
Production of Gasolines and Monocyclic Aromatic Hydrocarbons: From Fossil Raw Materials to Green Processes


Abstract
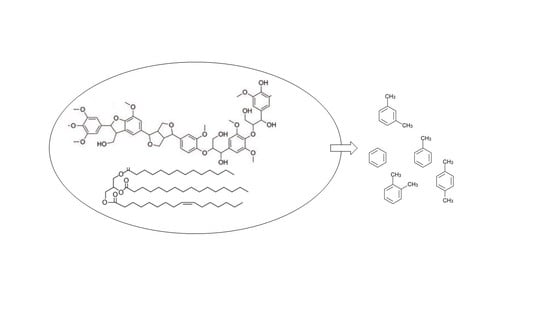
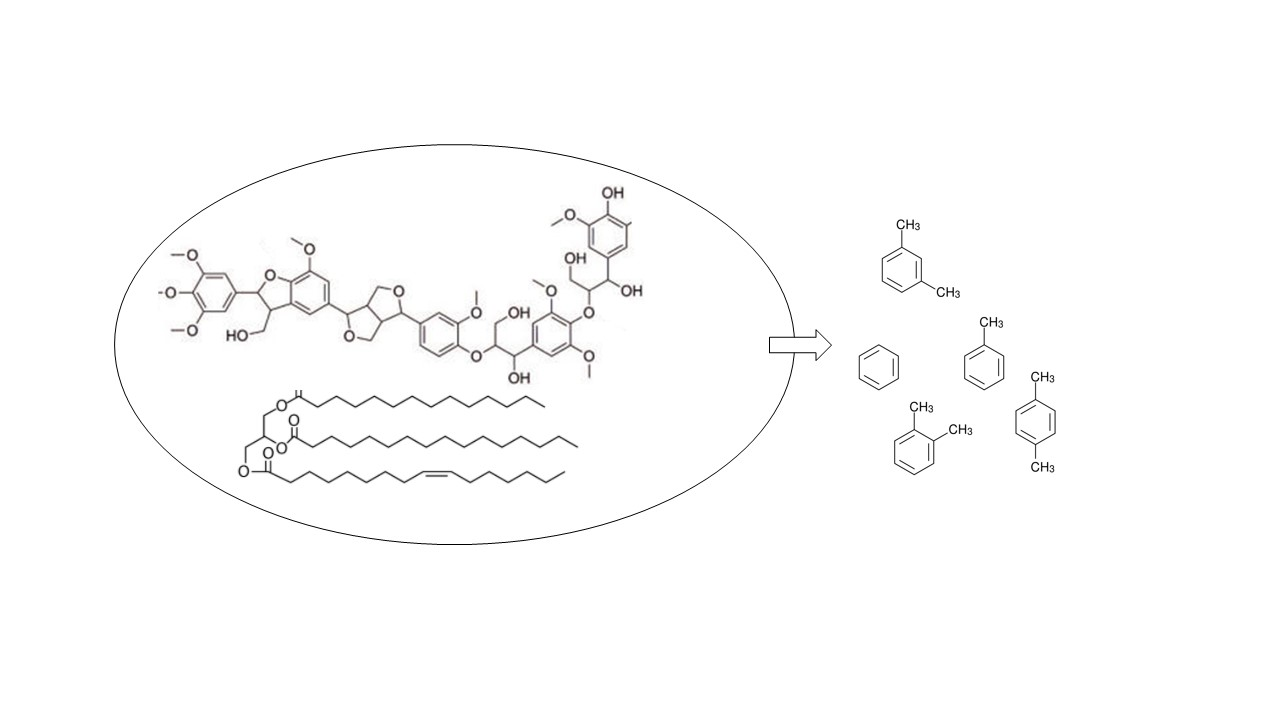
1. Introduction
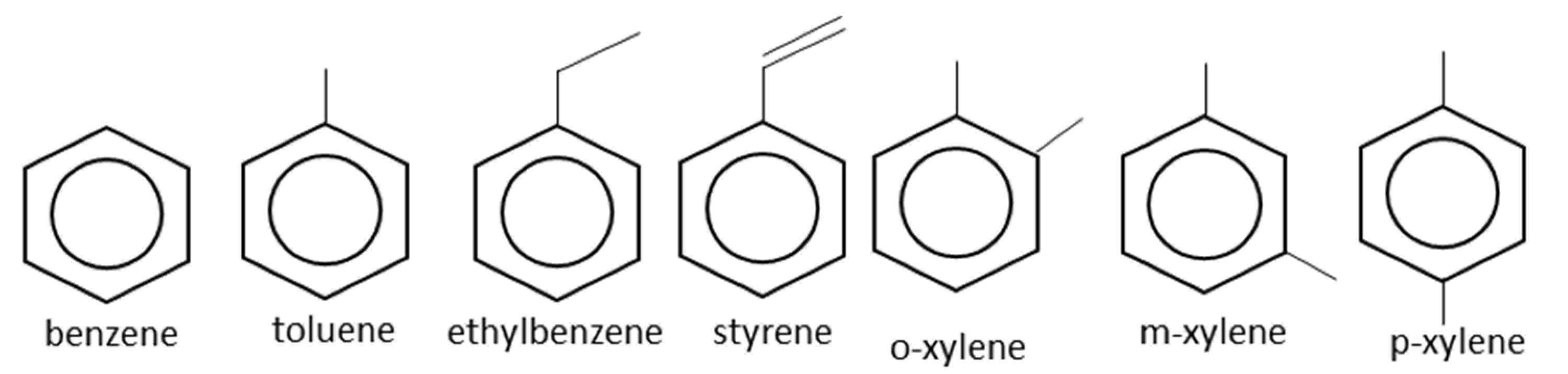
