
Fast Pyrolysis Oil Upgrading via HDO with Fe-Promoted Nb2O5-Supported Pd-Based Catalysts

 ,
, 
Abstract
:1. Introduction
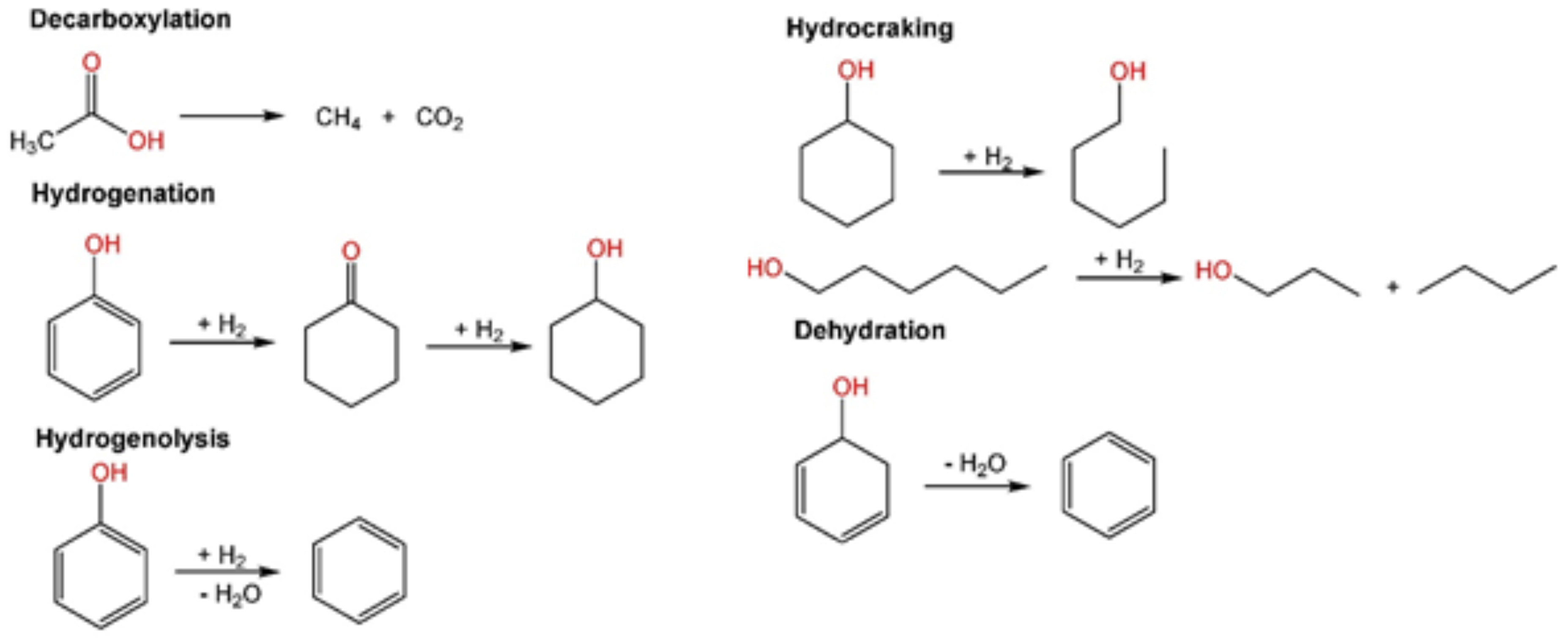
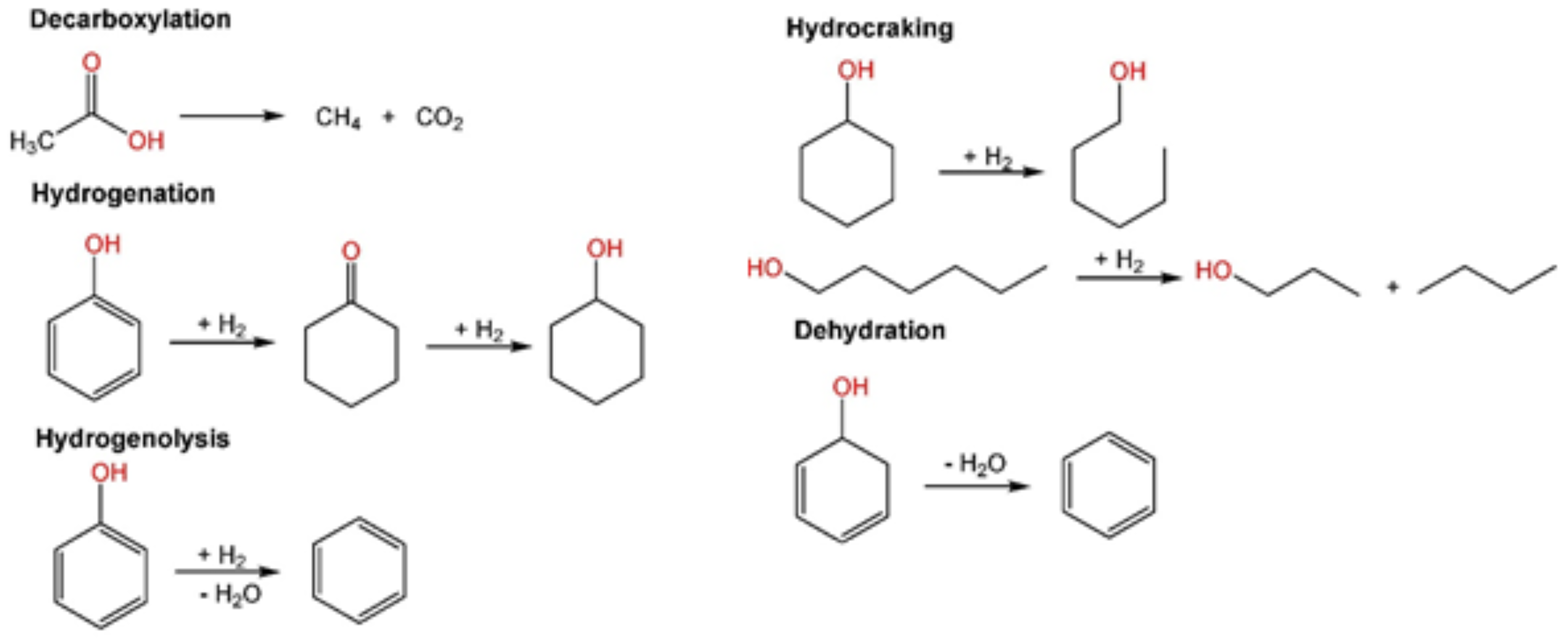
1.1. Catalytic Hydrodeoxygenation of Biomass-Derived Compounds
1.2. Niobium Oxide (Nb2O5) as a Solid Acid Support for HDO
1.3. Iron as Catalyst Promoter for HDO
2. Materials and Methods
2.1. Catalyst Preparation
2.2. Hydrodeoxygenation (HDO) of Fast Pyrolysis Bio-Oil
2.3. Characterization of the Gas Phase
2.4. Characterization of Feedstock and Liquid Products
2.5. Characterization of Catalysts and Solid Products
3. Results and Discussion
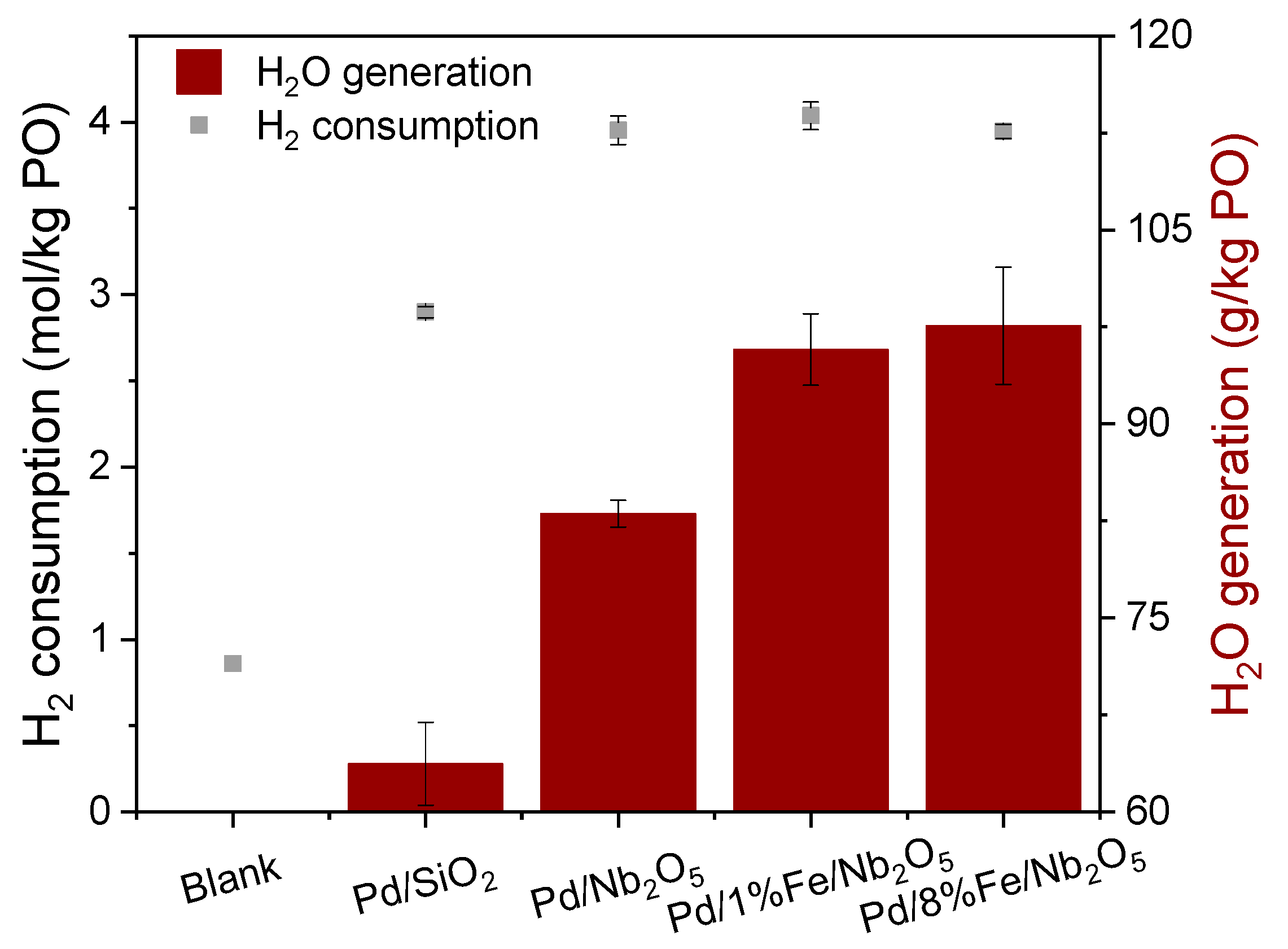
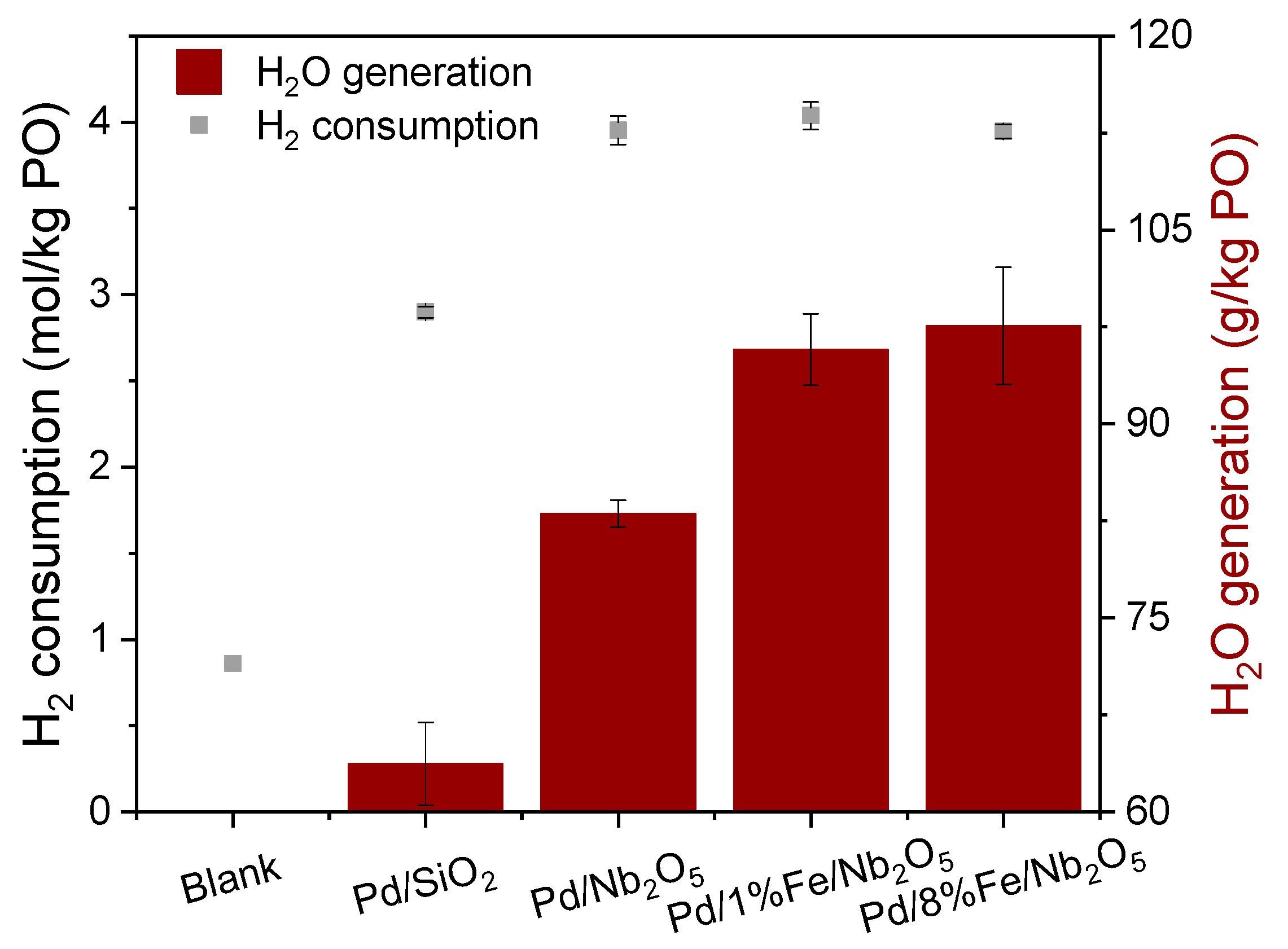
3.1. Hydrogen Consumption and Water Formation
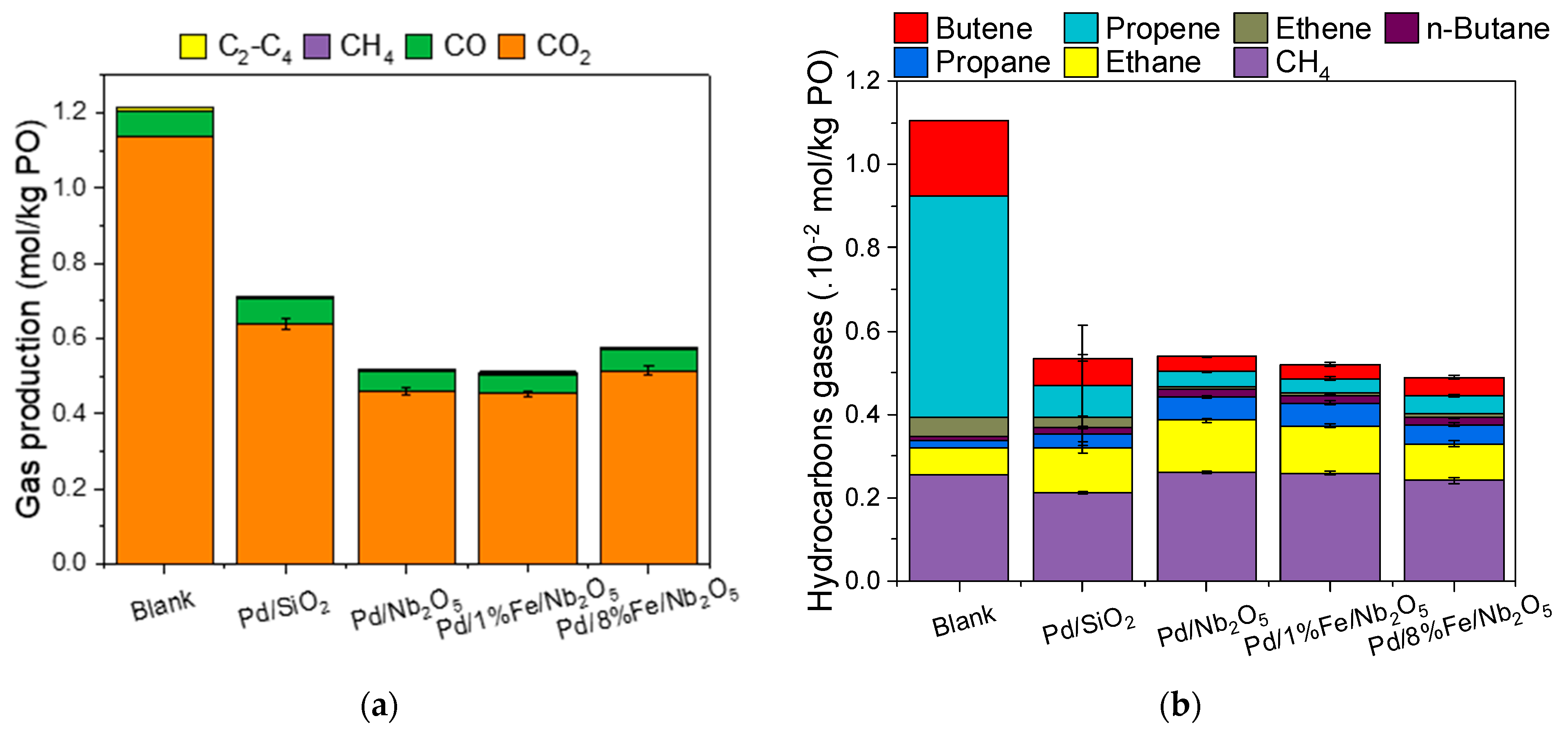
3.2. Characterization of Gas Phase Produced during HDO
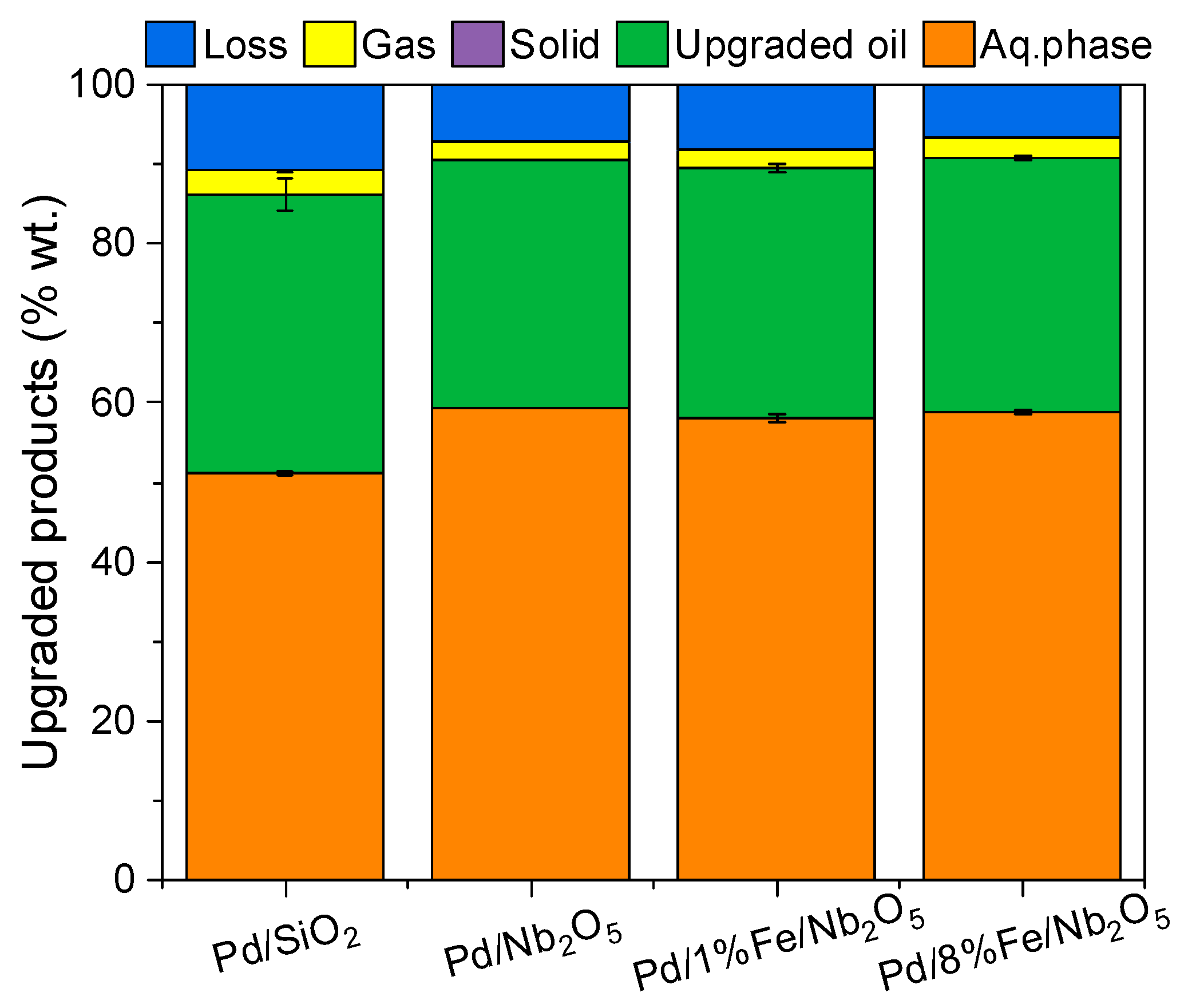
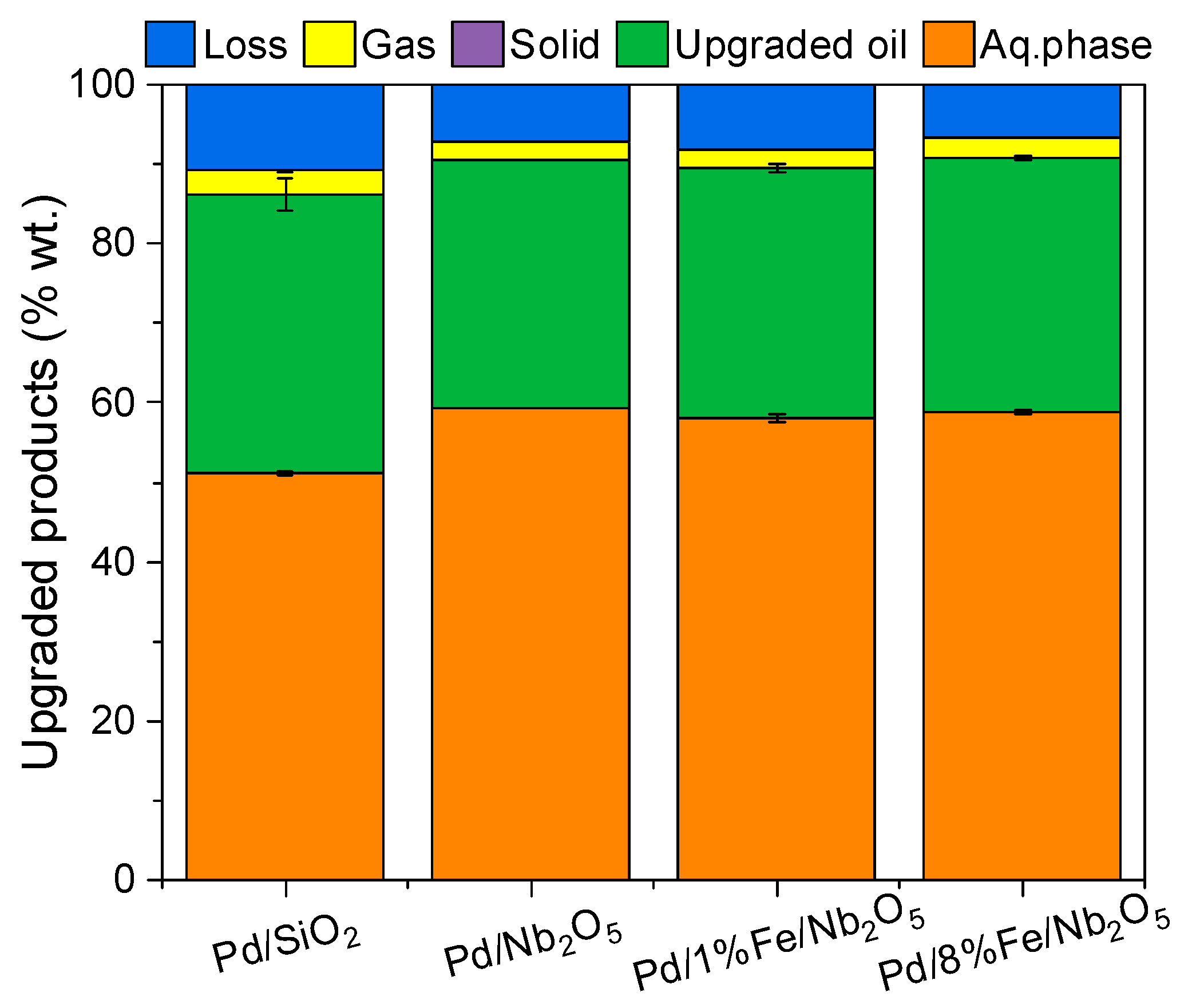
3.3. Product Distribution
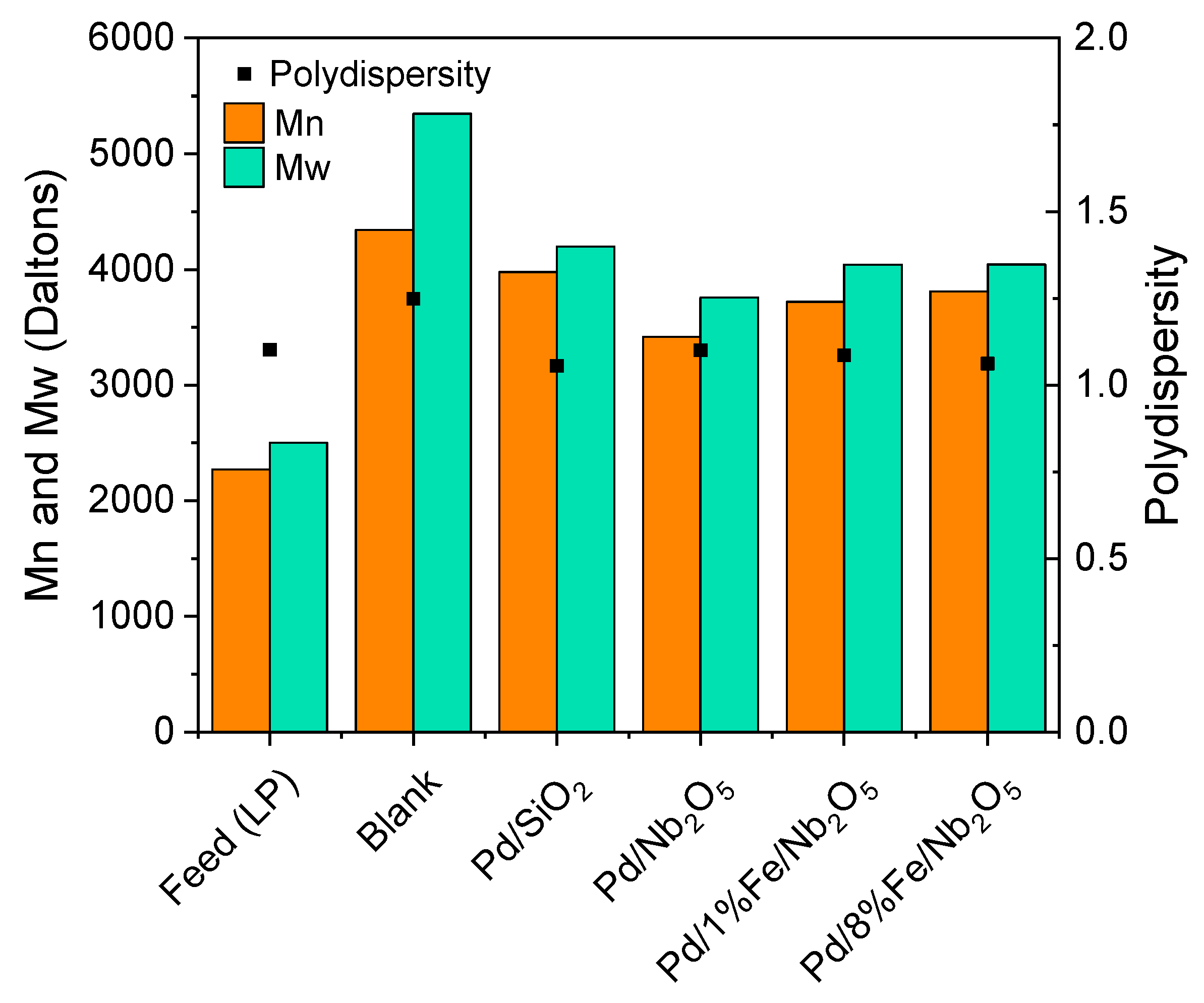
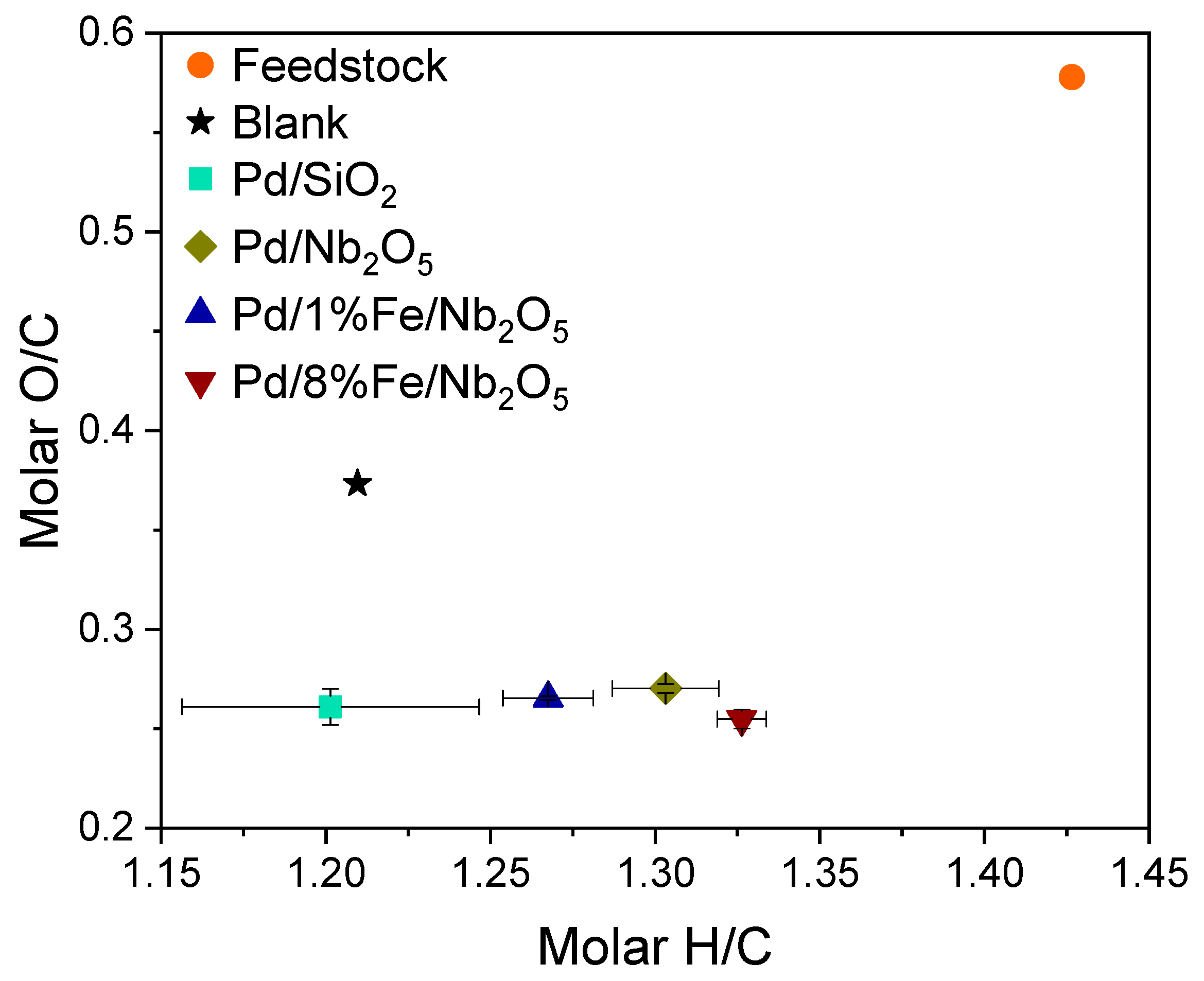
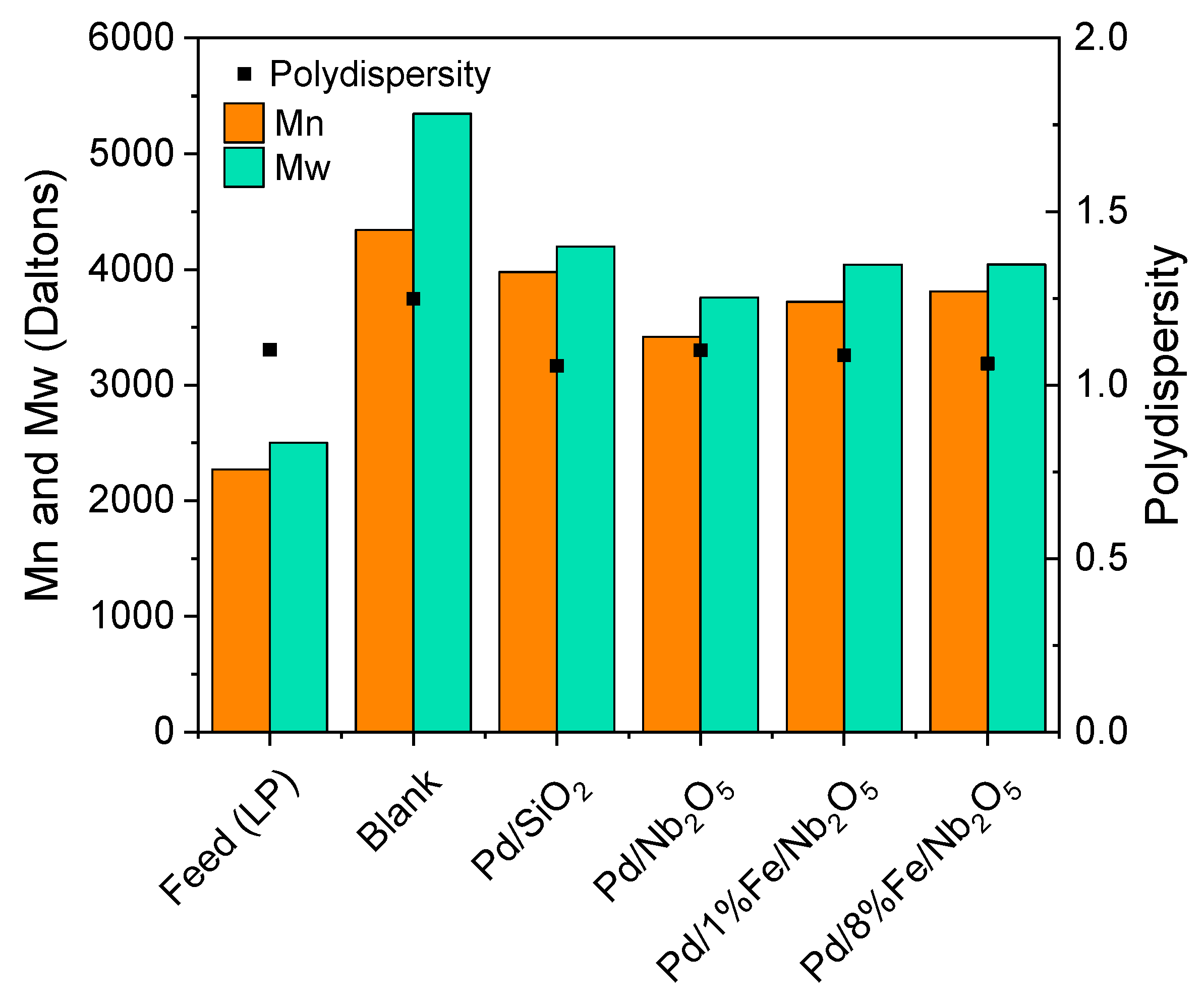
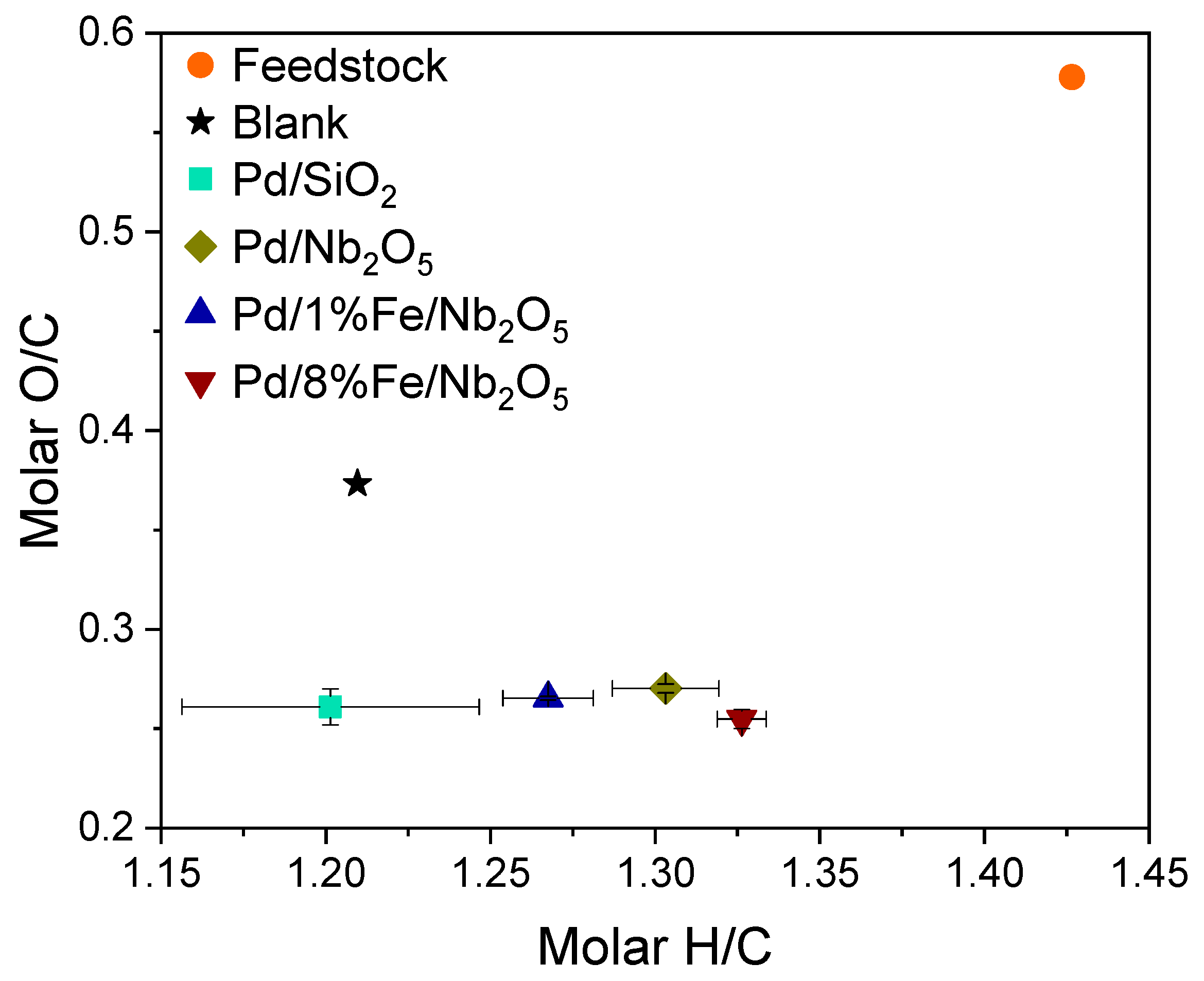
3.4. Product Characterization: Elemental Analysis, Water Content, High Heating Value and Molecular Weight
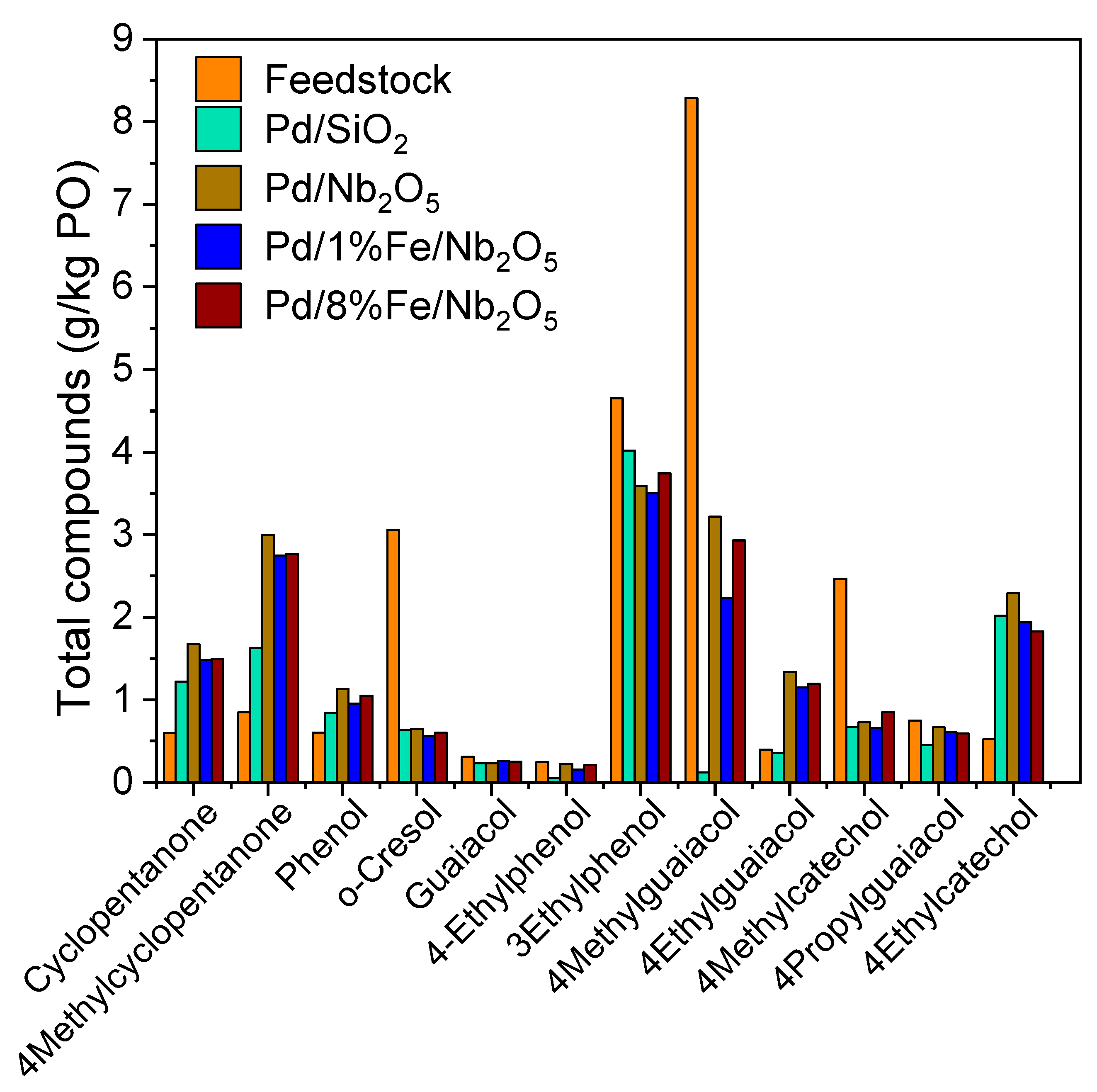
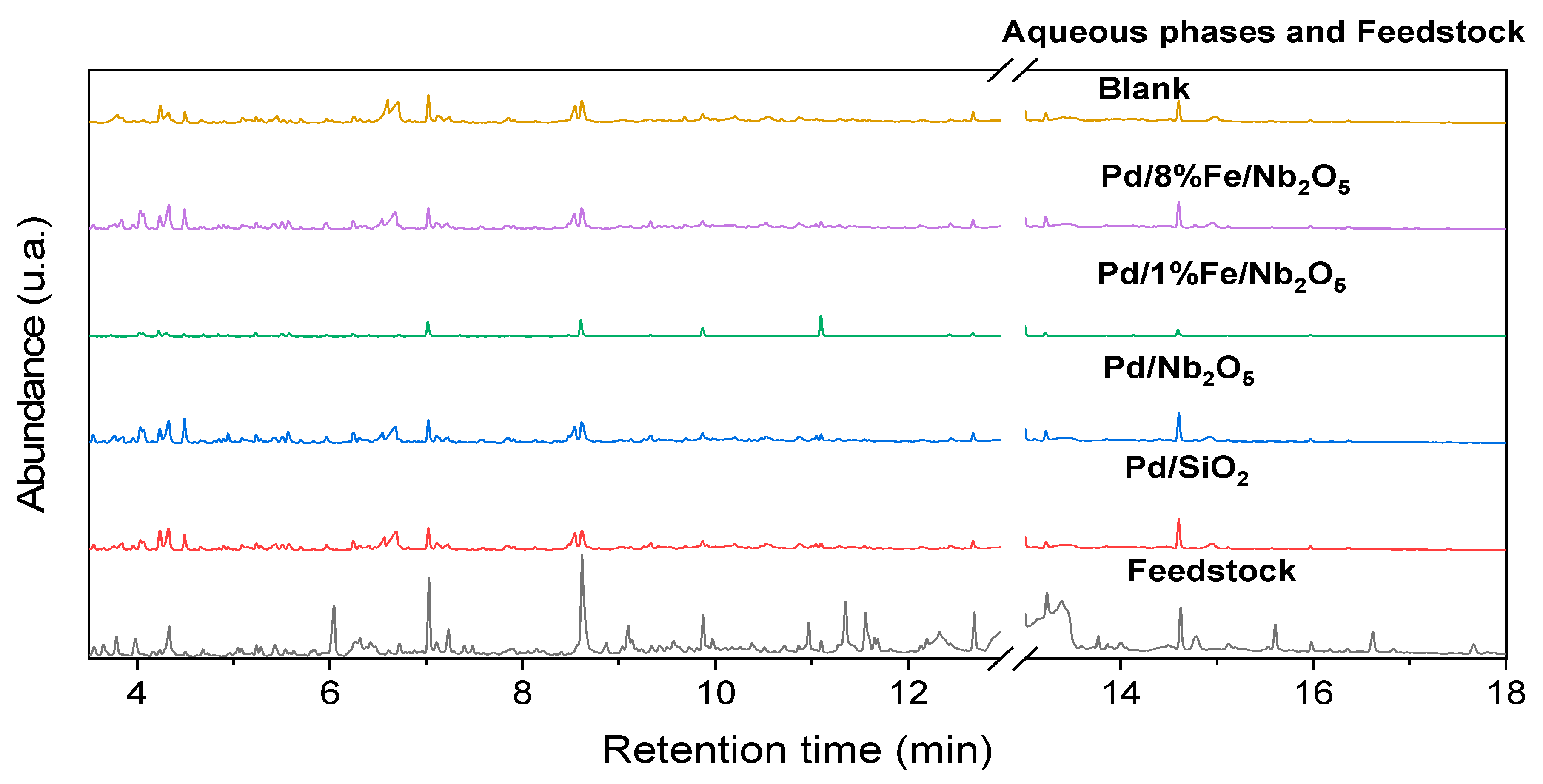
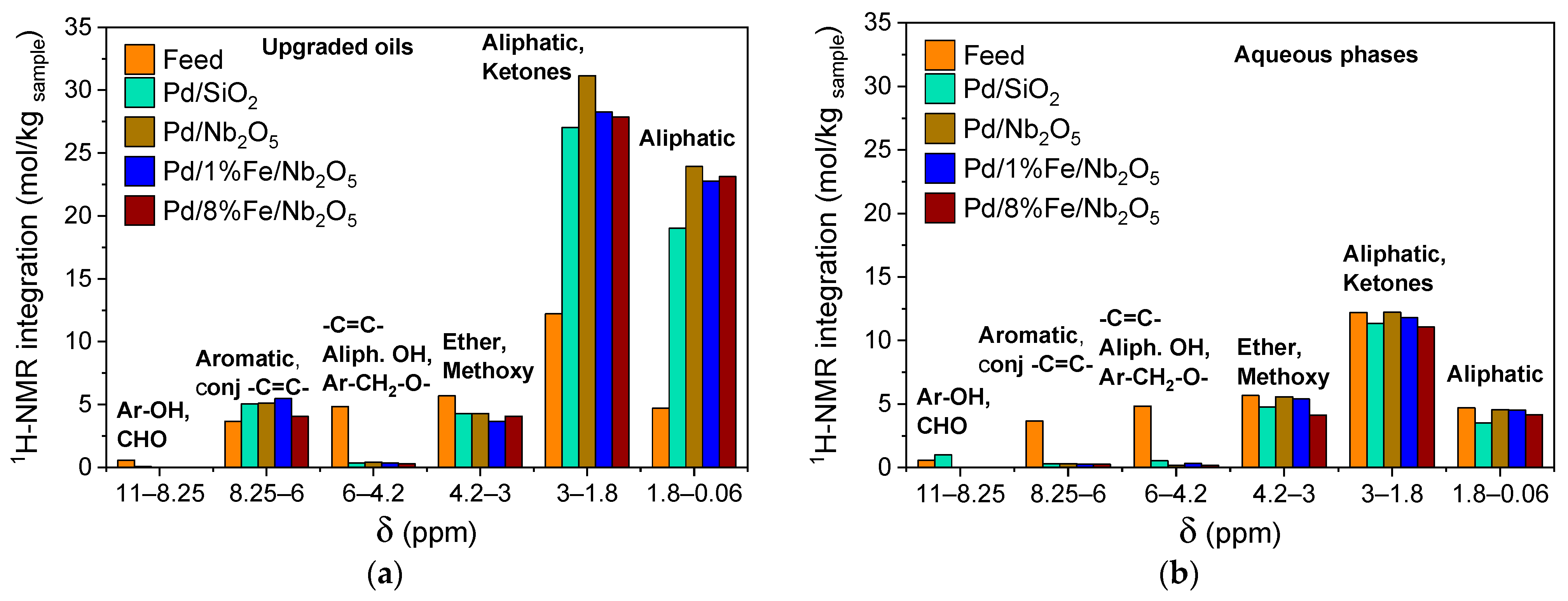
3.5. Product Characterization: Functional Groups via 1H-NMR and Chemical Composition via GC-MS/FID
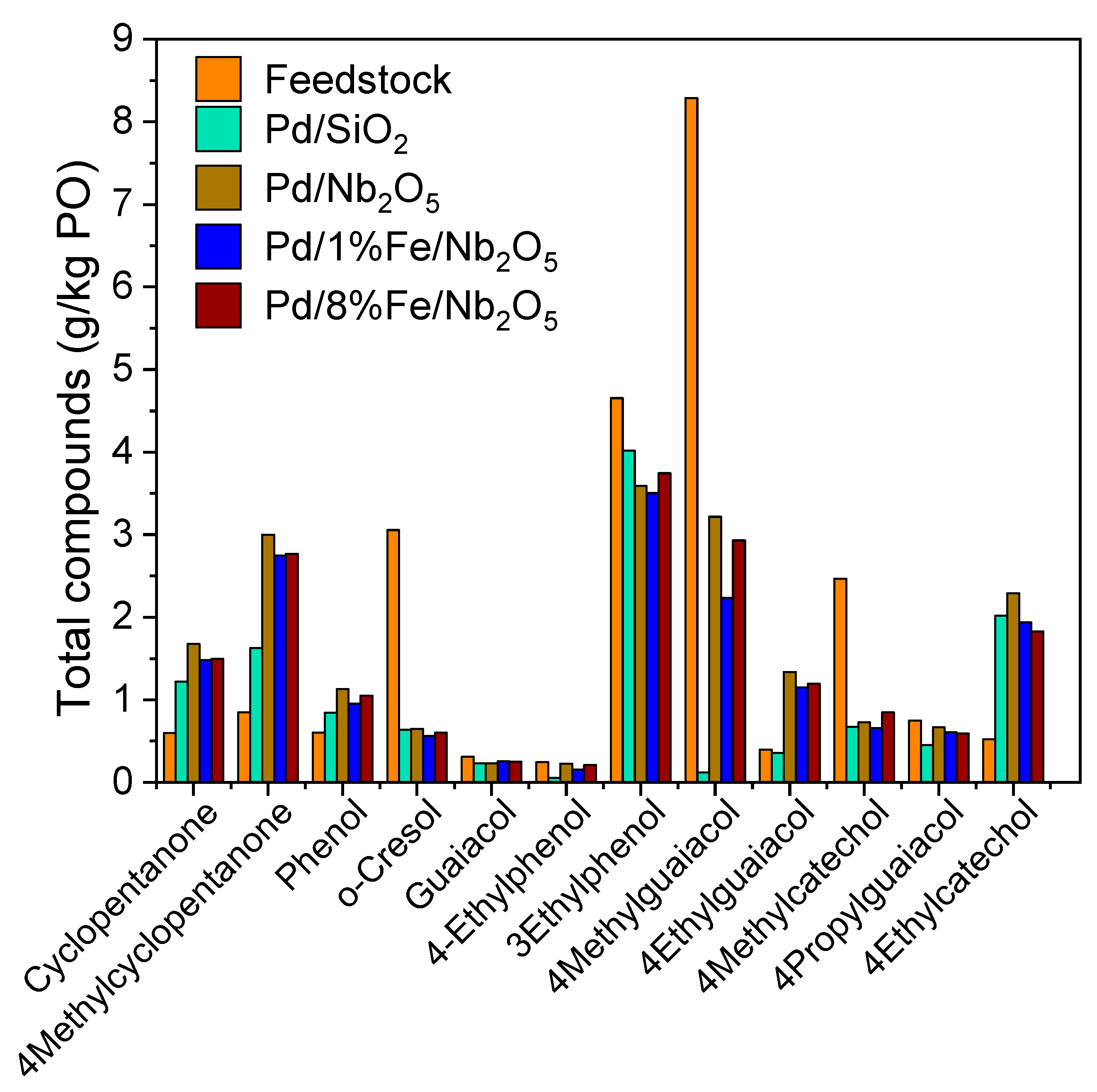
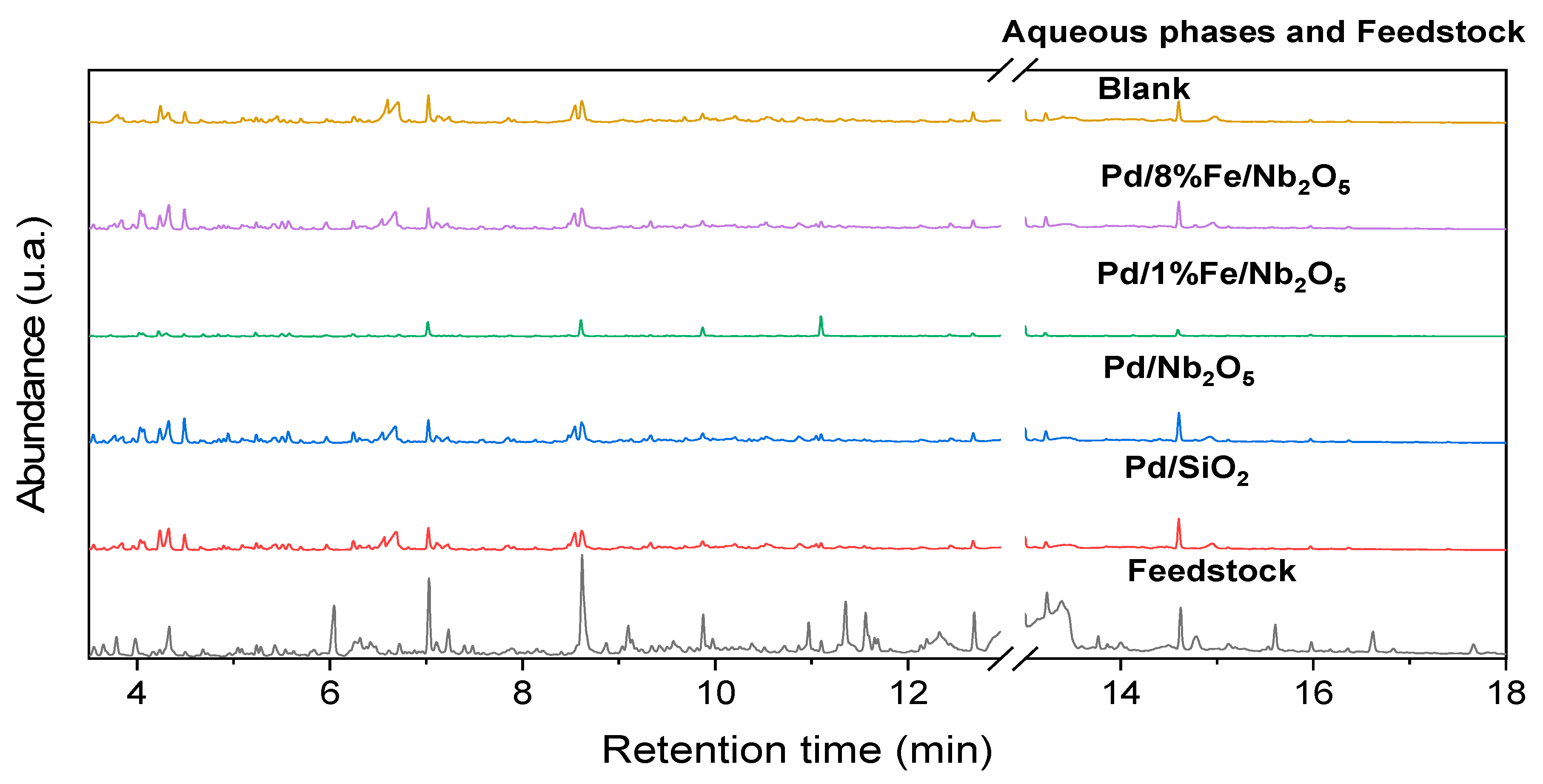
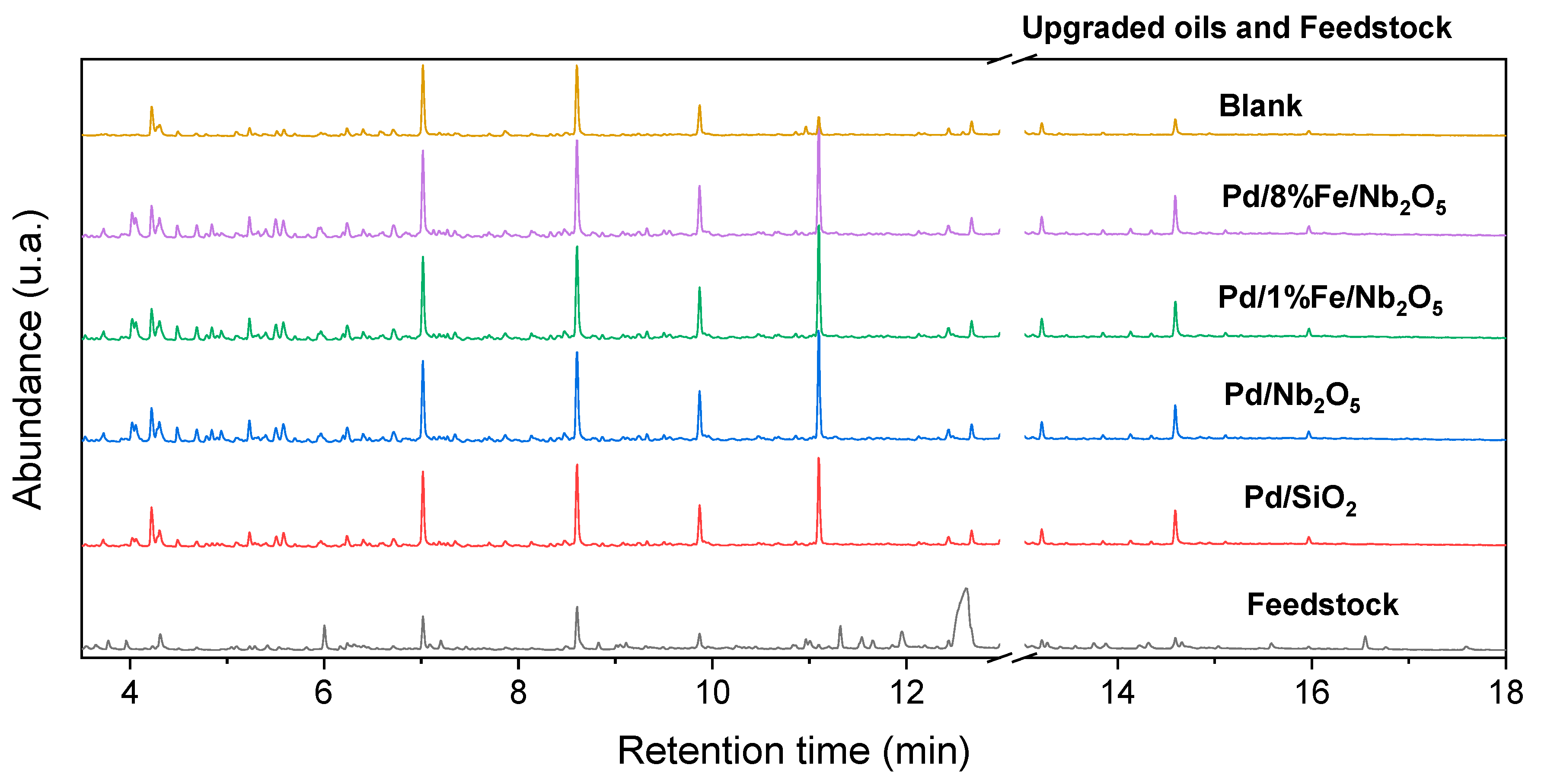
3.6. Product Characterization: Qualitative and Quantitative Analyses of Chemical Composition via GC-FID/MS
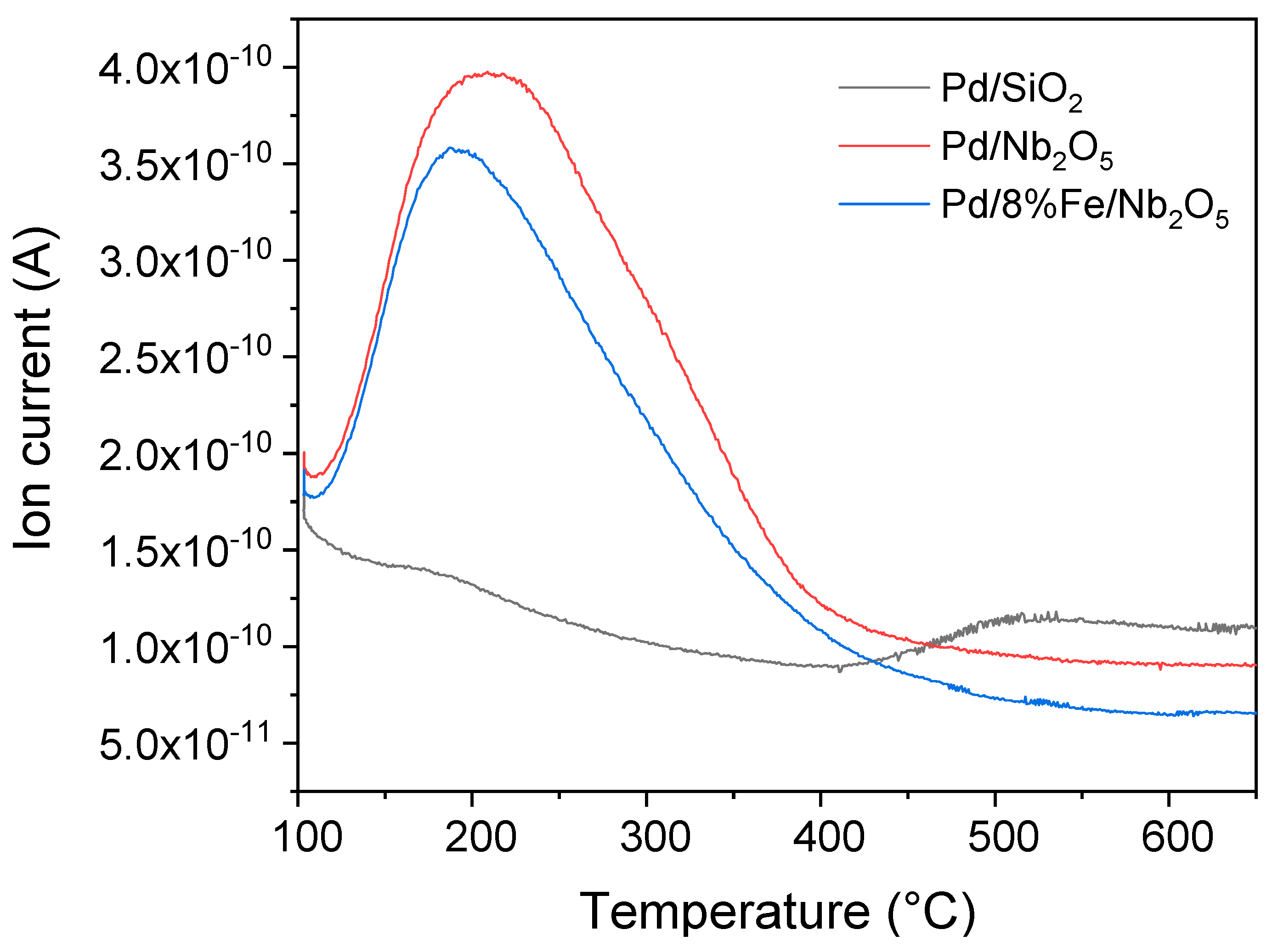
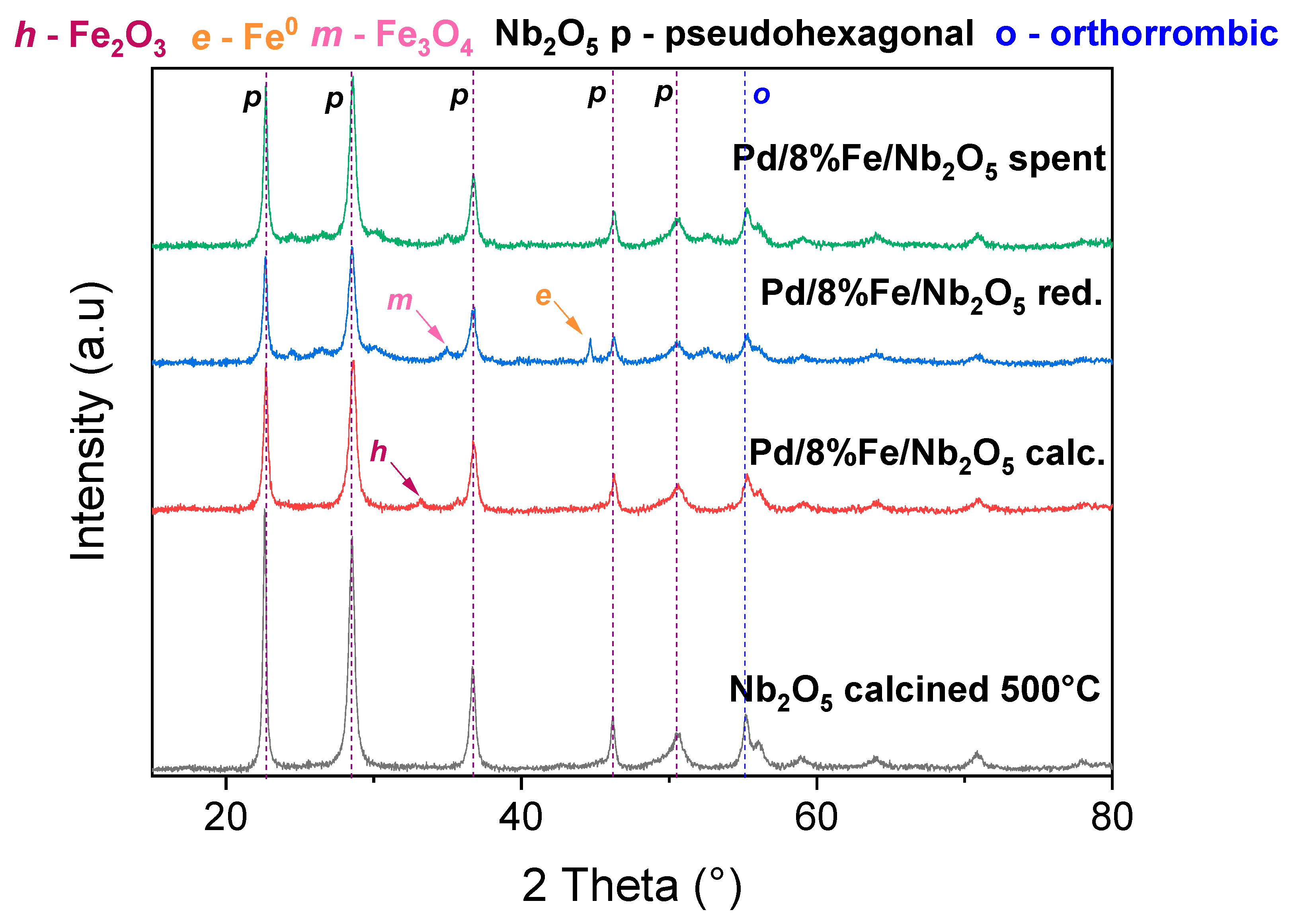
3.7. Characterization of Fresh and Spent Catalysts
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Bridgwater, A.V.; Meier, D.; Radlein, D. An overview of fast pyrolysis of biomass. Org. Geochem. 1999, 30, 1479–1493. [Google Scholar] [CrossRef]
- Schmitt, C.C.; Fonseca, F.G.; Fraga, M.M.C.; Wisniewski, A.; Karp, S.; José, H.M.; Rodrigues, R.C.L.B.; Moreira, R.; Hirayama, D.E.; Raffelt, K.; et al. Thermochemical and Catalytic Conversion Technologies for the Development of Brazilian Biomass Utilization. Catalysts 2021, 11, 1549. [Google Scholar] [CrossRef]
- Dahmen, N.; Henrich, E.; Dinjus, E.; Weirich, F. The bioliq® bioslurry gasification process for the production of biosynfuels, organic chemicals, and energy. Energy. Sustain. Soc. 2012, 2, 1–44. [Google Scholar] [CrossRef]
- IEA Bioenergy. Contribution of Bioenergy to the World’s Future Energy; IEA: Paris, France, 2007. [Google Scholar]
- IRENA. Global Renewables Outlook: Energy Transformation 2050; IRENA: Abu Dhabi, United Arab Emirates, 2020. [Google Scholar]
- IEA. World Energy Outlook 2019; IEA: Paris, France, 2019; p. 1. [Google Scholar]
- IEA. Renewables 2019—Analysis and Forecast to 2024; IEA: Paris, France, 2019; p. 204. [Google Scholar]
