
Structural and Electrochemical Properties of Layered P2-Na0.8Co0.8Ti0.2O2 Cathode in Sodium-Ion Batteries

 , ,
, ,  ,
,  ,
,
Abstract
:1. Introduction
2. Experimental Section
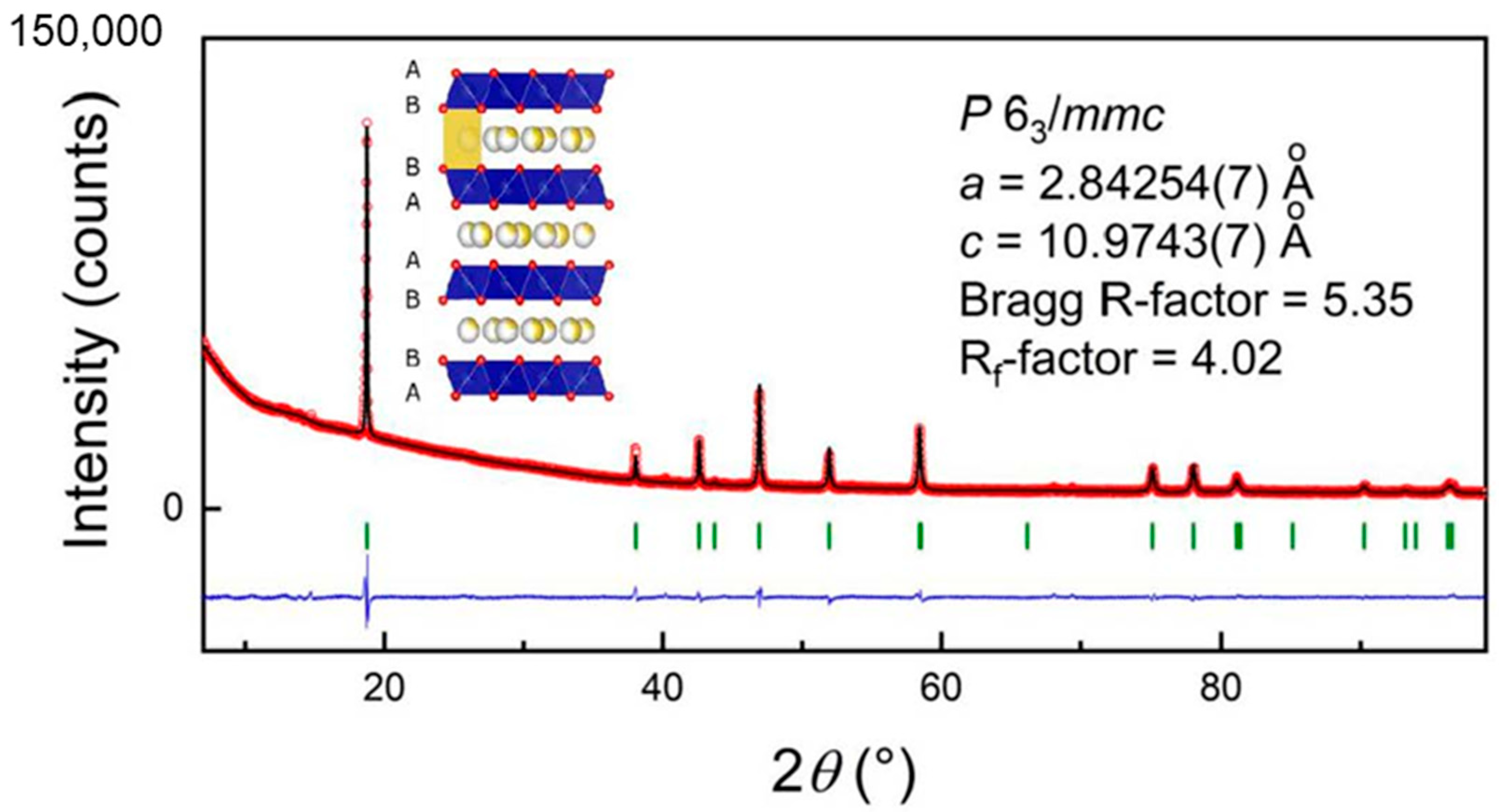
2.1. Sample Preparation and Pre-Characterization
2.2. Electrochemical Characterization
2.3. Operando X-ray Diffraction Studies
2.4. Operando XAS Studies
3. Results and Discussion
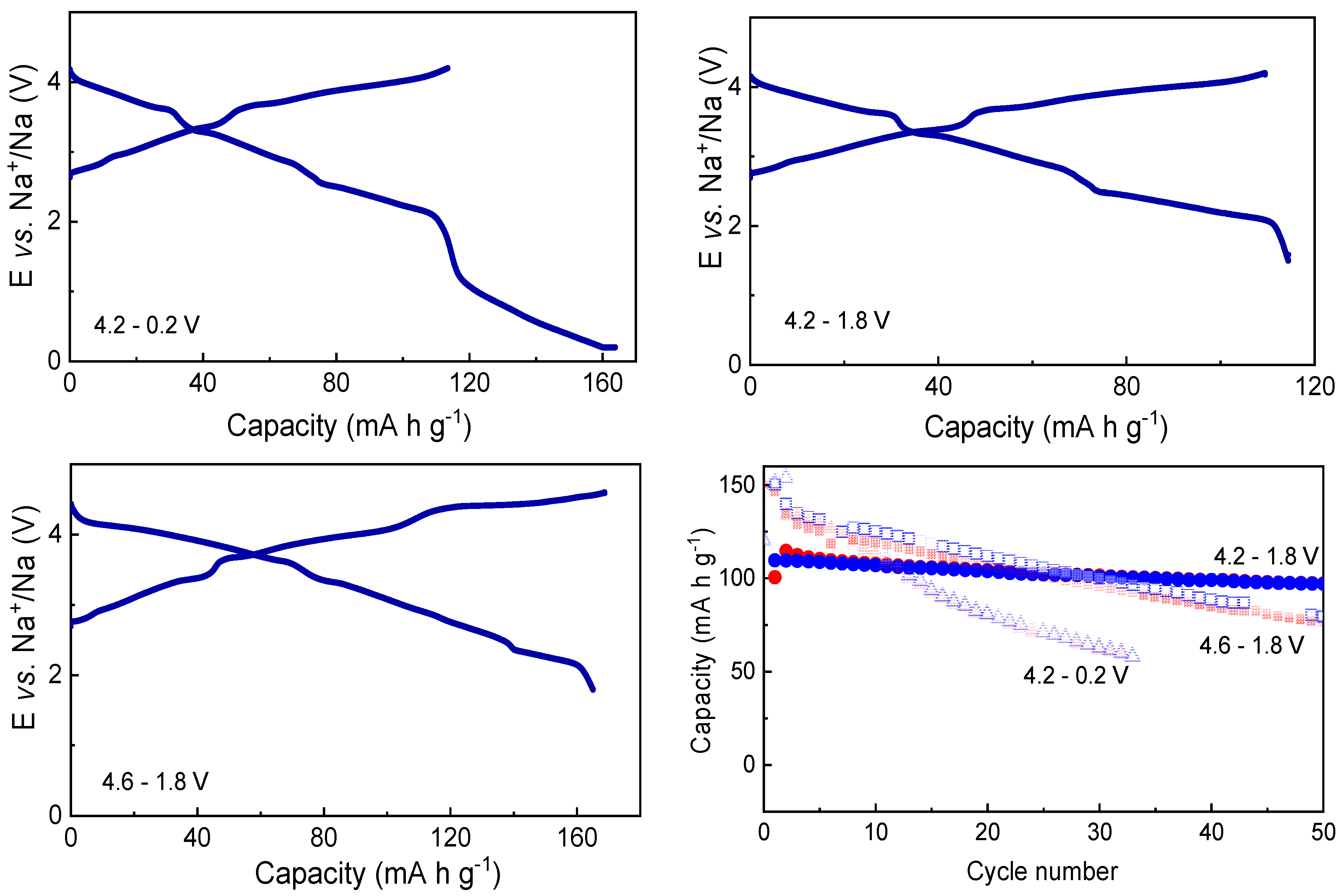
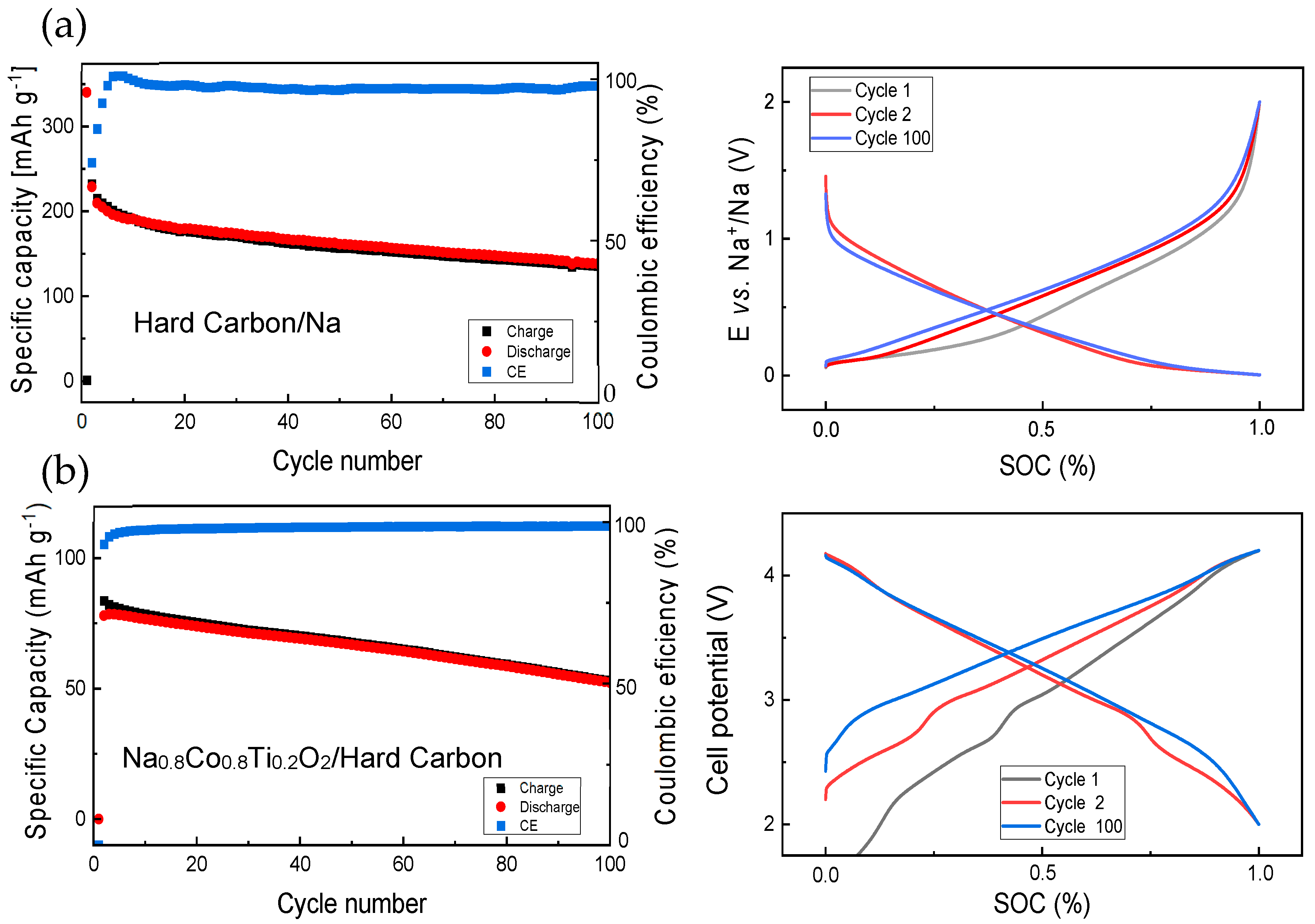
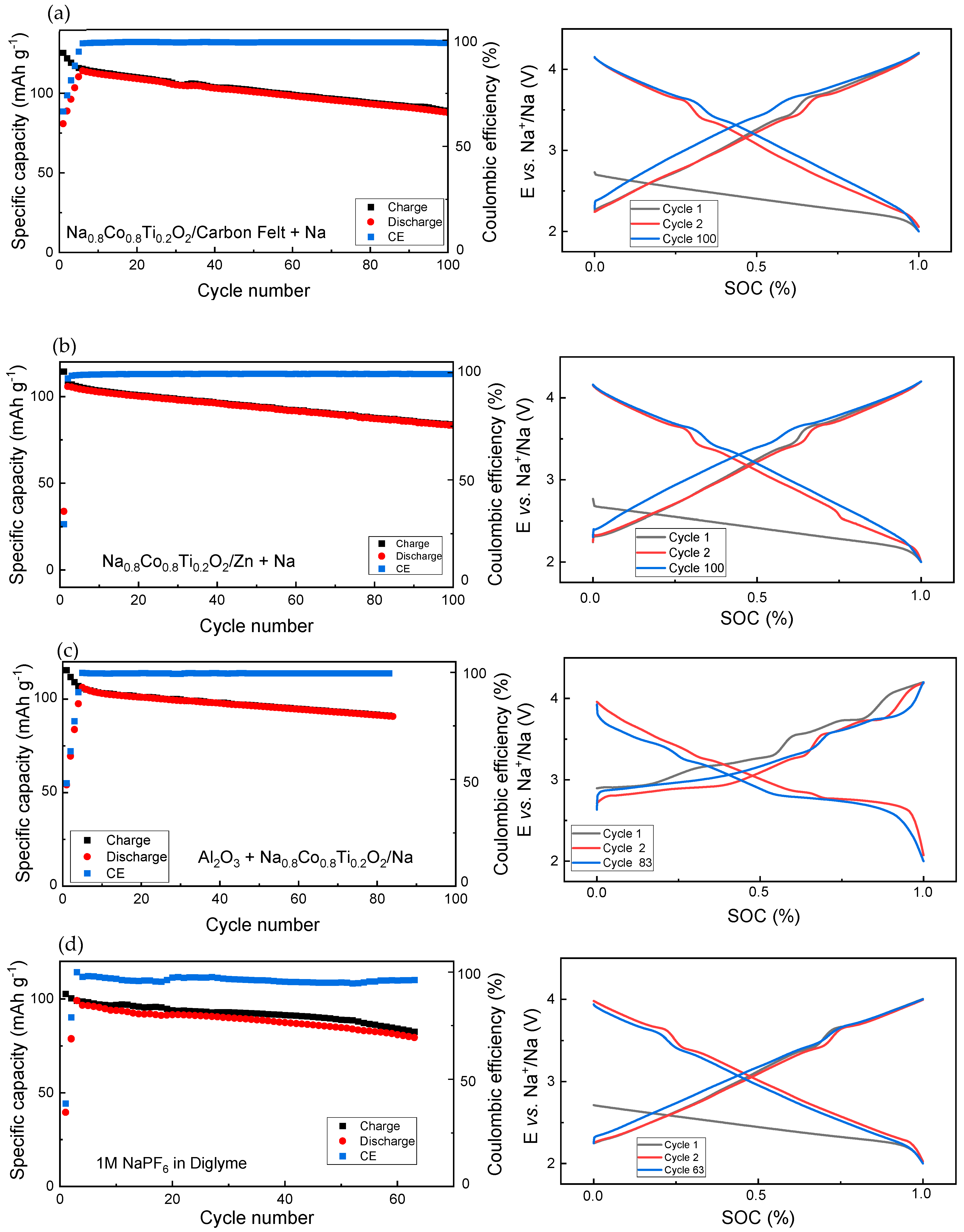
3.1. Electrochemical Studies
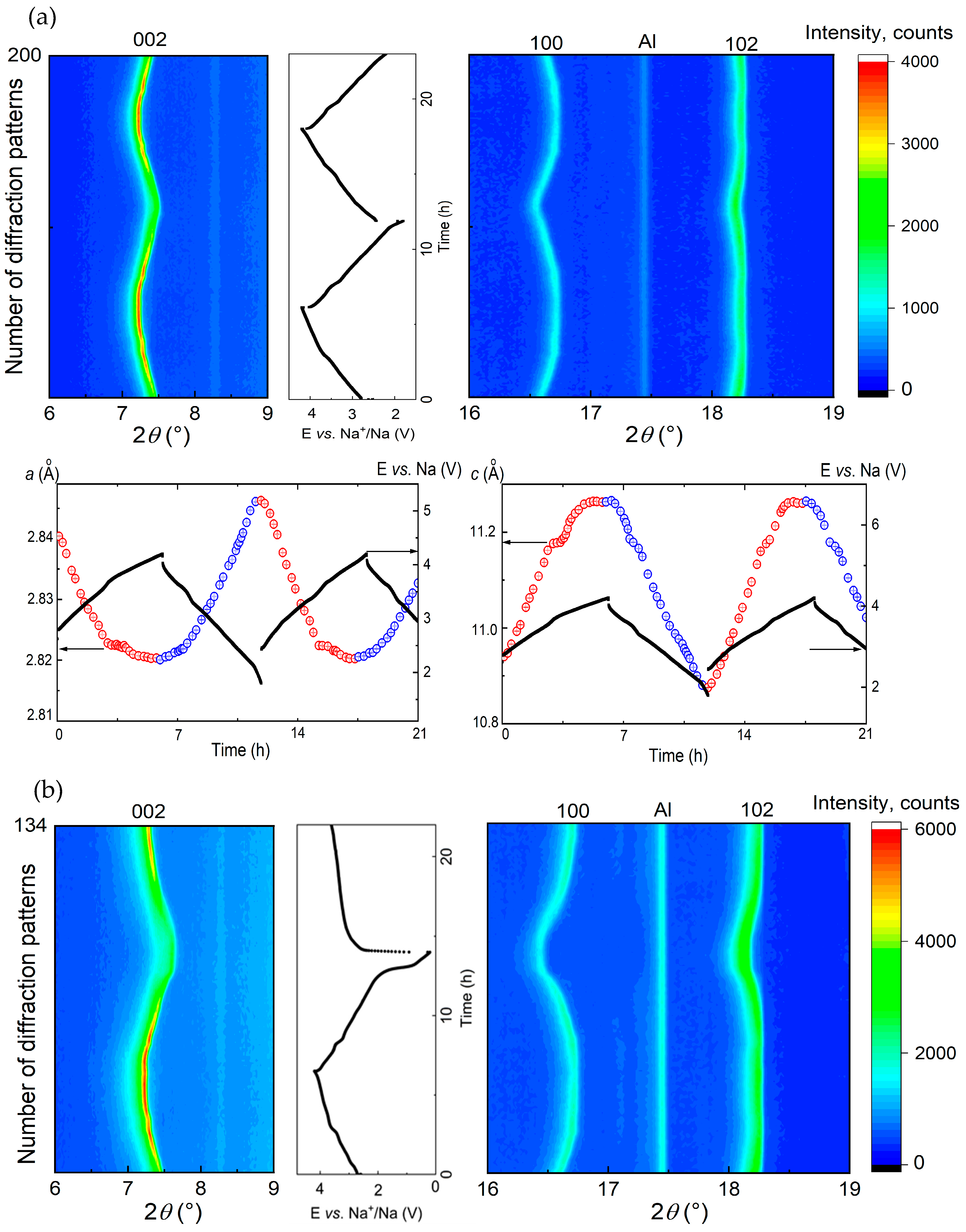
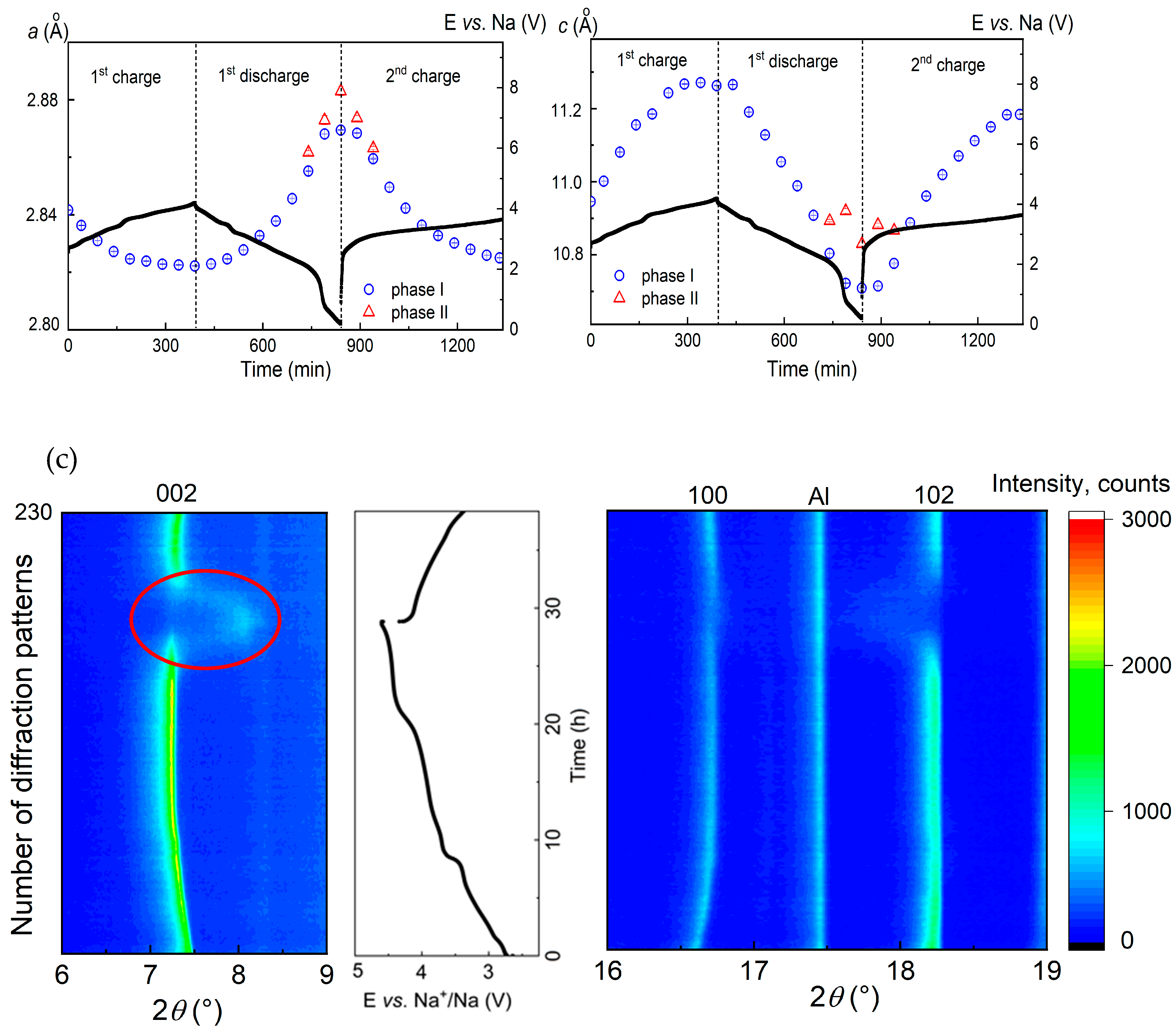
3.2. Structural Characterization Using Operando XRD Analysis
3.2.1. Voltage Window of 4.2–1.8 V vs. Na+/Na
3.2.2. Voltage Window of 4.2–0.2 V vs. Na+/Na
3.2.3. Voltage Window of 4.6–1.8 V vs. Na+/Na
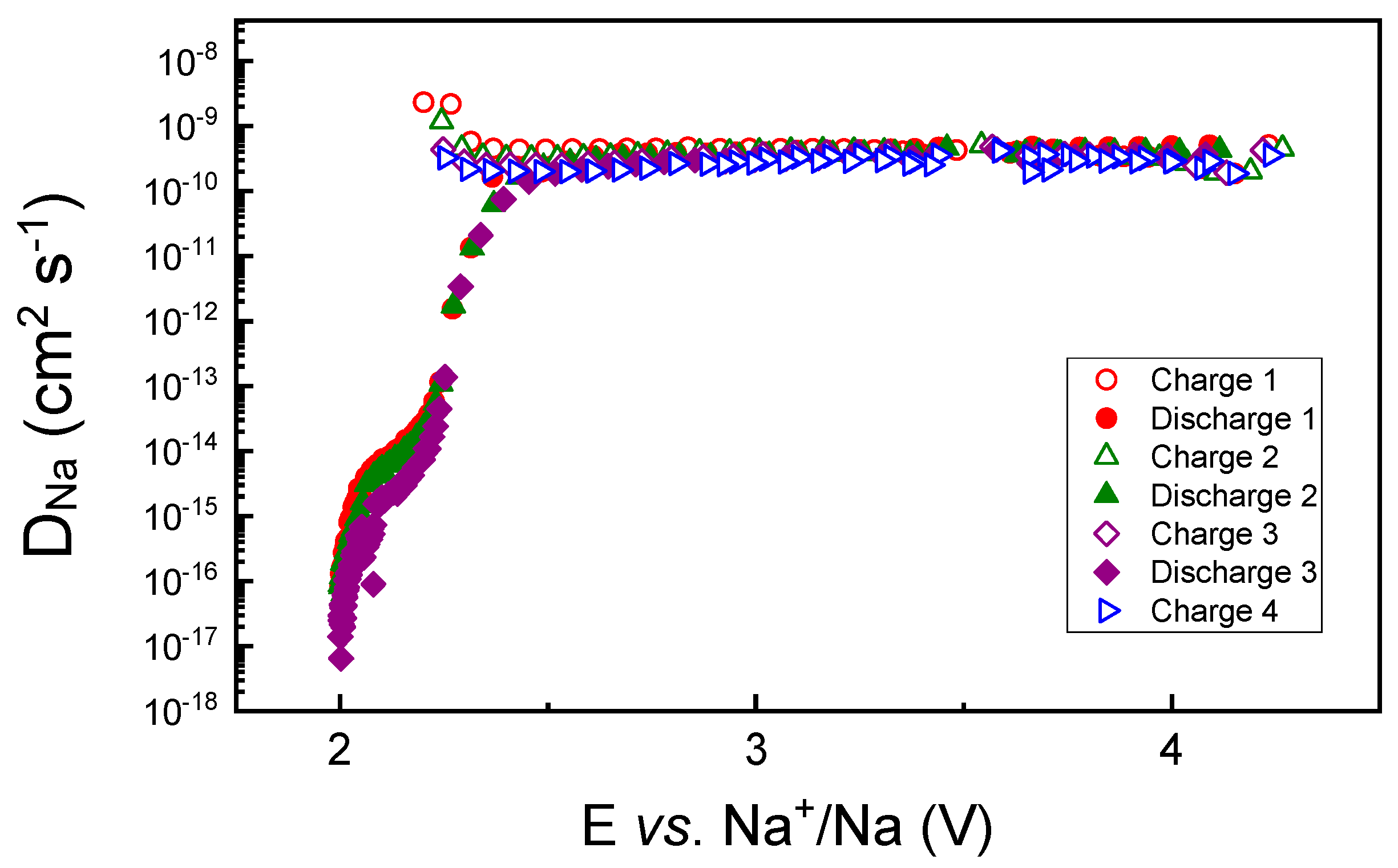
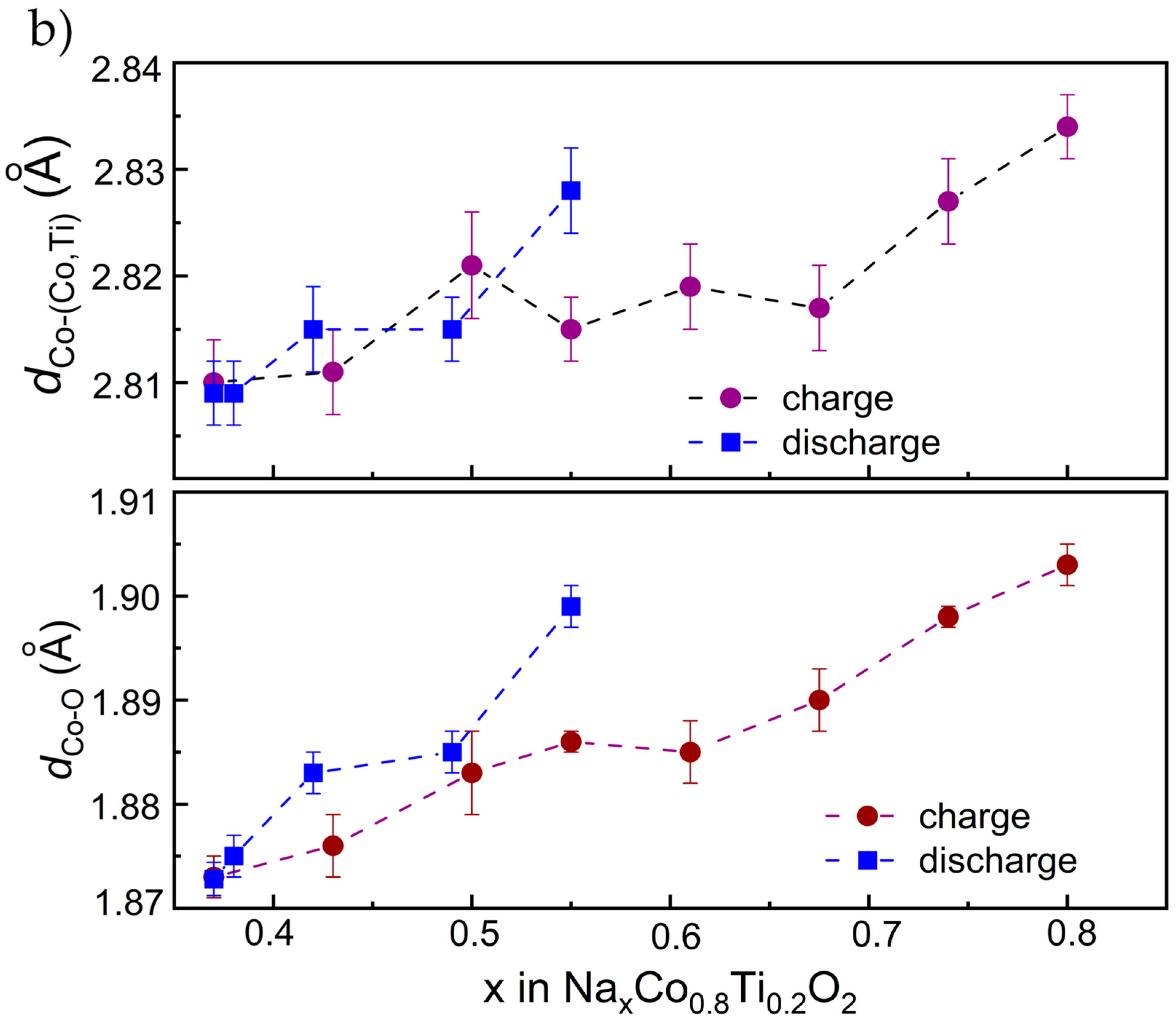
3.3. Structural Characterization Using Operando XAS Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Deng, J.; Luo, W.-B.; Chou, S.-L.; Liu, H.-K.; Dou, S.-X. Sodium-Ion Batteries: From Academic Research to Practical Commercialization. Adv. Energy Mater. 2018, 8, 1701428. [Google Scholar] [CrossRef]
- Hwang, J.-Y.; Myung, S.-T.; Sun, Y.-K. Sodium-ion batteries: Present and future. Chem. Soc. Rev. 2017, 46, 3529–3614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, A.; Song, J.; Vail, S.; Pan, W.; Barker, J.; Lu, Y. The Scale-up and Commercialization of Nonaqueous Na-Ion Battery Technologies. Adv. Energy Mater. 2018, 8, 1703137. [Google Scholar] [CrossRef]