2. Applications of Monocyclic Aromatic Hydrocarbons
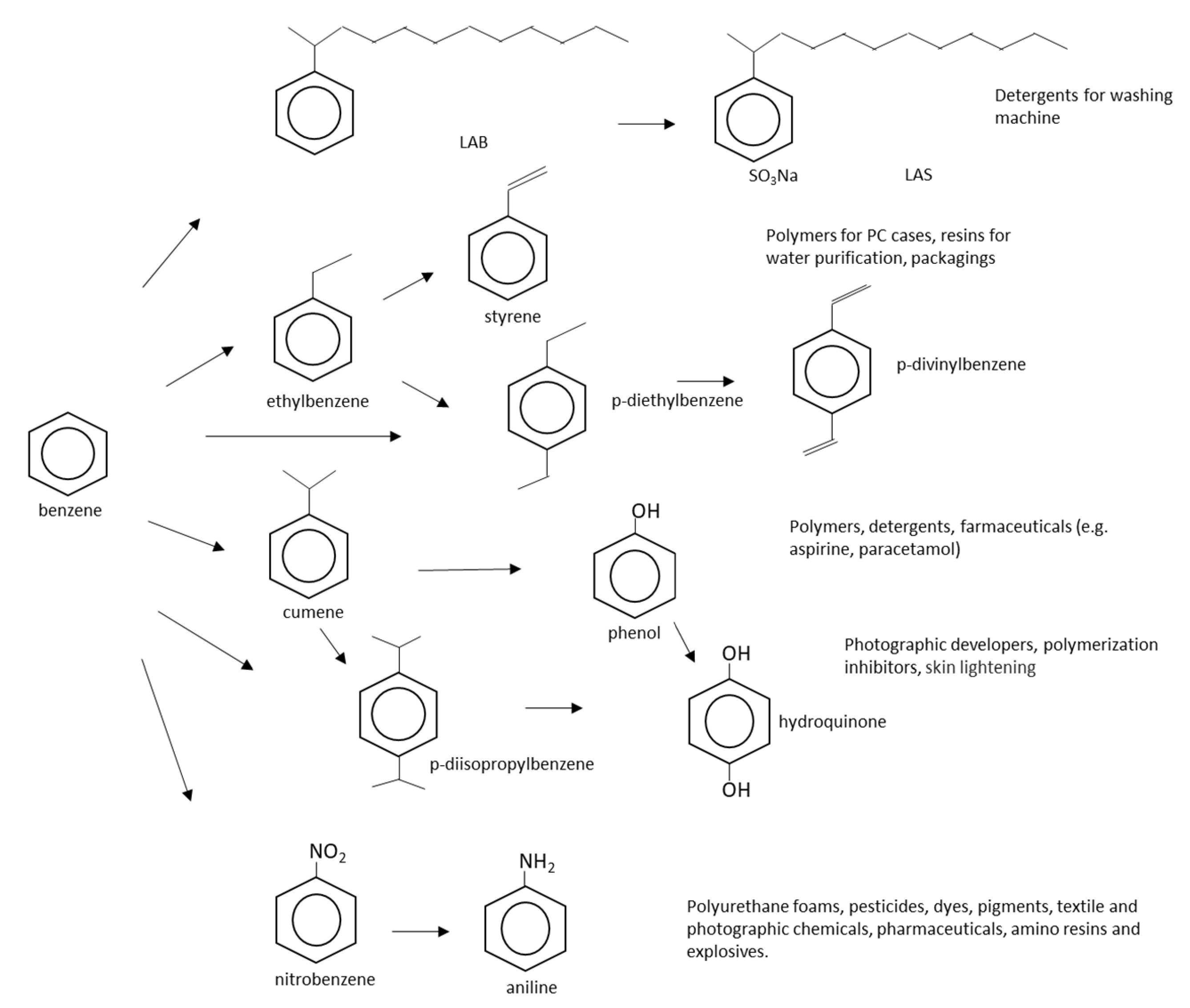
2.1. Benzene
2.2. Toluene
2.3. Ethylbenzene
2.4. Styrene
2.5. Mixed Xylenes
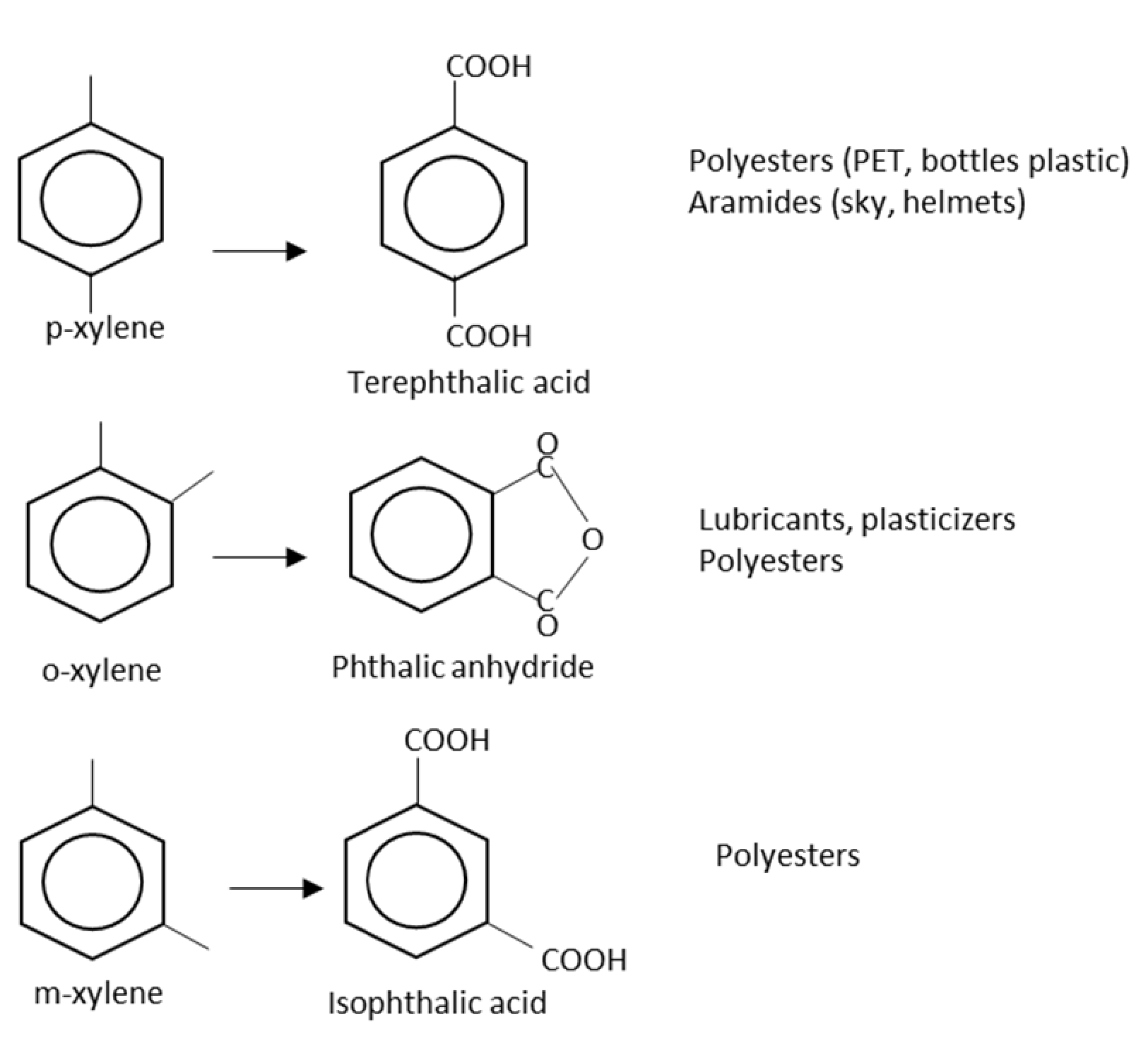
2.6. Ortho-Xylene