- Balan, V. Current Challenges in Commercially Producing Biofuels from Lignocellulosic Biomass. ISRN Biotechnol. 2014, 2014, 463074. [Google Scholar] [CrossRef]
- Wang, Z. 1.23 Energy and Air Pollution. Compr. Energy Syst. 2018, 1–5, 909–949. [Google Scholar]
- Tijmensen, M.J.A.; Faaij, A.P.C.; Hamelinck, C.N.; van Hardeveld, M.R.M. Exploration of the possibilities for production of Fischer Tropsch liquids and power via biomass gasification. Biomass Bioenergy 2002, 23, 129–152. [Google Scholar] [CrossRef]
- Glushkov, D.; Nyashina, G.; Shvets, A.; Pereira, A.; Ramanathan, A. Current Status of the Pyrolysis and Gasification Mechanism of Biomass. Energies 2022, 14, 7541. [Google Scholar] [CrossRef]
- Schmitt, C.C.; Reolon, M.B.G.; Zimmermann, M.; Raffelt, K.; Grunwaldt, J.-D.; Dahmen, N. Synthesis and Regeneration of Nickel-Based Catalysts for Hydrodeoxygenation of Beech Wood Fast Pyrolysis Bio-Oil. Catalysts 2018, 8, 449. [Google Scholar] [CrossRef] [Green Version]
- Mortensen, P.M.; Grunwaldt, J.-D.; Jensen, P.A.; Knudsen, K.G.; Jensen, A.D. A review of catalytic upgrading of bio-oil to engine fuels. Appl. Catal. A Gen. 2011, 407, 1–19. [Google Scholar] [CrossRef]
- Mohan, D.; Pittman, C.U., Jr.; Steele, P.H. Pyrolysis of Wood/Biomass for Bio-Oil: A Critical Review. Energy Fuels 2006, 20, 848–889. [Google Scholar] [CrossRef]
- Elliott, D.C. Biofuel from fast pyrolysis and catalytic hydrodeoxygenation. Curr. Opin. Chem. Eng. 2015, 9, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Mercader, F.D.M.; Groeneveld, M.; Kersten, S.; Venderbosch, R.; Hogendoorn, J. Pyrolysis oil upgrading by high pressure thermal treatment. Fuel 2010, 89, 2829–2837. [Google Scholar] [CrossRef]
- Schmitt, C.C. Catalytic Upgrading of Fast Pyrolysis Bio-Oils Applying Nickel-Based Catalysts. Ph.D. Thesis, KIT, Karlsruhe, Germany, 2021. [Google Scholar]
- Collard, F.X.; Blin, J. A review on pyrolysis of biomass constituents: Mechanisms and composition of the products obtained from the conversion of cellulose, hemicelluloses and lignin. Renew. Sustain. Energy Rev. 2014, 38, 594–608. [Google Scholar] [CrossRef]
- Sarangi, P.K.; Nanda, S.; Vo, D.-V.N. Technological Advancements in the Production and Application of Biomethanol. In Biorefinery of Alternative Resources: Targeting Green Fuels and Platform Chemicals; Springer: Singapore, 2020. [Google Scholar] [CrossRef]
- Pinheiro Pires, A.P.; Arauzo, J.; Fonts, I.; Domine, M.E.; Fernández Arroyo, A.; Garcia-Perez, M.E.; Montoya, J.; Chejne, F.; Pfromm, P.; Garcia-Perez, M. Challenges and Opportunities for Bio-Oil Refining: A Review. Energy Fuels 2019, 33, 4683–4720. [Google Scholar] [CrossRef]
- Jones, S.B.; Snowden-Swan, L.L. Production of Gasoline and Diesel from Biomass via Fast-Pyrolysis, Hydrotreating and Hydrocracking: 2012 State of Technology and Projections to 2017; PNNL Report No. 22684; Pacific Northwest National Laboratory: Richland, WA, USA, 2013. [Google Scholar]
