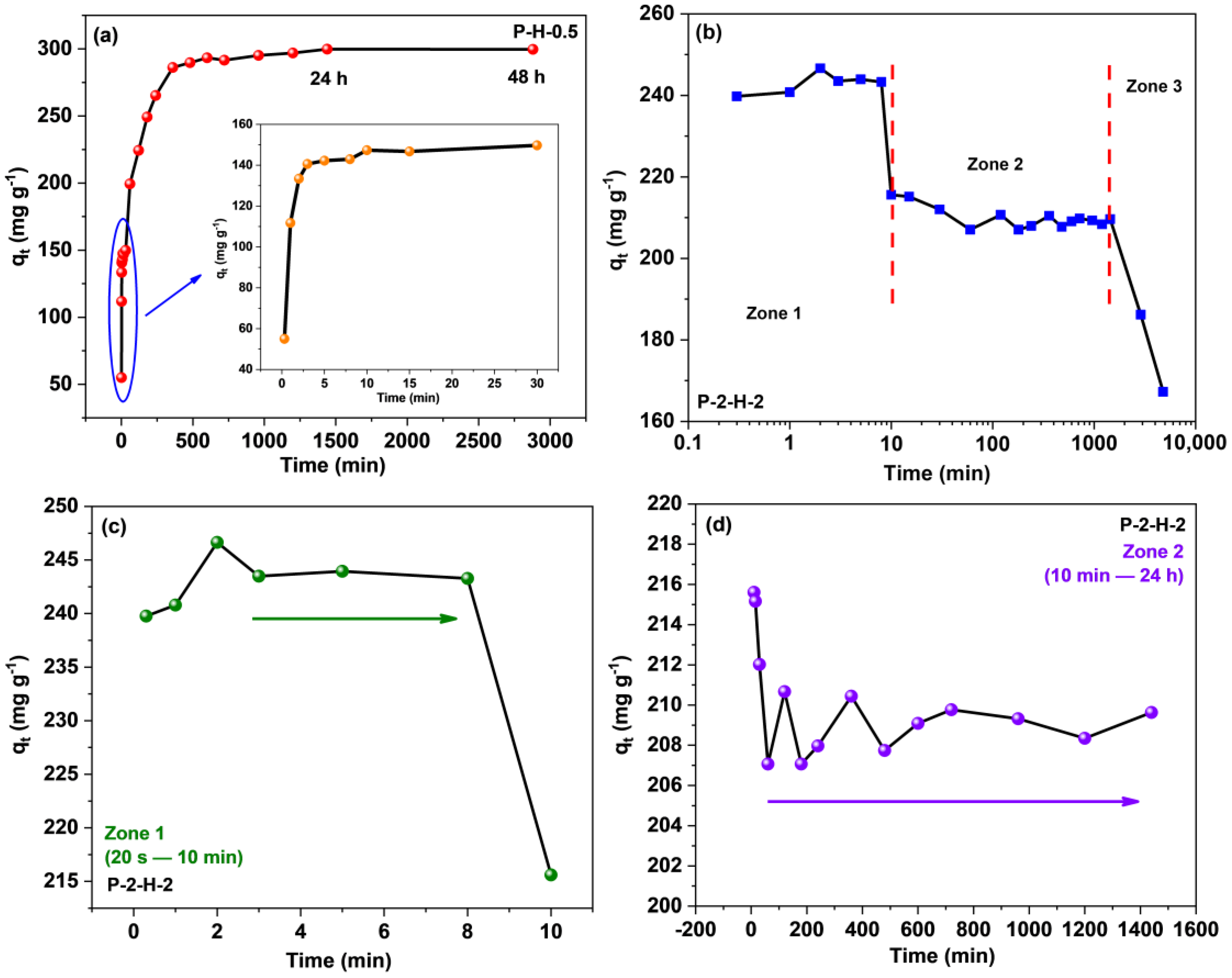
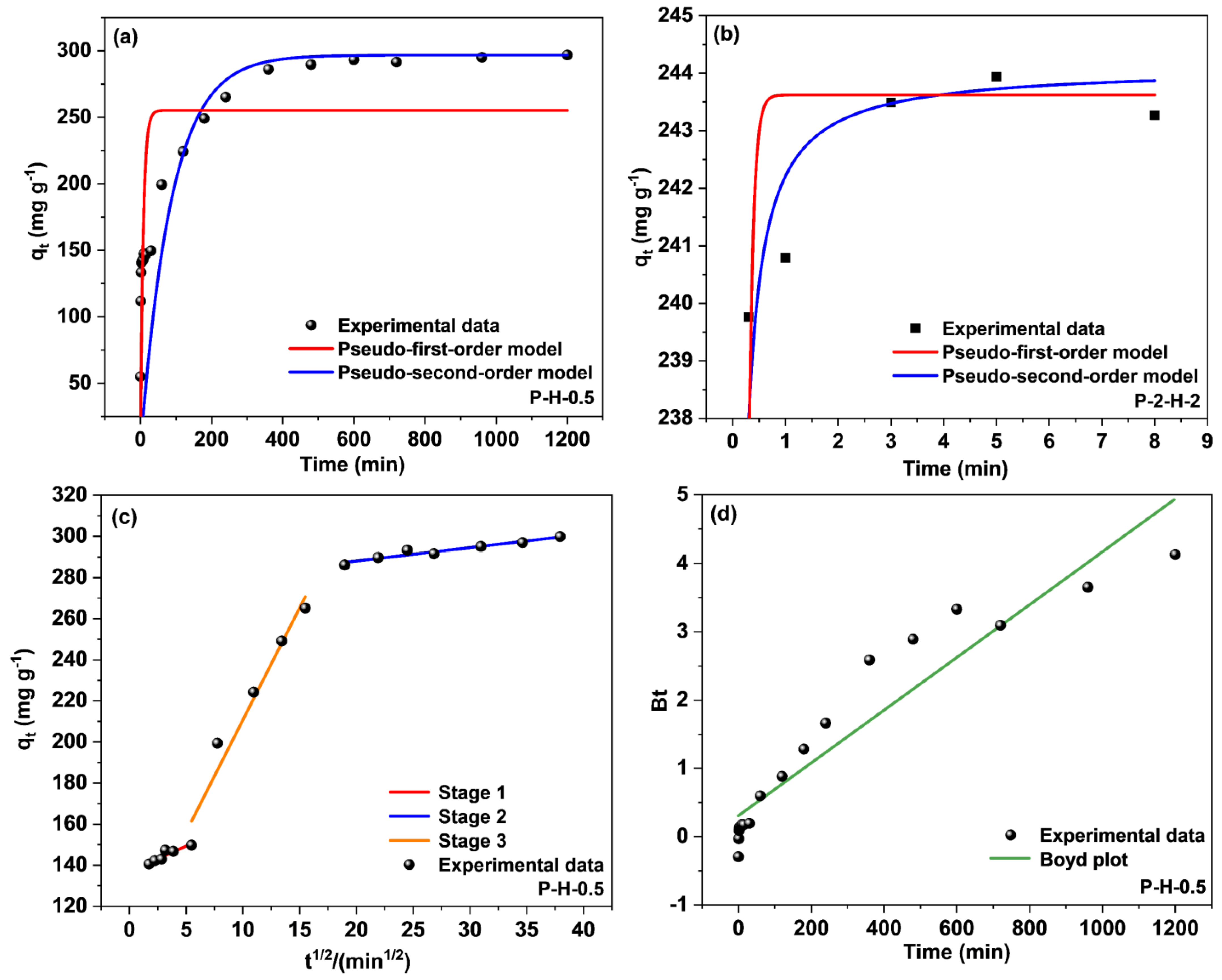
3.3.2. Contact Time Effect and Adsorption Kinetics Analysis
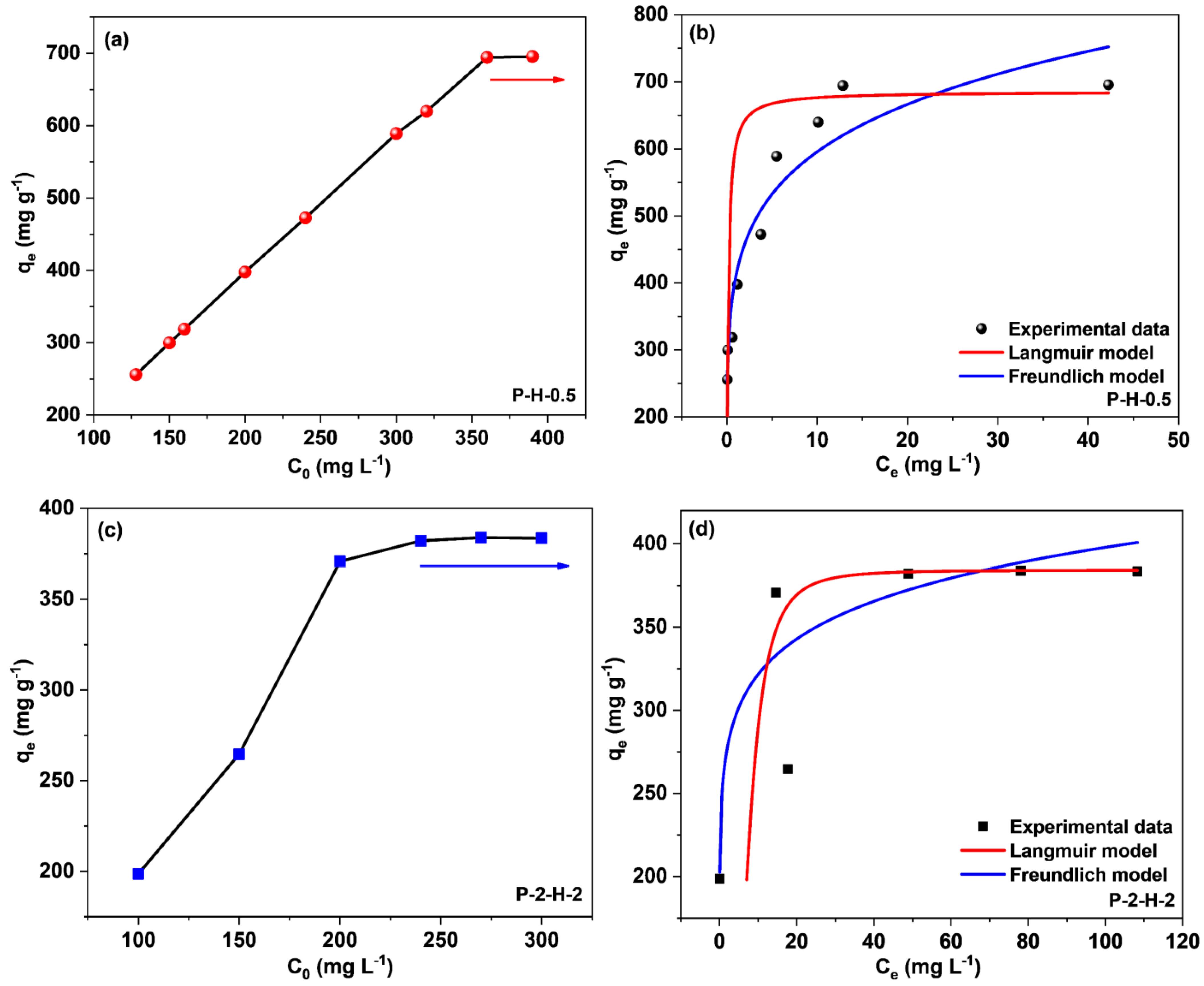
3.3.3. Initial Concentration Effect and Adsorption Isotherms Analysis
3.3.4. Adsorption Thermodynamics
3.3.5. Practical Application Strategies of PS–ACs
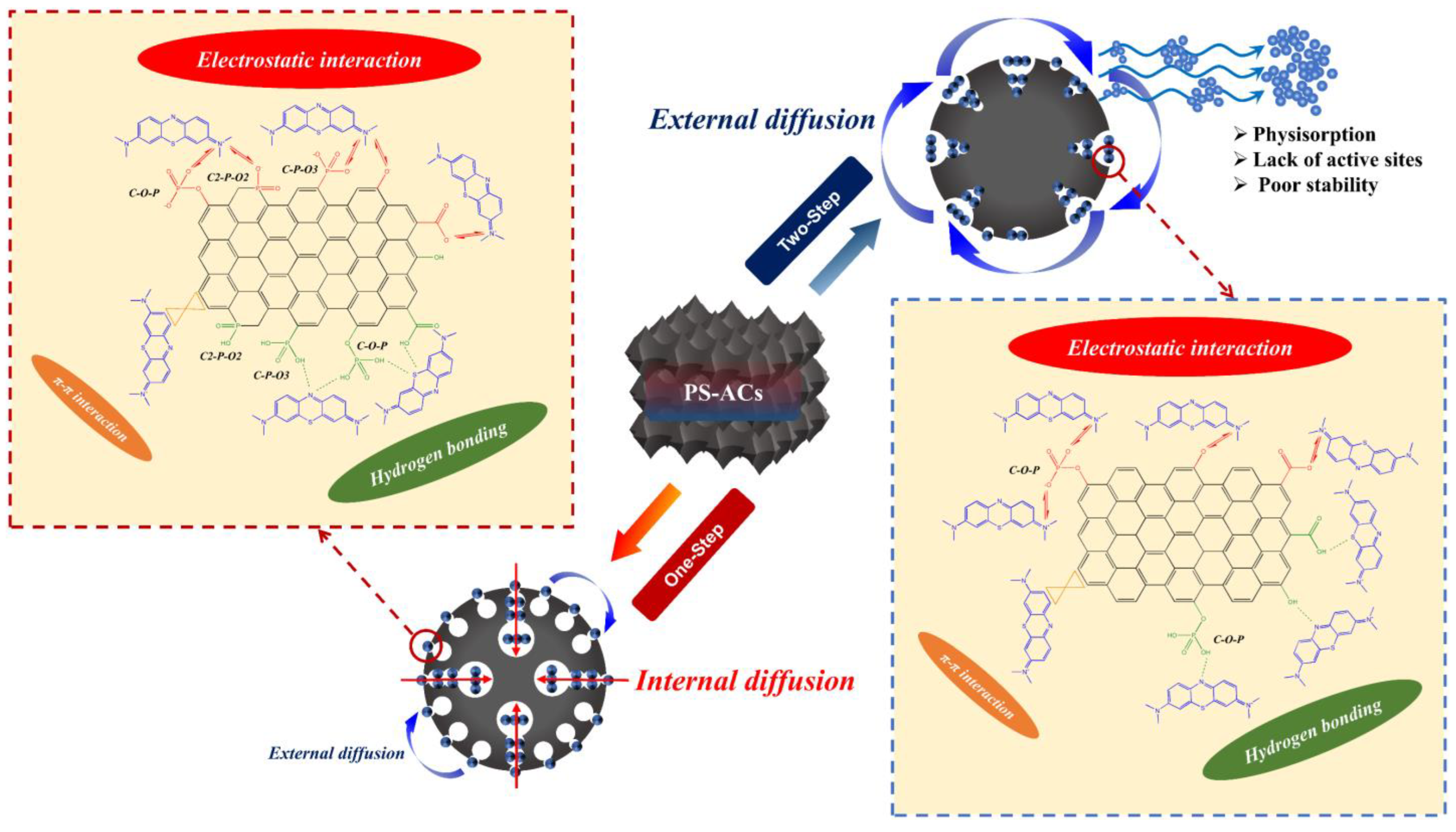
3.4. Summary of the Relevant Mechanism
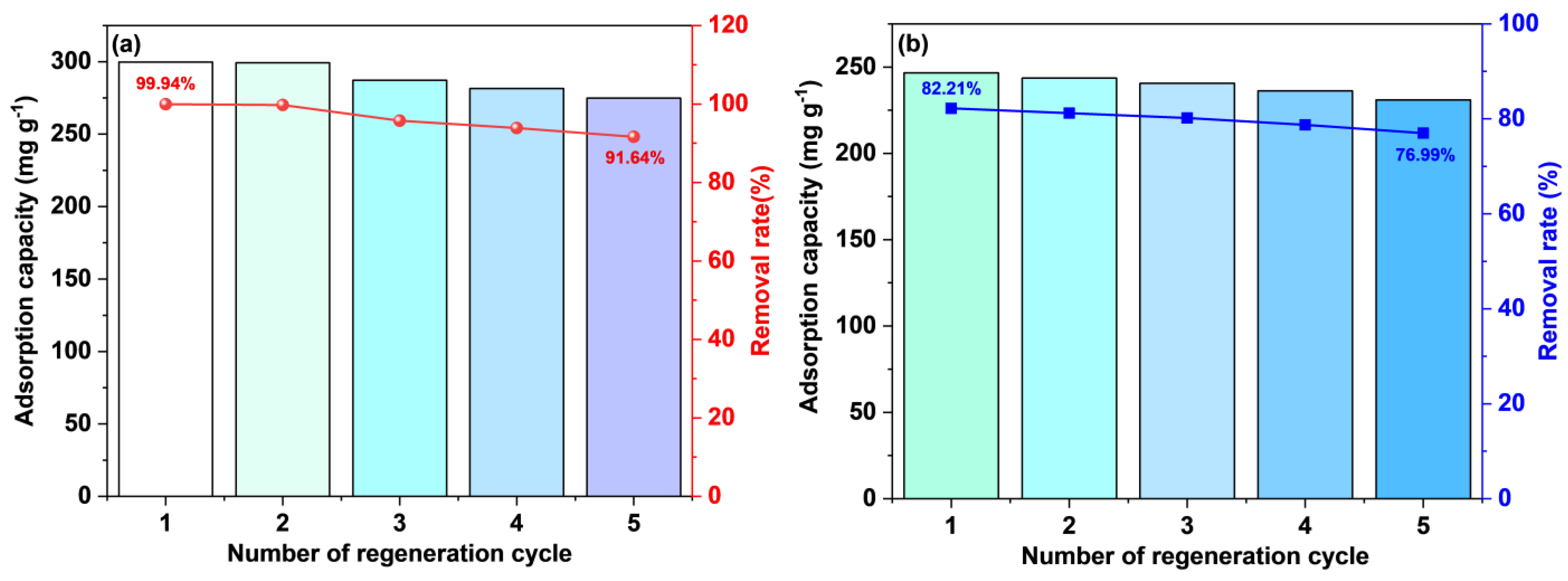
3.5. Reusability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hekmatnia, M.; Ardakani, A.F.; Isanezhad, A.; Monibi, H. A Novel Classification of Virtual Water Trade for the Sustainability of Global Freshwater Resources; Springer: Dordrecht, The Netherlands, 2023; ISBN 0123456789. [Google Scholar]
- Tkaczyk, A.; Mitrowska, K.; Posyniak, A. Synthetic Organic Dyes as Contaminants of the Aquatic Environment and Their Implications for Ecosystems: A Review. Sci. Total Environ. 2020, 717, 137222. [Google Scholar] [CrossRef] [PubMed]
- Rao, H.; Qi, W.; Su, R.; He, Z.; Peng, X. Mechanistic and Conformational Studies on the Interaction of Human Serum Albumin with Rhodamine B by NMR, Spectroscopic and Molecular Modeling Methods. J. Mol. Liq. 2020, 316, 113889. [Google Scholar] [CrossRef]
- Tran, T.H.; Le, A.H.; Pham, T.H.; Nguyen, D.T.; Chang, S.W.; Chung, W.J.; Nguyen, D.D. Adsorption Isotherms and Kinetic Modeling of Methylene Blue Dye onto a Carbonaceous Hydrochar Adsorbent Derived from Coffee Husk Waste. Sci. Total Environ. 2020, 725, 138325. [Google Scholar] [CrossRef] [PubMed]
- Caicedo, D.F.; Dos Reis, G.S.; Lima, E.C.; De Brum, I.A.S.; Thue, P.S.; Cazacliu, B.G.; Lima, D.R.; Dos Santos, A.H.; Dotto, G.L. Efficient Adsorbent Based on Construction and Demolition Wastes Functionalized with 3-Aminopropyltriethoxysilane (APTES) for the Removal Ciprofloxacin from Hospital Synthetic Effluents. J. Environ. Chem. Eng. 2020, 8, 103875. [Google Scholar] [CrossRef]
- Cheng, L.; Ji, Y.; Liu, X.; Mu, L.; Zhu, J. Sorption Mechanism of Organic Dyes on a Novel Self–Nitrogen–Doped Porous Graphite Biochar: Coupling DFT Calculations with Experiments. Chem. Eng. Sci. 2021, 242, 116739. [Google Scholar] [CrossRef]
- Sahu, S.; Pahi, S.; Tripathy, S.; Singh, S.K.; Behera, A.; Sahu, U.K.; Patel, R.K. Adsorption of Methylene Blue on Chemically Modified Lychee Seed Biochar: Dynamic, Equilibrium, and Thermodynamic Study. J. Mol. Liq. 2020, 315, 113743. [Google Scholar] [CrossRef]