2.7. Para-Xylene
2.8. Meta-Xylene
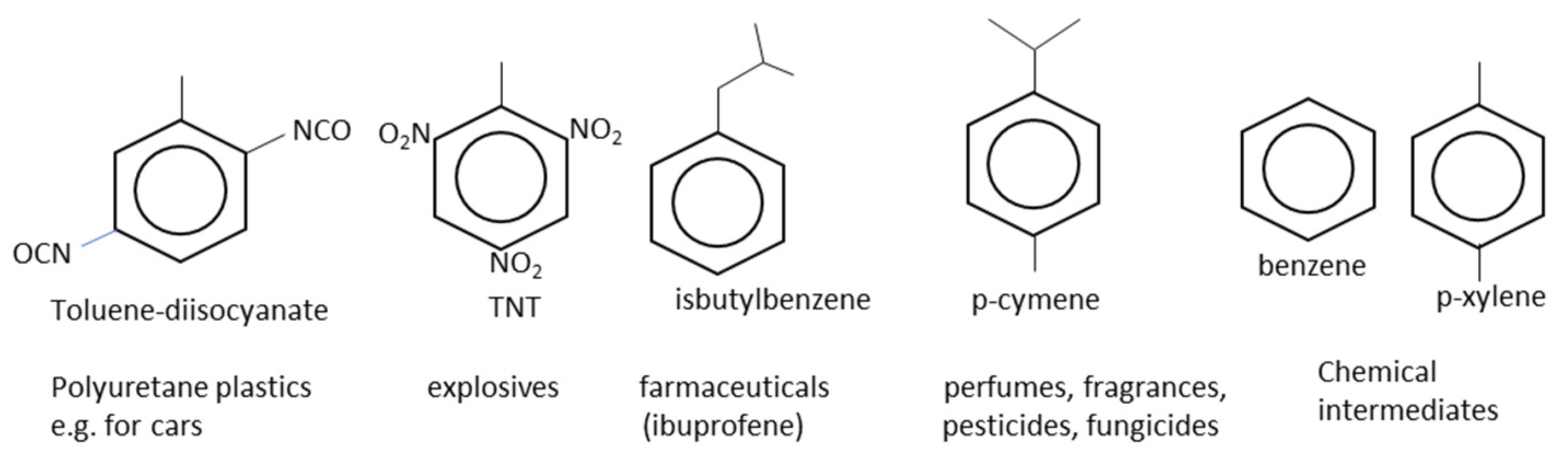
2.9. Other Industrially Relevant Monocyclic Aromatic Hydrocarbons
3. Manufacture of Monocyclic Aromatic Hydrocarbons and Gasolines from Fossil Raw Materials