- Delmas, C. Sodium and Sodium-Ion Batteries: 50 Years of Research. Adv. Energy Mater. 2018, 8, 1703137. [Google Scholar] [CrossRef]
- Kubota, K.; Kumakura, S.; Yoda, Y.; Kuroki, K.; Komaba, S. Electrochemistry and Solid-State Chemistry of NaMeO2 (Me = 3d Transition Metals). Adv. Energy Mater. 2018, 8, 1703415. [Google Scholar] [CrossRef]
- Sun, Y.; Guo, S.; Zhou, H. Adverse effects of interlayer-gliding in layered transition-metal oxides on electrochemical sodium-ion storage. Energy Environ. Sci. 2019, 12, 825–840. [Google Scholar] [CrossRef]
- Lei, Y.; Li, X.; Liu, L.; Ceder, G. Synthesis and Stoichiometry of Different Layered Sodium Cobalt Oxides. Chem. Mater. 2014, 26, 5288–5296. [Google Scholar] [CrossRef]
- Antolini, E.; Ferretti, M. Synthesis and Thermal Stability of LiCoO2. J. Solid State Chem. 1995, 117, 1–7. [Google Scholar] [CrossRef]
- Berthelot, R.; Carlier, D.; Delmas, C. Electrochemical investigation of the P2–NaxCoO2 phase diagram. Nature Mater. 2011, 10, 74–80. [Google Scholar] [CrossRef]
- Nazri, G.A.; Pistoia, G. (Eds.) Lithium Batteries: Science and Technology; Springer Science + Business Media: Berlin/Heidelberg, Germany, 2009; 708p. [Google Scholar]
- Kubota, K.; Asari, T.; Yoshida, H.; Yaabuuchi, N.; Shiiba, H.; Nakayama, M.; Komaba, S. Understanding the Structural Evolution and Redox Mechanism of a NaFeO2 –NaCoO2 Solid Solution for Sodium-Ion Batteries. Adv. Funct. Mater. 2016, 26, 6047–6059. [Google Scholar] [CrossRef]