- Wang, X.; Bayan, M.R.; Yu, M.; Ludlow, D.K.; Liang, X. Atomic Layer Deposition Surface Functionalized Biochar for Adsorption of Organic Pollutants: Improved Hydrophilia and Adsorption Capacity. Int. J. Environ. Sci. Technol. 2017, 14, 1825–1834. [Google Scholar] [CrossRef]
- Xiang, J.; Luo, B.X.; Li, J.M.; Mi, Y.; Tian, B.; Gong, S.J.; Zhou, Y.R.; Ma, T.W. Development of KOH and H3PO4–Modified Composite Biochar from Corn Straw and Activated Sludge for Removing Methylene Blue. Int. J. Environ. Sci. Technol. 2022, 20, 1673–1688. [Google Scholar] [CrossRef]
- Liu, X.; He, C.; Yu, X.; Bai, Y.; Ye, L.; Wang, B.; Zhang, L. Net–like Porous Activated Carbon Materials from Shrimp Shell by Solution–Processed Carbonization and H3PO4 Activation for Methylene Blue Adsorption. Powder Technol. 2018, 326, 181–189. [Google Scholar] [CrossRef]
- Dai, L.; Zhu, W.; He, L.; Tan, F.; Zhu, N.; Zhou, Q.; He, M.; Hu, G. Calcium–Rich Biochar from Crab Shell: An Unexpected Super Adsorbent for Dye Removal. Bioresour. Technol. 2018, 267, 510–516. [Google Scholar] [CrossRef]
- Zazycki, M.A.; Borba, P.A.; Silva, R.N.F.; Peres, E.C.; Perondi, D.; Collazzo, G.C.; Dotto, G.L. Chitin Derived Biochar as an Alternative Adsorbent to Treat Colored Effluents Containing Methyl Violet Dye. Adv. Powder Technol. 2019, 30, 1494–1503. [Google Scholar] [CrossRef]
- Mahdi, Z.; El Hanandeh, A.; Yu, Q. Influence of Pyrolysis Conditions on Surface Characteristics and Methylene Blue Adsorption of Biochar Derived from Date Seed Biomass. Waste Biomass Valorization 2017, 8, 2061–2073. [Google Scholar] [CrossRef]
- Han, Q.; Wang, J.; Goodman, B.A.; Xie, J.; Liu, Z. High Adsorption of Methylene Blue by Activated Carbon Prepared from Phosphoric Acid Treated Eucalyptus Residue. Powder Technol. 2020, 366, 239–248. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, J.; Li, D.; Liu, C.; Lu, Y.; Lin, X.; Zheng, Z. Insight into the KOH/KMnO4 Activation Mechanism of Oxygen–Enriched Hierarchical Porous Biochar Derived from Biomass Waste by in–Situ Pyrolysis for Methylene Blue Enhanced Adsorption. J. Anal. Appl. Pyrolysis 2021, 158, 105269. [Google Scholar] [CrossRef]
- Viswanathan, S.P.; Njazhakunnathu, G.V.; Neelamury, S.P.; Padmakumar, B.; Ambatt, T.P. The Efficiency of Aquatic Weed–Derived Biochar in Enhanced Removal of Cationic Dyes from Aqueous Medium. Biomass Convers. Biorefinery 2022. [Google Scholar] [CrossRef]