- Han, Y.; Gholizadeh, M.; Tran, C.-C.; Kaliaguine, S.; Li, C.-Z.; Olarte, M.; Garcia-Perez, M. Hydrotreatment of pyrolysis bio-oil: A review. Fuel Process. Technol. 2019, 195, 106140. [Google Scholar] [CrossRef]
- Jin, W.; Pérez, L.P.; Shen, D.; Sepúlveda-Escribano, A.; Gu, S.; Reina, T.R. Catalytic Upgrading of Biomass Model Compounds: Novel Approaches and Lessons Learnt from Traditional Hydrodeoxygenation—A Review. ChemCatChem 2018, 11, 924–960. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Kwon, E.E.; Kim, Y.T.; Jung, S.; Kim, H.J.; Huber, G.W.; Lee, J. Recent advances in hydrodeoxygenation of biomass-derived oxygenates over heterogeneous catalysts. Green Chem. 2019, 21, 3715–3743. [Google Scholar] [CrossRef]
- Shafaghat, H.; Rezaei, P.S.; Daud, W.M.A.W. Catalytic hydrodeoxygenation of simulated phenolic bio-oil to cycloalkanes and aromatic hydrocarbons over bifunctional metal/acid catalysts of Ni/HBeta, Fe/HBeta and NiFe/HBeta. J. Ind. Eng. Chem. 2016, 35, 268–276. [Google Scholar] [CrossRef]
- Leal, G.F.; Lima, S.; Graça, I.; Carrer, H.; Barrett, D.; Teixeira-Neto, E.; Curvelo, A.A.S.; Rodella, C.; Rinaldi, R. Design of Nickel Supported on Water-Tolerant Nb2O5 Catalysts for the Hydrotreating of Lignin Streams Obtained from Lignin-First Biorefining. iScience 2019, 15, 467–488. [Google Scholar] [CrossRef] [Green Version]
- Mäki-Arvela, P.; Murzin, D.Y. Hydrodeoxygenation of Lignin-Derived Phenols: From Fundamental Studies towards Industrial Applications. Catalysts 2017, 7, 265. [Google Scholar] [CrossRef] [Green Version]
- Tran, N.; Uemura, Y.; Trinh, T.; Ramli, A. Hydrodeoxygenation of Guaiacol over Pd–Co and Pd–Fe Catalysts: Deactivation and Regeneration. Processes 2021, 9, 430. [Google Scholar] [CrossRef]
- Si, Z.; Zhang, X.; Wang, C.; Ma, L.; Dong, R. An Overview on Catalytic Hydrodeoxygenation of Pyrolysis Oil and Its Model Compounds. Catalysts 2017, 7, 169. [Google Scholar] [CrossRef] [Green Version]
- Robinson, A.M.; Hensley, J.E.; Medlin, J. Bifunctional Catalysts for Upgrading of Biomass-Derived Oxygenates: A Review. ACS Catal. 2016, 6, 5026–5043. [Google Scholar] [CrossRef]
- Lugo-José, Y.K.; Monnier, J.R.; Williams, C.T. Gas-phase, catalytic hydrodeoxygenation of propanoic acid, over supported group VIII noble metals: Metal and support effects. Appl. Catal. A Gen. 2014, 469, 410–418. [Google Scholar] [CrossRef]
- Resende, K.A.; Hori, C.; Noronha, F.B.; Shi, H.; Gutierrez, O.Y.; Camaioni, D.M.; Lercher, J. Aqueous phase hydrogenation of phenol catalyzed by Pd and PdAg on ZrO2. Appl. Catal. A Gen. 2017, 548, 128–135. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, K.; Nolte, M.W.; Choi, Y.S.; Brown, R.C.; Shanks, B.H. Catalytic Deoxygenation of Bio-Oil Model Compounds over Acid–Base Bifunctional Catalysts. ACS Catal. 2016, 6, 2608–2621. [Google Scholar] [CrossRef]