- Guo, S.; Sun, Y.; Yi, J.; Zhu, K.; Liu, P.; Zhu, Y.; Zhu, G.-Z.; Chen, M.; Ishida, M.; Zhou, H. Understanding sodium-ion diffusion in layered P2 and P3 oxides via experiments and first-principles calculations: A bridge between crystal structure and electrochemical performance. NPG Asia Mater. 2016, 8, e266. [Google Scholar] [CrossRef] [Green Version]
- Sabi, N.; Sarapulova, A.; Indris, S.; Ehrenberg, H.; Alami, J.; Saadoune, I. Effect of Titanium Substitution in a P2-Na2/3Co0.95Ti0.05O2 Cathode Material on the Structural and Electrochemical Properties. ACS Appl. Mater. Interfaces 2017, 9, 37778–37785. [Google Scholar] [CrossRef]
- Sabi, N.; Doubaji, S.; Hashimoto, K.; Komaba, S.; Amine, K.; Solhy, A.; Manoun, B.; Bilal, E.; Saadoune, I. Layered P2-Na2/3Co1/2Ti1/2O2 as a high-performance cathode material for sodium-ion batteries. J. Power Sources 2017, 342, 998–1005. [Google Scholar] [CrossRef]
- Maletti, S.; Giebeler, L.; Oswald, S.; Tsirlin, A.A.; Senyshyn, A.; Michaelis, A.; Mikhailova, D. Irreversible made reversible: Increasing electrochemical capacity by understanding the structural transformations of NaxCo0.5Ti0.5O2. ACS Appl. Mater. Interfaces 2018, 10, 36108–36119. [Google Scholar] [CrossRef]
- Sabi, N.; Sarapulova, A.; Indris, S.; Dsoke, S.; Zhao, Z.; Dahbi, M.; Ehrenberg, H.; Saadoune, I. Evidence of a Pseudo-Capacitive Behavior Combined with an Insertion/Extraction Reaction Upon Cycling of the Positive Electrode Material P2-NaxCo0.9Ti0.1O2 for Sodium-ion Batteries. ChemElectroChem 2019, 6, 892–903. [Google Scholar] [CrossRef]