3.1. Monocyclic Aromatics in Crude Oil and in Commercial Gasolines
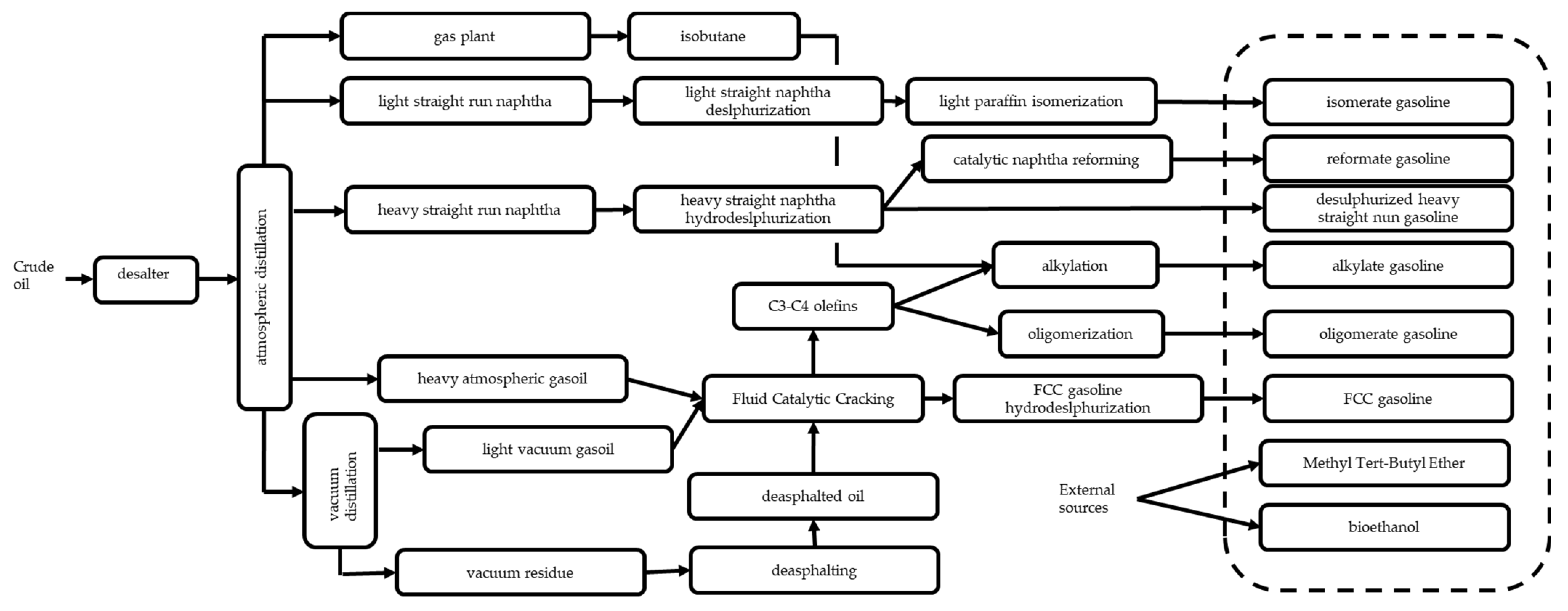
3.2. Conventional Production of Commercial Gasolines and Monocyclic Aromatics from Oil
3.3. The Naphtha Catalytic Reforming Process