- Hmid, A.; Mondelli, D.; Fiore, S.; Fanizzi, F.P.; Al Chami, Z.; Dumontet, S. Production and Characterization of Biochar from Three–Phase Olive Mill Waste through Slow Pyrolysis. Biomass Bioenergy 2014, 71, 330–339. [Google Scholar] [CrossRef]
- Bridgwater, A.V. Review of Fast Pyrolysis of Biomass and Product Upgrading. Biomass Bioenergy 2012, 38, 68–94. [Google Scholar] [CrossRef]
- Wang, Z.; Wu, J.; He, T.; Wu, J. Corn Stalks Char from Fast Pyrolysis as Precursor Material for Preparation of Activated Carbon in Fluidized Bed Reactor. Bioresour. Technol. 2014, 167, 551–554. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, X.; Qi, F.; Shi, H.; Zhang, Y.; Ma, P. Efficient Preparation of P–doped Carbon with Ultra–High Mesoporous Ratio from Furfural Residue for Dye Removal. Sep. Purif. Technol. 2022, 292, 120954. [Google Scholar] [CrossRef]
- Fu, Y.; Shen, Y.; Zhang, Z.; Ge, X.; Chen, M. Activated Bio–Chars Derived from Rice Husk via One– and Two–Step KOH–Catalyzed Pyrolysis for Phenol Adsorption. Sci. Total Environ. 2019, 646, 1567–1577. [Google Scholar] [CrossRef]
- Gupta, V.K.; Pathania, D.; Sharma, S.; Singh, P. Preparation of Bio–Based Porous Carbon by Microwave Assisted Phosphoric Acid Activation and Its Use for Adsorption of Cr(VI). J. Colloid Interface Sci. 2013, 401, 125–132. [Google Scholar] [CrossRef]
- Abu El-Rub, Z.; Bramer, E.A.; Brem, G. Experimental Comparison of Biomass Chars with Other Catalysts for Tar Reduction. Fuel 2008, 87, 2243–2252. [Google Scholar] [CrossRef]
- Inguanzo, M.; Menéndez, J.A.; Fuente, E.; Pis, J.J. Reactivity of Pyrolyzed Sewage Sludge in Air and CO2. J. Anal. Appl. Pyrolysis 2001, 58–59, 943–954. [Google Scholar] [CrossRef]
- Tezcan Un, U.; Ates, F. Low-Cost Adsorbent Prepared from Poplar Sawdust for Removal of Disperse Orange 30 Dye from Aqueous Solutions. Int. J. Environ. Sci. Technol. 2019, 16, 899–908. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, X.; Yang, R.; Li, G.; Hu, C. The Role of H3PO4 in the Preparation of Activated Carbon from NaOH–Treated Rice Husk Residue. RSC Adv. 2015, 5, 32626–32636. [Google Scholar] [CrossRef]
- Crombie, K.; Mašek, O. Pyrolysis Biochar Systems, Balance between Bioenergy and Carbon Sequestration. GCB Bioenergy 2015, 7, 349–361. [Google Scholar] [CrossRef]