- Kang, S.M.; Park, J.-H.; Jin, A.; Jung, Y.H.; Mun, J.; Sung, Y.-E. Na+/Vacancy Disordered P2-Na0.67Co1−xTixO2: High-Energy and High-Power Cathode Materials for Sodium Ion Batteries. ACS Appl. Mater. Interfaces 2018, 10, 3562. [Google Scholar] [CrossRef]
- Sabi, N.; Sarapulova, A.; Indris, S.; Dsoke, S.; Trouillet, V.; Mereacre, L.; Ehrenberg, H.; Saadoune, I. Investigation of “Na2/3Co2/3Ti1/3O2” as a multi-phase positive electrode material for sodium batteries. J. Power Sources 2021, 481, 229120. [Google Scholar] [CrossRef]
- Watanabe, E.; Zhao, W.; Sugahara, A.; de Boisse, B.M.; Lander, L.; Asakura, D.; Okamoto, Y.; Mizokawa, T.; Okubo, M.; Yamada, A. Redox-Driven Spin Transition in a Layered Battery Cathode Material. Chem. Mat. 2019, 31, 23582365. [Google Scholar] [CrossRef]
- Kim, S.; Ma, X.; Ong, S.P.; Ceder, G. A comparison of destabilization mechanisms of the layered NaxMO2 and LixMO2 compounds upon alkali de-intercalation. Phys. Chem. Chem. Phys. 2012, 14, 15571–15578. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Adair, K.R.; Sun, X. Recent developments and insights into the understanding of Na metal anodes for Na-metal batteries. Energy Environ. Sci. 2018, 11, 2673–2695. [Google Scholar] [CrossRef]
- Eshetu, G.G.; Elia, G.A.; Armand, M.; Forsyth, M.; Komaba, S.; Rojo, T.; Passerini, S. Electrolytes and Interphases in Sodium-Based Rechargeable Batteries: Recent Advances and Perspectives. Adv. Energy Mater. 2020, 10, 2000093. [Google Scholar] [CrossRef] [Green Version]