- Dixon, D.J. Bifunctional catalysis. Beilstein J. Org. Chem. 2016, 12, 1079–1080. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, A.; Kaila, R.; Honkela, M.; Slioor, R.; Krause, A. Hydrodeoxygenation of guaiacol on noble metal catalysts. Catal. Today 2009, 147, 239–246. [Google Scholar] [CrossRef]
- Tanabe, K.; Hölderich, W.F. Industrial application of solid acid-base catalysts. Appl. Catal. A Gen. 1999, 181, 399–434. [Google Scholar] [CrossRef]
- Noronha, F.; Frydman, A.; Aranda, D.; Perez, C.; Soares, R.; Morawek, B.; Castner, D.; Campbell, C.; Frety, R.; Schmal, M. The promoting effect of noble metal addition on niobia-supported cobalt catalysts. Catal. Today 1996, 28, 147–157. [Google Scholar] [CrossRef]
- Nowak, I.; Ziolek, M. Niobium Compounds: Preparation, Characterization, and Application in Heterogeneous Catalysis. Chem. Rev. 1999, 99, 3603–3624. [Google Scholar] [CrossRef] [PubMed]
- Passos, F.B.; Aranda, D.A.G.; Soares, R.R.; Schmal, M. Effect of preparation method on the properties of Nb2O5 promoted platinum catalysts. Catal. Today 1998, 43, 3–9. [Google Scholar] [CrossRef]
- Tanabe, K. Catalytic applications of niobium compounds. Niobium Sci. Technol. 2001, 78, 269–290. [Google Scholar]
- Barrios, A.M.; Teles, C.A.; De Souza, P.M.; Rabelo-Neto, R.C.; Jacobs, G.; Davis, B.H.; Borges, L.E.; Noronha, F.B. Hydrodeoxygenation of phenol over niobia supported Pd catalyst. Catal. Today 2017, 302, 115–124. [Google Scholar] [CrossRef]
- Jing, Y.; Xin, Y.; Guo, Y.; Liu, X.; Wang, Y. Highly efficient Nb2O5 catalyst for aldol condensation of biomass-derived carbonyl molecules to fuel precursors. Chin. J. Catal. 2019, 40, 1168–1177. [Google Scholar] [CrossRef]
- Teles, C.A.; De Souza, P.M.; Rabelo-Neto, R.C.; Griffin, M.B.; Mukarakate, C.; Orton, K.A.; Resasco, D.E.; Noronha, F.B. Catalytic upgrading of biomass pyrolysis vapors and model compounds using niobia supported Pd catalyst. Appl. Catal. B Environ. 2018, 238, 38–50. [Google Scholar] [CrossRef]
- Dong, L.; Shao, Y.; Han, X.; Liu, X.; Xia, Q.; Parker, S.F.; Cheng, Y.Q.; Daemen, L.L.; Ramirez-Cuesta, A.J.; Wang, Y.; et al. Comparison of two multifunctional catalysts [M/Nb2O5 (M = Pd, Pt)] for one-pot hydrodeoxygenation of lignin. Catal. Sci. Technol. 2018, 8, 6129–6136. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Karim, A.; Zhang, H.; Kovarik, L.; Li, X.S.; Hensley, A.; McEwen, J.-S.; Wang, Y. Carbon-supported bimetallic Pd–Fe catalysts for vapor-phase hydrodeoxygenation of guaiacol. J. Catal. 2013, 306, 47–57. [Google Scholar] [CrossRef]
- Kim, J.K.; Lee, J.K.; Kang, K.H.; Song, J.C.; Song, I.K. Selective cleavage of C–O bond in benzyl phenyl ether to aromatics over Pd–Fe bimetallic catalyst supported on ordered mesoporous carbon. Appl. Catal. A Gen. 2015, 498, 142–149. [Google Scholar] [CrossRef]
- Channiwala, S.A.; Parikh, P.P. A unified correlation for estimating HHV of solid, liquid and gaseous fuels. Fuel 2002, 81, 1051–1063. [Google Scholar] [CrossRef]