- Chen, D.; Li, Y.; Cen, K.; Luo, M.; Li, H.; Lu, B. Pyrolysis Polygeneration of Poplar Wood: Effect of Heating Rate and Pyrolysis Temperature. Bioresour. Technol. 2016, 218, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Eom, I.Y.; Lee, S.M.; Choi, D.; Yeo, H.; Choi, I.G.; Choi, J.W. Investigation of Physicochemical Properties of Biooils Produced from Yellow Poplar Wood (Liriodendron tulipifera) at Various Temperatures and Residence Times. J. Anal. Appl. Pyrolysis 2011, 92, 2–9. [Google Scholar] [CrossRef]
- Prahas, D.; Kartika, Y.; Indraswati, N.; Ismadji, S. Activated Carbon from Jackfruit Peel Waste by H3PO4 Chemical Activation: Pore Structure and Surface Chemistry Characterization. Chem. Eng. J. 2008, 140, 32–42. [Google Scholar] [CrossRef]
- Jagtoyen, M.; Derbyshire, F. Activated Carbons from Yellow Poplar and White Oak by H3PO4 Activation. Carbon 1998, 36, 1085–1097. [Google Scholar] [CrossRef]
- Ahmed, M.J.; Theydan, S.K. Optimization of Microwave Preparation Conditions for Activated Carbon from Albizia Lebbeck Seed Pods for Methylene Blue Dye Adsorption. J. Anal. Appl. Pyrolysis 2014, 105, 199–208. [Google Scholar] [CrossRef]
- Deng, H.; Yang, L.; Tao, G.; Dai, J. Preparation and Characterization of Activated Carbon from Cotton Stalk by Microwave Assisted Chemical Activation−Application in Methylene Blue Adsorption from Aqueous Solution. J. Hazard. Mater. 2009, 166, 1514–1521. [Google Scholar] [CrossRef]
- Dural, M.U.; Cavas, L.; Papageorgiou, S.K.; Katsaros, F.K. Methylene Blue Adsorption on Activated Carbon Prepared from Posidonia oceanica (L.) Dead Leaves: Kinetics and Equilibrium Studies. Chem. Eng. J. 2011, 168, 77–85. [Google Scholar] [CrossRef]
- Pezoti, O.; Cazetta, A.L.; Souza, I.P.A.F.; Bedin, K.C.; Martins, A.C.; Silva, T.L.; Almeida, V.C. Adsorption Studies of Methylene Blue onto ZnCl2–Activated Carbon Produced from Buriti Shells (Mauritia flexuosa L.). J. Ind. Eng. Chem. 2014, 20, 4401–4407. [Google Scholar] [CrossRef]
- Wu, M.; Guo, Q.; Fu, G. Preparation and Characteristics of Medicinal Activated Carbon Powders by CO2 Activation of Peanut Shells. Powder Technol. 2013, 247, 188–196. [Google Scholar] [CrossRef]
- Zhang, H.; Liao, W.; Zhou, X.; Shao, J.; Chen, Y.; Zhang, S.; Chen, H. Coeffect of Pyrolysis Temperature and Potassium Phosphate Impregnation on Characteristics, Stability, and Adsorption Mechanism of Phosphorus–Enriched Biochar. Bioresour. Technol. 2022, 344, 126273. [Google Scholar] [CrossRef]
- Spokas, K.A. Review of the Stability of Biochar in Soils: Predictability of O:C Molar Ratios. Carbon Manag. 2010, 1, 289–303. [Google Scholar] [CrossRef]