- Hasa, I.; Hassoun, J.; Passerini, S. Nanostructured Na-ion and Li-ion anodes for battery application: A comparative overview. Nano Res. 2017, 10, 3942–3969. [Google Scholar] [CrossRef]
- Lu, Q.; Omar, A.; Ding, L.; Oswald, S.; Hantusch, M.; Giebeler, L.; Nielsch, K.; Mikhailova, D. A facile method to stabilize sodium metal anodes towards high-performance sodium batteries. Mater. Chem. A 2021, 9, 9038. [Google Scholar] [CrossRef]
- Weppner, W.; Huggins, R.A. Determination of the kinetic parameters of mixed-conducting electrodes and application to the system Li3Sb. J. Electrochem. Soc. 1977, 124, 1569–1578. [Google Scholar] [CrossRef]
- Lu, Q.; Omar, A.; Hantusch, M.; Oswald, S.; Ding, L.; Nielsch, K.; Mikhailova, D. Dendrite-free and corrosion-resistant sodium metal anode for sodium batteries. submitted.
- Roisnel, T.; Rodriguez-Carvajal, J. WinPLOTR: A Windows tool for powder diffraction pattern analysis. Mater. Sci. Forum 2001, 378–381, 118–123. [Google Scholar] [CrossRef] [Green Version]
- Herklotz, M.; Weiss, J.; Ahrens, E.; Yavuz, M.; Mereacre, L.; Kiziltas-Yavuz, N.; Draeger, C.; Ehrenberg, H.; Eckert, J.; Fauth, F.; et al. A novel high-throughput setup for in situ powder diffraction on coin cell batteries. J. Appl. Cryst. 2016, 49, 340–345. [Google Scholar] [CrossRef]
- Maletti, S.; Sarapulova, A.; Schökel, A.; Mikhailova, D. Operando Studies on the NaNi0.5Ti0.5O2 Cathode for Na-Ion Batteries: Elucidating Titanium as a Structure Stabilizer. ACS Appl. Mater. Interfaces 2019, 11, 33923–33930. [Google Scholar] [CrossRef] [PubMed]