3.4. Co-Production of Aromatics in Petrochemical Plants: The Steam Cracking Processes
3.5. Recovery from Coal Benzol
3.6. Methanol to Gasoline (MTG) and Methanol to Aromatics (MTA) Processes
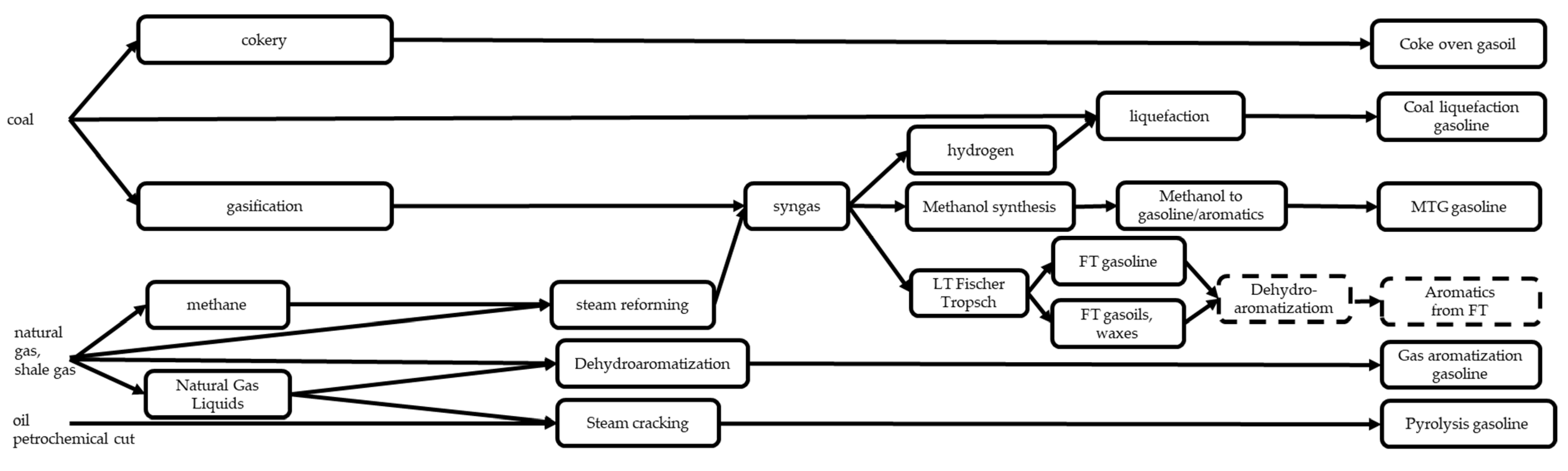
3.7. Other Processes Producing Gasolines from Coal