- Kaouah, F.; Boumaza, S.; Berrama, T.; Trari, M.; Bendjama, Z. Preparation and Characterization of Activated Carbon from Wild Olive Cores (Oleaster) by H3PO4 for the Removal of Basic Red 46. J. Clean. Prod. 2013, 54, 296–306. [Google Scholar] [CrossRef]
- Qin, Q.; Xu, Y. Enhanced Nitrobenzene Adsorption in Aqueous Solution by Surface Silylated MCM-41. Microporous Mesoporous Mater. 2016, 232, 143–150. [Google Scholar] [CrossRef]
- Li, H.Z.; Zhang, Y.N.; Guo, J.Z.; Lv, J.Q.; Huan, W.W.; Li, B. Preparation of Hydrochar with High Adsorption Performance for Methylene Blue by Co-Hydrothermal Carbonization of Polyvinyl Chloride and Bamboo. Bioresour. Technol. 2021, 337, 125442. [Google Scholar] [CrossRef]
- Zuo, S.; Liu, J.; Yang, J.; Cai, X. Effects of the Crystallinity of Lignocellulosic Material on the Porosity of Phosphoric Acid–Activated Carbon. Carbon 2009, 47, 3578–3580. [Google Scholar] [CrossRef]
- Yang, T.; Lua, A.C. Textural and Chemical Properties of Zinc Chloride Activated Carbons Prepared from Pistachio–Nut Shells. Mater. Chem. Phys. 2006, 100, 438–444. [Google Scholar] [CrossRef]
- Yang, Z.; Gleisner, R.; Mann, D.H.; Xu, J.; Jiang, J.; Zhu, J.Y. Lignin Based Activated Carbon Using H3PO4 Activation. Polymers 2020, 12, 2829. [Google Scholar] [CrossRef]
- Zulfiqar, M.; Rabat, N.E.; Bahadar, A.; Lashari, N.; Mahnashi, M.H.; Alqarni, A.O. Development of Elaeis Guineensis/Polyvinyl Alcohol/Carbon Nanotube Composites for Efficient Adsorption of Dye: Experimental and Theoretical Approach. Int. J. Environ. Sci. Technol. 2022, 19, 6499–6520. [Google Scholar] [CrossRef]
- Bernardino, C.A.R.; Mahler, C.F.; Veloso, M.C.C.; Romeiro, G.A. Preparation of Biochar from Sugarcane By-Product Filter Mud by Slow Pyrolysis and Its Use Like Adsorbent. Waste Biomass Valorization 2017, 8, 2511–2521. [Google Scholar] [CrossRef]
- Puziy, A.M.; Poddubnaya, O.I.; Martínez-Alonso, A.; Suárez-García, F.; Tascón, J.M.D. Surface Chemistry of Phosphorus–Containing Carbons of Lignocellulosic Origin. Carbon 2005, 43, 2857–2868. [Google Scholar] [CrossRef]
- Tran, T.H.; Le, H.H.; Pham, T.H.; Nguyen, D.T.; La, D.D.; Chang, S.W.; Lee, S.M.; Chung, W.J.; Nguyen, D.D. Comparative Study on Methylene Blue Adsorption Behavior of Coffee Husk–Derived Activated Carbon Materials Prepared Using Hydrothermal and Soaking Methods. J. Environ. Chem. Eng. 2021, 9, 105362. [Google Scholar] [CrossRef]