- Rehr, J.J.; Leon, J.d.; Zabinsky, S.I.; Albers, R.C. Theoretical X-ray Absorption Fine Structure Standards. J. Am. Chem. Soc. 1991, 113, 5135–5140. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, D.-W.; Lv, W.; Qin, L.; Niu, S.; Zhang, S.; Cao, T.; Kang, F.; Yang, Q.-H. Ethers Illume Sodium-Based Battery Chemistry: Uniqueness, Surprise, and Challenges. Adv. Energy Mater. 2018, 8, 1801361. [Google Scholar] [CrossRef]
- Paulsen, J.M.; Dahn, J.R. Studies of the layered manganese bronzes, Na2/3[Mn1−xMx]O2 with M=Co, Ni, Li, and Li2/3[Mn1−xMx]O2 prepared by ion-exchange. Solid State Ionics 1999, 126, 3–24. [Google Scholar] [CrossRef]
- Somerville, J.W.; Sobkowiak, A.; Tapia-Ruiz, N.; Billaud, J.; Lozano, J.G.; House, R.A.; Gallington, L.C.; Ericsson, T.; Häggström, L.; Roberts, M.R.; et al. Nature of the “Z”-phase in layered Na-ion battery cathodes. Energy Environ. Sci. 2019, 12, 2223–2232. [Google Scholar] [CrossRef] [Green Version]
- Newville, M. Fundamentals of XAFS, Revision 1.7; University of Chicago: Chicago, IL, USA, 2004. [Google Scholar]
- Vitova, T.; Mangold, S.; Paulmann, C.; Gospodinov, M.; Marinova, V.; Mihailova, B. X–ray absorption spectroscopy of Ru-doped relaxor ferroelectrics with a perovskite-type structure. Phys. Rev. B 2014, 89, 144112. [Google Scholar] [CrossRef] [Green Version]
- Poltavets, V.V.; Croft, M.; Greenblatt, M. Charge transfer, hybridization and local inhomogeneity effects in NaxCoO2·yH2O: An x-ray absorption spectroscopy study. Phys. Rev. B 2006, 74, 125103. [Google Scholar] [CrossRef] [Green Version]