- Joseph, J.; Baker, C.; Mukkamala, S.; Beis, S.H.; Wheeler, M.C.; DeSisto, W.J.; Jensen, B.L.; Frederick, B.G. Chemical Shifts and Lifetimes for Nuclear Magnetic Resonance (NMR) Analysis of Biofuels. Energy Fuels 2010, 24, 5153–5162. [Google Scholar] [CrossRef]
- Shao, Y.; Xia, Q.; Liu, X.; Lu, G.; Wang, Y. Pd/Nb2O5/SiO2 Catalyst for the Direct Hydrodeoxygenation of Biomass-Related Compounds to Liquid Alkanes under Mild Conditions. ChemSusChem 2015, 8, 1761–1767. [Google Scholar] [CrossRef]
- Nakajima, K.; Hirata, J.; Kim, M.; Gupta, N.K.; Murayama, T.; Yoshida, A.; Hiyoshi, N.; Fukuoka, A.; Ueda, W. Facile Formation of Lactic Acid from a Triose Sugar in Water over Niobium Oxide with a Deformed Orthorhombic Phase. ACS Catal. 2018, 8, 283–290. [Google Scholar] [CrossRef]
- Kon, K.; Onodera, W.; Takakusagi, S.; Shimizu, K.I. Hydrodeoxygenation of fatty acids and triglycerides by Pt-loaded Nb2O5catalysts. Catal. Sci. Technol. 2014, 4, 3705–3712. [Google Scholar] [CrossRef]
- Boscagli, C.; Raffelt, K.; Zevaco, T.A.; Olbrich, W.; Otto, T.N.; Sauer, J.; Grunwaldt, J.-D. Mild hydrotreatment of the light fraction of fast-pyrolysis oil produced from straw over nickel-based catalysts. Biomass Bioenergy 2015, 83, 525–538. [Google Scholar] [CrossRef] [Green Version]
- French, R.J.; Stunkel, J.; Baldwin, R.M. Mild Hydrotreating of Bio-Oil: Effect of Reaction Severity and Fate of Oxygenated Species. Energy Fuels 2011, 25, 3266–3274. [Google Scholar] [CrossRef]
- Mercader, F.D.M.; Groeneveld, M.J.; Kersten, S.R.A.; Geantet, C.; Toussaint, G.; Way, N.W.J.; Schaverien, C.J.; Hogendoorn, K.J.A. Hydrodeoxygenation of pyrolysis oil fractions: Process understanding and quality assessment through co-processing in refinery units. Energy Environ. Sci. 2011, 4, 985–997. [Google Scholar] [CrossRef]
- Venderbosch, R.H.; Ardiyanti, A.R.; Wildschut, J.; Oasmaa, A.; Heeres, H.J. Stabilization of biomass-derived pyrolysis oils. J. Chem. Technol. Biotechnol. 2010, 85, 674–686. [Google Scholar] [CrossRef]
- Mercader, F.D.M.; Koehorst, P.J.J.; Heeres, H.; Kersten, S.R.A.; Hogendoorn, J.A. Competition between hydrotreating and polymerization reactions during pyrolysis oil hydrodeoxygenation. AIChE J. 2010, 57, 3160–3170. [Google Scholar] [CrossRef]
- Haynes, W.M. CRC Handbook of Chemistry and Physics; CRC Press: New York, NY, USA, 2014. [Google Scholar]
- Dean, J.A. Lange’s Handbook of Chemistry, 15th ed.; McGraw-Hill: New York, NY, USA, 1998. [Google Scholar]
- Meiboom, S. Nuclear Magnetic Resonance Study of the Proton Transfer in Water. J. Chem. Phys. 1961, 34, 375–388. [Google Scholar] [CrossRef]
- Bouxin, F.P.; Zhang, X.; Kings, I.N.; Lee, A.F.; Simmons, M.J.H.; Wilson, K.; Jackson, S.D. Mechanistic Aspects of Hydrodeoxygenation of p-Methylguaiacol over Rh/Silica and Pt/Silica. Org. Process Res. Dev. 2018, 22, 1586–1589. [Google Scholar] [CrossRef]
- Schmitt, C.C.; Moreira, R.; Neves, R.C.; Richter, D.; Funke, A.; Raffelt, K.; Grunwaldt, J.-D.; Dahmen, N. From agriculture residue to upgraded product: The thermochemical conversion of sugarcane bagasse for fuel and chemical products. Fuel Process. Technol. 2019, 197, 106199. [Google Scholar] [CrossRef]