- Guo, Z.; Zhang, J.; Liu, H. Ultra–High Rhodamine B Adsorption Capacities from an Aqueous Solution by Activated Carbon Derived from: Phragmites Australis Doped with Organic Acid by Phosphoric Acid Activation. RSC Adv. 2016, 6, 40818–40827. [Google Scholar] [CrossRef]
- Puziy, A.M.; Poddubnaya, O.I.; Socha, R.P.; Gurgul, J.; Wisniewski, M. XPS and NMR Studies of Phosphoric Acid Activated Carbons. Carbon 2008, 46, 2113–2123. [Google Scholar] [CrossRef]
- Ye, X.N.; Lu, Q.; Wang, X.; Guo, H.Q.; Cui, M.S.; Dong, C.Q.; Yang, Y.P. Catalytic Fast Pyrolysis of Cellulose and Biomass to Selectively Produce Levoglucosenone Using Activated Carbon Catalyst. ACS Sustain. Chem. Eng. 2017, 5, 10815–10825. [Google Scholar] [CrossRef]
- Ma, J.; Shen, Y.; Shen, C.; Wen, Y.; Liu, W. Al–Doping Chitosan–Fe(III) Hydrogel for the Removal of Fluoride from Aqueous Solutions. Chem. Eng. J. 2014, 248, 98–106. [Google Scholar] [CrossRef]
- Zhou, Y.; Sun, L.; Wang, H.; Liang, W.; Yang, J.; Wang, L.; Shuang, S. Investigation on the Uptake and Release Ability of β–Cyclodextrin Functionalized Fe3O4 Magnetic Nanoparticles by Methylene Blue. Mater. Chem. Phys. 2016, 170, 83–89. [Google Scholar] [CrossRef]
- Lomenech, C.; Hurel, C.; Messina, L.; Schembri, M.; Tosi, P.; Orange, F.; Georgi, F.; Mija, A.; Kuzhir, P. A Humins–Derived Magnetic Biochar for Water Purification by Adsorption and Magnetic Separation. Waste Biomass Valorization 2021, 12, 6497–6512. [Google Scholar] [CrossRef]
- Basha, S.; Murthy, Z.V.P.; Jha, B. Sorption of Hg(II) onto Carica Papaya: Experimental Studies and Design of Batch Sorber. Chem. Eng. J. 2009, 147, 226–234. [Google Scholar] [CrossRef]
- Kumar, K.V.; Ramamurthi, V.; Sivanesan, S. Modeling the Mechanism Involved during the Sorption of Methylene Blue onto Fly Ash. J. Colloid Interface Sci. 2005, 284, 14–21. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Y.; Li, S.; Zhong, W.; Wei, W. Enhanced Methylene Blue Adsorption onto Activated Reed–Derived Biochar by Tannic Acid. J. Mol. Liq. 2018, 268, 658–666. [Google Scholar] [CrossRef]
- Rashid, J.; Tehreem, F.; Rehman, A.; Kumar, R. Synthesis Using Natural Functionalization of Activated Carbon from Pumpkin Peels for Decolourization of Aqueous Methylene Blue. Sci. Total Environ. 2019, 671, 369–376. [Google Scholar] [CrossRef]
- Boukhemkhem, A.; Rida, K. Improvement Adsorption Capacity of Methylene Blue onto Modified Tamazert Kaolin. Adsorpt. Sci. Technol. 2017, 35, 753–773. [Google Scholar] [CrossRef]