- Reddy, B.V.R.; Ravikumar, R.; Nithya, C.; Gopukumar, S. High performance NaxCoO2 as a cathode material for rechargeable sodium batteries. J. Mater. Chem. A 2015, 3, 18059–18063. [Google Scholar] [CrossRef]
- Yoshida, H.; Yabuuchi, N.; Komada, S. NaFe0.5Co0.5O2 as high energy and power positive electrode for Na-ion batteries. Electrochem. Commun. 2013, 34, 60–63. [Google Scholar] [CrossRef] [Green Version]
- Vassilaras, P.; Toumar, A.J.; Ceder, G. Electrochemical properties of NaNi1/3Co1/3Fe1/3O2 as a cathode material for Na-ion batteries. Electrochem. Commun. 2014, 38, 79–81. [Google Scholar] [CrossRef]
- Yang, P.; Zhang, C.; Li, M.; Yang, X.; Wang, C.; Bie, X.; Wei, Y.; Chen, G.; Du, F. P2-NaCo0.5Mn0.5O2 as a Positive Electrode Materia lfor Sodium-Ion Batteries. ChemPhysChem 2015, 16, 3408–3412. [Google Scholar] [CrossRef]
- Lu, Q.; Wang, X.; Omar, A.; Mikhailova, D. 3D Ni/Na metal anode for improved sodium metal batteries. Mater. Lett. 2020, 275, 128206. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atom | Wyckoff-Position | x | y | z | Occupancy |
|---|---|---|---|---|---|
| O | 4f | 0.66667 | 0.33333 | 0.0885(6) | 1 |
| Co | 2a | 0 | 0 | 0 | 0.8 |
| Ti | 2a | 0 | 0 | 0 | 0.2 |
| Na(1) | 2b | 0 | 0 | 0.25 | 0.20(1) |
| Na(2) | 2c | 0.33333 | 0.66667 | 0.25 | 0.60(1) |
| Material | Capacity (after N Cycles) | Current Density (C-Rate) | Potential Range, V | Reference |
|---|---|---|---|---|
| P2-NaxCoO2 | 110 (50) | 0.1 | 2.0–4.2 | [37] |