- Dutta, S.; Bhat, N.S. Catalytic Transformation of Biomass-Derived Furfurals to Cyclopentanones and Their Derivatives: A Review. ACS Omega 2021, 6, 35145–35172. [Google Scholar] [CrossRef]
- Girisuta, B.; Janssen, L.P.B.M.; Heeres, H.J. A kinetic study on the decomposition of 5-hydroxymethylfurfural into levulinic acid. Green Chem. 2006, 8, 701–709. [Google Scholar] [CrossRef] [Green Version]
- Ren, D.; Song, Z.; Li, L.; Liu, Y.; Jin, F.; Huo, Z. Production of 2,5-hexanedione and 3-methyl-2-cyclopenten-1-one from 5-hydroxymethylfurfural. Green Chem. 2016, 18, 3075–3081. [Google Scholar] [CrossRef]
- Li, X.; Wan, W.; Chen, J.G.; Wang, T. Selective Hydrogenation of Biomass-Derived 2(5H)-Furanone to γ-Butyrolactone over Ni-Based Bimetallic Catalysts. ACS Sustain. Chem. Eng. 2018, 6, 16039–16046. [Google Scholar] [CrossRef]
- Mukundan, S.; Wahab, A.; Atanda, L.; Konarova, M.; Beltramini, J. Highly active and robust Ni–MoS2supported on mesoporous carbon: A nanocatalyst for hydrodeoxygenation reactions. RSC Adv. 2019, 9, 17194–17202. [Google Scholar] [CrossRef] [Green Version]
- Olcese, R.; Bettahar, M.M.; Malaman, B.; Ghanbaja, J.; Tibavizco, L.; Petitjean, D.; Dufour, A. Gas-phase hydrodeoxygenation of guaiacol over iron-based catalysts. Effect of gases composition, iron load and supports (silica and activated carbon). Appl. Catal. B Environ. 2013, 129, 528–538. [Google Scholar] [CrossRef]
- Hong, Y.; Zhang, H.; Sun, J.; Ayman, K.M.; Hensley, A.J.R.; Gu, M.; Engelhard, M.H.; McEwen, J.-S.; Wang, Y. Synergistic Catalysis between Pd and Fe in Gas Phase Hydrodeoxygenation of m-Cresol. ACS Catal. 2014, 4, 3335–3345. [Google Scholar] [CrossRef]
- Yang, Y.; Tan, M.; Garcia, A.; Zhang, Z.; Lin, J.; Wan, S.; McEwen, J.-S.; Wang, S.; Wang, Y. Controlling the Oxidation State of Fe-Based Catalysts through Nitrogen Doping toward the Hydrodeoxygenation of m-Cresol. ACS Catal. 2020, 10, 7884–7893. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, J.; Zhang, L.; Tan, M.; Lin, J.; Wan, S.; Wang, S.; Wang, Y. Enhanced Antioxidation Stability of Iron-Based Catalysts via Surface Decoration with ppm Platinum. ACS Sustain. Chem. Eng. 2018, 6, 14010–14016. [Google Scholar] [CrossRef]
- Popov, A.; Kondratieva, E.; Goupil, J.M.; Mariey, L.; Bazin, P.; Gilson, J.-P.; Travert, A.; Maugé, F. Bio-oils Hydrodeoxygenation: Adsorption of Phenolic Molecules on Oxidic Catalyst Supports. J. Phys. Chem. C 2010, 114, 15661–15670. [Google Scholar] [CrossRef]
- Ko, E.; Weissman, J. Structures of niobium pentoxide and their implications on chemical behavior. Catal. Today 1990, 8, 27–36. [Google Scholar] [CrossRef]
- Munoz, M.; de Pedro, Z.M.; Menendez, N.; Casas, J.A.; Rodriguez, J.J. A ferromagnetic γ-alumina-supported iron catalyst for CWPO. Application to chlorophenols. Appl. Catal. B Environ. 2013, 136–137, 218–224. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C (wt.%) | H (wt.%) | N (wt.%) | O (wt.%) | H2O (wt.%) | HHV (MJ/kg) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| wb. | db. | wb. | db. | wb. | db. | wb. | db. | wb. | wb. | db. | |