- Koyuncu, H.; Kul, A.R. Biosorption Study for Removal of Methylene Blue Dye from Aqueous Solution Using a Novel Activated Carbon Obtained from Nonliving Lichen (Pseudevernia furfuracea (L.) Zopf.). Surf. Interfaces 2020, 19, 100527. [Google Scholar] [CrossRef]
- Lian, F.; Cui, G.; Liu, Z.; Duo, L.; Zhang, G.; Xing, B. One–Step Synthesis of a Novel N–Doped Microporous Biochar Derived from Crop Straws with High Dye Adsorption Capacity. J. Environ. Manag. 2016, 176, 61–68. [Google Scholar] [CrossRef]
- Doǧan, M.; Alkan, M.; Demirbaş, Ö.; Özdemir, Y.; Özmetin, C. Adsorption Kinetics of Maxilon Blue GRL onto Sepiolite from Aqueous Solutions. Chem. Eng. J. 2006, 124, 89–101. [Google Scholar] [CrossRef]
- Yu, P.; Luo, Z.; Wang, Q.; Fang, M.; Zhou, J.; Wang, W.; Liang, X.; Cai, W. Activated Carbon–Based CO2uptake Evaluation at Different Temperatures: The Correlation Analysis and Coupling Effects of the Preparation Conditions. J. CO2 Util. 2020, 40, 101214. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SSA a (m2·g−1) | VTotal b (cm3·g−1) | VMicro c (cm3·g−1) | D1 d (nm) | D2 e (nm) |
|---|---|---|---|---|---|
| P–H–0.5 | 1004.15 | 0.66 | 0.11 | 2.63 | 3.04 |
| P–2–H–2 | 3.65 | 4.5 × 10−3 | 1.6 × 10−4 | 4.95 | 10.29 |
| P–H–0.5–S | 927.75 | 0.62 | 0.12 | 2.68 | 3.42 |
| P–2–H–2–S | 2.44 | 2.5 × 10−3 | 9.1 × 10−4 | 4.14 | 23.93 |
| PC–2 | 194.46 | 0.11 | 7.3 × 10−2 | 2.34 | 3.75 |
| Sample | Parameters | PFO | PSO | Parameters | Intra–Particle Diffusion | ||
|---|---|---|---|---|---|---|---|
| Stage 1 | Stage 2 | Stage 3 | |||||
| P–H–0.5 | qe, cal (mg g−1) a | 254.09 | 298.81 | C (mg g−1) | 137.04 | 101.75 | 275.23 |
| k1 (min−1) or k2 (g mg−1 min−1) | 0.13 | 1.61 × 10−3 | kp (mg g−1 min−1/2) | 2.46 | 10.91 | 0.64 | |
| R2 | 0.5698 | 0.9698 | R2 | 0.823 | 0.9431 | 0.9192 | |
| P–2–H–2 | qe, cal (mg g−1) | 243.63 | 243.75 | – | – | – | – |
| k1 (min−1) or k2 (g mg−1 min−1) | 13.81 | 0.69 | – | – | – | – | |
| R2 | 0.4176 | 0.9391 | – | – | – | – | |
| Sample | Parameters | Langmuir | Parameters | Freundlich |
|---|---|---|---|---|
| P–H–0.5 | qm (mg g−1) a | 685.43 | n | 6.18 |
| b (L mg−1) | 7.47 | kF (L g−1) | 410.29 | |
| R2 | 0.9087 | R2 | 0.8368 | |
| P–2–H–2 | qm (mg g−1) | 384.25 | n | 10.76 |
| b (L mg−1) | 0.028 | kF (L g−1) | 252.75 | |
| R2 | 0.9569 | R2 | 0.7758 |
| Feedstock | Preparation Method | Activating Reagent | Pyrolysis/Activation Temperature (K) | Residence Time (min) | Adsorption Capacity (mg·g–1) | Adsorption Time (min) | Reference |
|---|---|---|---|---|---|---|---|