| O3-NaCoO2 | 130 (30) | 0.05 | 2.0–4.0 | [38] |
| O3-NaFe0.5Co0.5O2 | 140 (50) | 0.05 | 2.5–4.0 | [38] |
| O3-NaNi0.3Co0.3Fe0.3O2 | 135 (20) | 0.2 | 2.0–4.2 | [39] |
| P2-NaCo0.5Mn0.5O2 | 120 (30) | 0.1 | 1.5–4.15 | [40] |
| P2-Na0.67Co0.95Ti0.05O2 | 106 (100) | 0.1 | 2.0–4.2 | [13] |
| P2-Na0.67Co0.5Ti0.5O2 | 100 (50) | 0.1 | 2.0–4.2 | [14] |
| Na3V2(PO4)3 | 75 (200) | 1.0 | 1.5–3.5 | [41] |
| P2-Na0.8Co0.8Ti0.2O2 | 100 (50) | 0.1 | 2.0–4.2 | This work |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pohle, B.; Gorbunov, M.V.; Lu, Q.; Bahrami, A.; Nielsch, K.; Mikhailova, D. Structural and Electrochemical Properties of Layered P2-Na0.8Co0.8Ti0.2O2 Cathode in Sodium-Ion Batteries. Energies 2022, 15, 3371. https://doi.org/10.3390/en15093371
Pohle B, Gorbunov MV, Lu Q, Bahrami A, Nielsch K, Mikhailova D. Structural and Electrochemical Properties of Layered P2-Na0.8Co0.8Ti0.2O2 Cathode in Sodium-Ion Batteries. Energies. 2022; 15(9):3371. https://doi.org/10.3390/en15093371
Chicago/Turabian StylePohle, Björn, Mikhail V. Gorbunov, Qiongqiong Lu, Amin Bahrami, Kornelius Nielsch, and Daria Mikhailova. 2022. "Structural and Electrochemical Properties of Layered P2-Na0.8Co0.8Ti0.2O2 Cathode in Sodium-Ion Batteries" Energies 15, no. 9: 3371. https://doi.org/10.3390/en15093371
APA StylePohle, B., Gorbunov, M. V., Lu, Q., Bahrami, A., Nielsch, K., & Mikhailova, D. (2022). Structural and Electrochemical Properties of Layered P2-Na0.8Co0.8Ti0.2O2 Cathode in Sodium-Ion Batteries. Energies, 15(9), 3371. https://doi.org/10.3390/en15093371