| Feedstock | 32.9 | 52.8 | 8.1 | 6.2 | 0.1 | 0.2 | 58.9 | 40.7 | 37.7 | 14.9 | 21.6 |
| Upgraded oil | |||||||||||
| Blank | 10.0 | - | 6.3 | - | 0.1 | - | 31.1 | - | - | - | 26.0 |
| Pd/SiO2 | 64.2 ± 0.2 | 69.0 ± 0.8 | 7.2 ± 0.1 | 6.9 ± 0.2 | 0.1 ± 0.0 | 0.1 ± 0.0 | 28.6 ± 0.1 | 24.0 ± 0.6 | 7.0 ± 0.7 | 27.9 ± 0.1 | 29.7 ± 0.1 |
| Pd/Nb2O5 | 61.8 ± 0.2 | 67.8 ± 0.1 | 7.7 ± 0.1 | 7.4 ± 0.1 | 0.3 ± 0.0 | 0.3 ± 0.0 | 30.2 ± 0.1 | 24.5 ± 0.2 | 8.8 ± 0.3 | 27.5 ± 0.1 | 29.8 ± 0.2 |
| Pd/1%Fe/Nb2O5 | 62.1 ± 0.2 | 68.3 ± 0.1 | 7.6 ± 0.1 | 7.2 ± 0.1 | 0.3 ± 0.0 | 0.3 ± 0.0 | 30.0 ± 0.2 | 24.2 ± 0.1 | 9.1 ± 0.3 | 27.5 ± 0.1 | 29.8 ± 0.1 |
| Pd/8%Fe/Nb2O5 | 62.8 ± 0.2 | 68.7 ± 0.3 | 7.9 ± 0.0 | 7.5 ± 0.0 | 0.3 ± 0.0 | 0.3 ± 0.0 | 29.0 ± 0.2 | 23.4 ± 0.3 | 8.6 ± 0.2 | 28.2 ± 0.1 | 30.5 ± 0.1 |
| Aqueous phase | |||||||||||
| Blank | 12.2 | 45.5 | 9.9 | 6.6 | 1.7 | 6.3 | 23.8 | 41.6 | 73.2 | 13.4 | 19.3 |
| Pd/SiO2 | 13.9 ± 0.8 | 48.5 ± 3.1 | 9.6 ± 0.0 | 5.8 ± 0.0 | 0.3 ± 0.0 | 1.0 ± 0.0 | 76.4 ± 0.9 | 44.7 ± 3.1 | 71.4 ± 0.1 | 8.3 ± 0.4 | 19.1 ± 1.4 |
| Pd/Nb2O5 | 15.3 ± 0.1 | 48.7 ± 0.4 | 9.9 ± 0.1 | 7.5 ± 0.2 | 0.3 ± 0.0 | 1.0 ± 0.0 | 74.4 ± 0.1 | 42.8 ± 0.5 | 68.5 ± 0.2 | 9.3 ± 0.01 | 21.4 ± 0.3 |
| Pd/1%Fe/Nb2O5 | 15.1 ± 0.1 | 48.7 ± 0.4 | 9.6 ± 0.0 | 6.3 ± 0.1 | 0.3 ± 0.0 | 1.0 ± 0.1 | 75.0 ± 0.1 | 44.1 ± 0.4 | 68.9 ± 0.4 | 8.8 ± 0.0 | 19.8 ± 0.1 |
| Pd/8%Fe/Nb2O5 | 14.9 ± 0.4 | 49.5 ± 2.0 | 9.9 ± 0.6 | 7.1 ± 0.9 | 0.3 ± 0.0 | 1.0 ± 0.0 | 74.9 ± 0.6 | 42.4 ± 2.4 | 69.8 ± 0.6 | 9.1 ± 0.5 | 21.2 ± 1.7 |
| Integration Range (ppm) | Proton Assignments |
|---|---|
| 12.5–11.0 | carboxylic acids |
| 11.0–8.25 | -CHO, aromatic OH |
| 8.25–6.0 | aromatic, conjugated -C=C- |
| 6.0–4.8 | aliphatic OH, -C=C-, Ar-CH2-O- |
| 4.8–3.0 | ether, methoxy |
| 3.0–1.8 | CH2C=O, aliphatics, CH3 of acetic acid, CH2 of propanoic acid |
| 1.8–0.1 | aliphatic |
| Retention Time | Chemical Compound | Retention Time | Chemical Compound |
|---|---|---|---|
| 3.305 | 2-furaldehyde | 7.398 | maltol |
| 3.392 | 2-methyl-cyclopentanone | 7.482 | 3-ethyl-2-hydroxy-2-cyclopenten-1-one |
| 3.414 | 2-hydroxyethyl ester acetic acid | 7.87 | 2,4-dimethyl-phenol |
| 3.467 | 3-methyl-cyclopentanone | 8.604 | 4-methylguaiacol |
| 3.653 | 1-acetyloxy- 2-propanone | 8.622 | catechol (1,2-Benzenediol) |
| 4.057 | 4-hydroxy-3-hexanone | 8.868 | 1,4;3,6-dianhydro-.α.-D-glucopyranose |
| 4.222 | 2-methyl-2-cyclopenten-1-one | 9.031 | 2,3-anhydro-d-galactosan |
| 4.281 | 1-(2-furanyl)-1-ethanone | 9.098 | 5-hydroxymethylfuraldehyde |
| 4.3 | γ-butyrolactone | 9.132 | 1,6-anhydro-β.-D-glucopyranose |
| 4.336 | 2(5H)-furanone | 9.338 | 2,3-anhydro-d-mannosan |
| 4.485 | 2,5-hexanedione | 9.564 | 3-methyl-catechol |
| 4.657 | 2-hydroxyethyl ester propanoic acid | 9.731 | hydroquinone (1,4-Benzenediol) |