| PS | One–step | H3PO4 | 723 | 30 | 695.54 | 240 | This study |
| PS | Two–step | H3PO4 | 823 + 723 | 120 + 120 | 383.91 | 2 | This study |
| Reeds | Two–step | HNO3 | 773 + 298 | 120 + 720 | 48.8 | 720 | [57] |
| Pumpkin peels | One–step | – | 523 | 60 | 80.8 | 30 | [58] |
| Tamazert kaolin | Three–step | Air + HCl + NaOH | 1073 + 353 + 353 | 300 + 420 + 420 | 111 | 5 | [59] |
| Lychee seed | Two–step | KOH | 973 + Microwave activation (600 W) | 10 | 124.5 | 120 | [7] |
| Cotton stalk | One–step | ZnCl2 | Microwave activation (560 W) | 9 | 193.5 | 120 | [33] |
| Peanut shell | Two–step | CO2 | 773 + 1173 | 60 + 300 | 225.8 | 180 | [36] |
| Lichen | One–step | ZnCl2 | 1041 | 60 | 243.9 | <70 | [60] |
| Buriti fruits | Two–step | ZnCl2 + CO2 | 773 + 973 | 120 + 90 | 274.6 | 80 | [35] |
| Posidonia oceanica | One–step | ZnCl2 | 773 | 120 | 285.7 | 60 | [34] |
| Albizia lebbeck seed pods | One–step | KOH | Microwave activation (620 W) | 8 | 381.2 | 270 | [32] |
| Corn straw | One–step | NH3 | 1073 | 180 | 436 | 720 | [61] |
| Sample | T (K) | ΔG0 (kJ mol−1) | ΔH0 (kJ mol−1) | ΔS0 (kJ mol−1 K−1) | R2 |
|---|---|---|---|---|---|
| P–H–0.5 | 298 | −20.24 | −137.67 | −0.402 | 0.8952 |
| 308 | −10.13 | ||||
| 318 | −9.06 | ||||
| P–2–H–2 | 298 | −5.51 | −15.24 | −3.3 × 10−2 | 0.9696 |
| 308 | −4.89 | ||||
| 318 | −4.39 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, C.; Luo, Z.; Yu, P.; Wang, Q. Comparative Study on the Performance and Mechanism of Adsorption–Oriented Phosphorus–Modified High–Efficiency and Durable Activated Biochar from Fast Pyrolysis. Energies 2023, 16, 5363. https://doi.org/10.3390/en16145363
Sun C, Luo Z, Yu P, Wang Q. Comparative Study on the Performance and Mechanism of Adsorption–Oriented Phosphorus–Modified High–Efficiency and Durable Activated Biochar from Fast Pyrolysis. Energies. 2023; 16(14):5363. https://doi.org/10.3390/en16145363
Chicago/Turabian StyleSun, Chenhao, Zhongyang Luo, Peng Yu, and Qinhui Wang. 2023. "Comparative Study on the Performance and Mechanism of Adsorption–Oriented Phosphorus–Modified High–Efficiency and Durable Activated Biochar from Fast Pyrolysis" Energies 16, no. 14: 5363. https://doi.org/10.3390/en16145363
APA StyleSun, C., Luo, Z., Yu, P., & Wang, Q. (2023). Comparative Study on the Performance and Mechanism of Adsorption–Oriented Phosphorus–Modified High–Efficiency and Durable Activated Biochar from Fast Pyrolysis. Energies, 16(14), 5363. https://doi.org/10.3390/en16145363