| 4.68 | 5-methyl-2(5H)-furanone | 9.865 | 4-ethylguaiacol |
| 4.69 | 2-ethyl-cyclopentanone | 9.973 | 4-methyl-catechol |
| 4.84 | 2-methyl-cyclohexanone | 10.719 | 2-methyl-hydroquinone |
| 4.899 | γ-valerolactone | 10.866 | syringol |
| 5.045 | 5-methyl-2-furaldehyde | 10.965 | eugenol or vinyl guaiacol |
| 5.086 | 3-methyl-2-cyclopenten-1-one | 11.096 | 4-propylguaiacol |
| 5.24 | phenol | 11.277 | 4-ethylcatechol |
| 5.282 | 3-methyl-2(5H)-furanone | 11.547 | 3-Hydroxy-4-methoxy-benzaldehyde |
| 5.43 | 2-hydroxy-γ-butyrolactone | 11.652 | Eugenol |
| 5.569 | 2-hydroxy-cyclohexanone | 12.132 | 4-Methyl-syringol |
| 5.627 | 2,5-dihydro-3,5-dimethyl-2-furanone | 12.192 | Isoeugenol |
| 5.966 | 3,6-heptanedione | 12.671 | 1-3-hydroxy-4-methoxyphenyl-ethanone (Acetoguaiacone) |
| 6.211 | propyl-ester acetic acid | ||
| 6.318 | 4-methyl-2(5H)-furanone | 13.224 | 1-4-hydroxy-3-methoxyphenyl- 2-propanone (Guaiacyl acetone) |
| 6.4 | x-cresol (x = o, m, p) (methyl-phenol) | ||
| 6.542 | 4-oxopentanoic acid | 13.768 | 1’hydroxy-eugenol |
| 6.711 | x-cresol (x = o, m, p) (methyl-phenol) | 13.863 | 1-4-hydroxy-3-methoxyphenyl-1-propanone (Propioguaiacone) |
| 7.016 | guaiacol (methoxy-phenol) | ||
| 7.34 | 2,5-octanedione |
| Pd (wt.%) | Fe (wt.%) | C–Coke (wt.%) | Surface Area (m2/g) | |
|---|---|---|---|---|
| Support | ||||
| SiO2 | - | - | - | 245.1 |
| Nb2O5 | - | - | - | 25.9 |
| Fresh catalysts | ||||
| Pd/SiO2 | 0.9 | - | - | 220.5 |
| Pd/Nb2O5 | 1.0 (1.4 *) | - | - | 38.8 |
| Pd/1%Fe/Nb2O5 | 1.0 | 0.9 | - | 37.6 |
| Pd/8%Fe/Nb2O5 | 1.0 (1.4 *) | 7.8 (15.2 *) | - | 33.3 |
| Spent catalysts | ||||
| Pd/SiO2 | 0.7 | - | 1.7 | - |
| Pd/Nb2O5 | 0.9 | 0.1 | 2.8 | - |
| Pd/1%Fe/Nb2O5 | 0.9 | 0.3 | 3.0 | - |
| Pd/8%Fe/Nb2O5 | 1.0 | 3.8 | 2.6 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campos Fraga, M.M.; Lacerda de Oliveira Campos, B.; Hendrawidjaja, H.; Carriel Schmitt, C.; Raffelt, K.; Dahmen, N. Fast Pyrolysis Oil Upgrading via HDO with Fe-Promoted Nb2O5-Supported Pd-Based Catalysts. Energies 2022, 15, 4762. https://doi.org/10.3390/en15134762
Campos Fraga MM, Lacerda de Oliveira Campos B, Hendrawidjaja H, Carriel Schmitt C, Raffelt K, Dahmen N. Fast Pyrolysis Oil Upgrading via HDO with Fe-Promoted Nb2O5-Supported Pd-Based Catalysts. Energies. 2022; 15(13):4762. https://doi.org/10.3390/en15134762
Chicago/Turabian StyleCampos Fraga, Mariana Myriam, Bruno Lacerda de Oliveira Campos, Handoyo Hendrawidjaja, Caroline Carriel Schmitt, Klaus Raffelt, and Nicolaus Dahmen. 2022. "Fast Pyrolysis Oil Upgrading via HDO with Fe-Promoted Nb2O5-Supported Pd-Based Catalysts" Energies 15, no. 13: 4762. https://doi.org/10.3390/en15134762
APA StyleCampos Fraga, M. M., Lacerda de Oliveira Campos, B., Hendrawidjaja, H., Carriel Schmitt, C., Raffelt, K., & Dahmen, N. (2022). Fast Pyrolysis Oil Upgrading via HDO with Fe-Promoted Nb2O5-Supported Pd-Based Catalysts. Energies, 15(13), 4762. https://doi.org/10.3390/en15134762