3.8. Aromatics from Other Sources
4. Separation of Monocyclic Aromatic Hydrocarbons from Naphthas
4.1. Distillation
4.2. Liquid-Liquid Extraction (LLE)
4.3. Extractive Distillation (ED)
4.4. Azeotropic Distillation
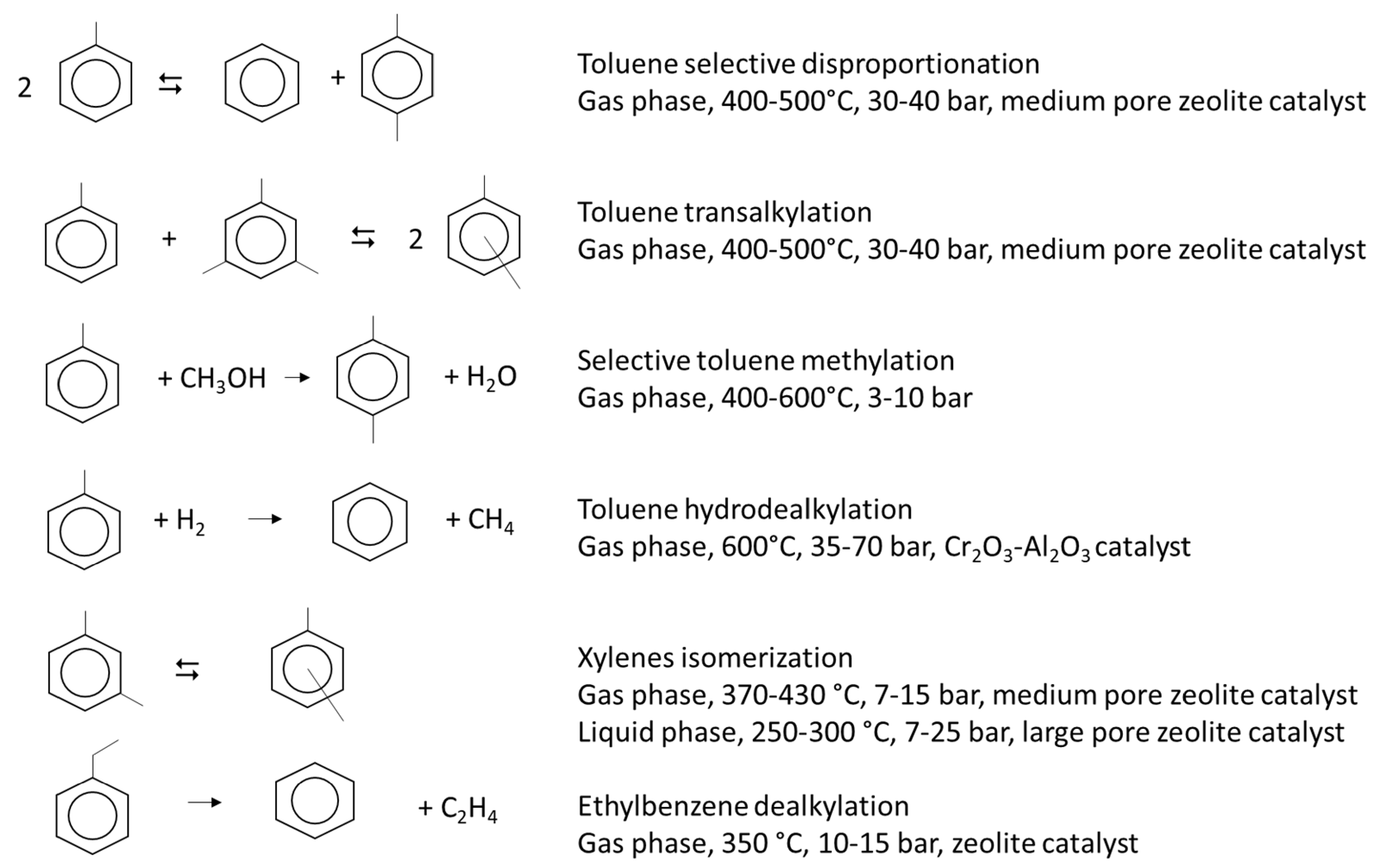
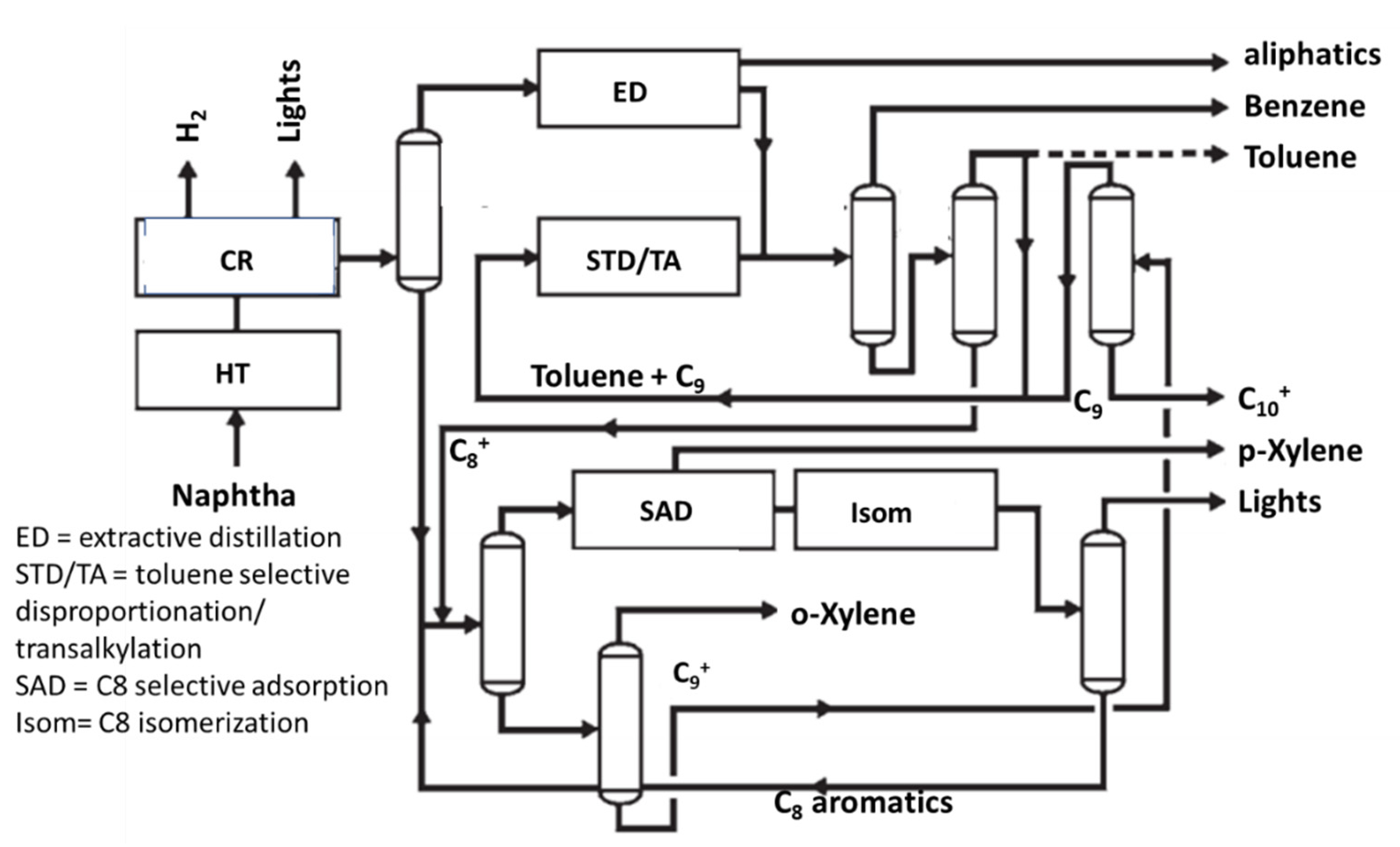
5. The Aromatics Loops
6. Potential Green Processes for the Production of Monocyclic Aromatic Hydrocarbons
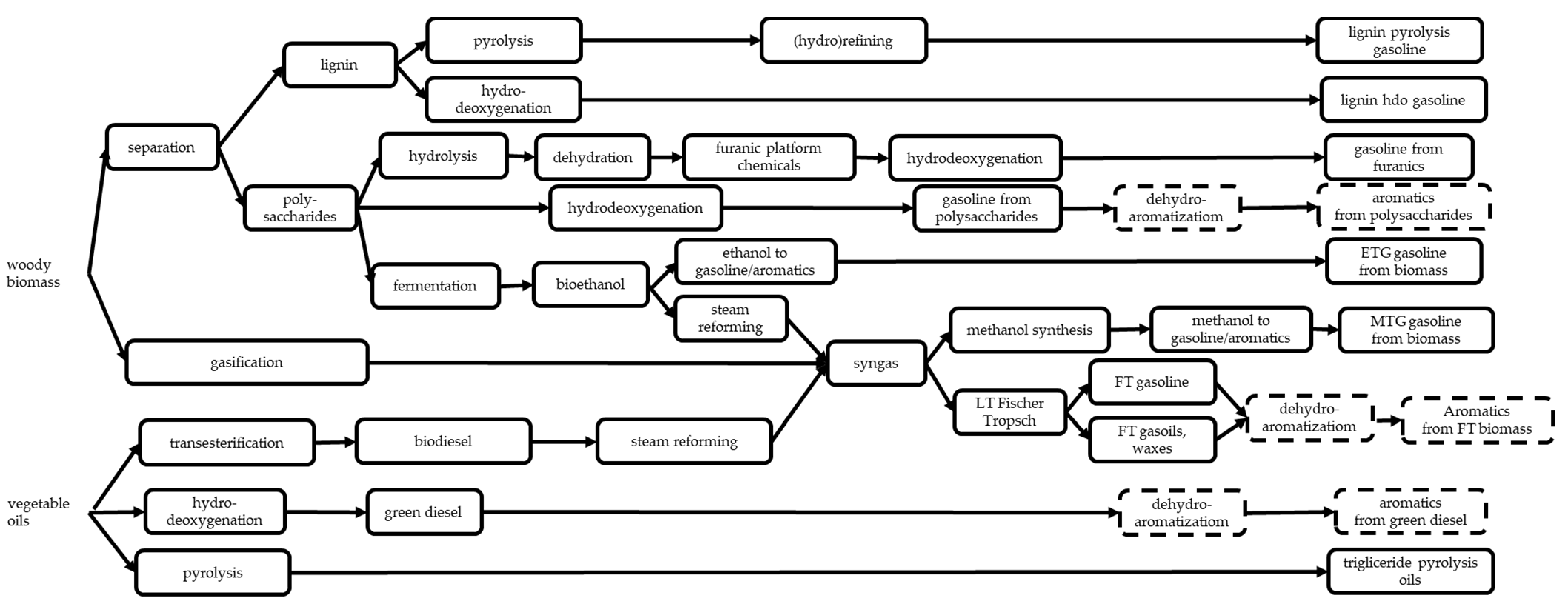
6.1. Biomass vs. Fossil Raw Materials
6.2. Biomethanol to Gasoline and Aromatics
6.2.1. Processes for Producing Biomethanol
6.2.2. Conversion of Biomethanol to Gasoline and Aromatics
6.3. Bioethanol to Gasolines and Aromatics
6.4. Hydrocarbons from Vegetable Oils
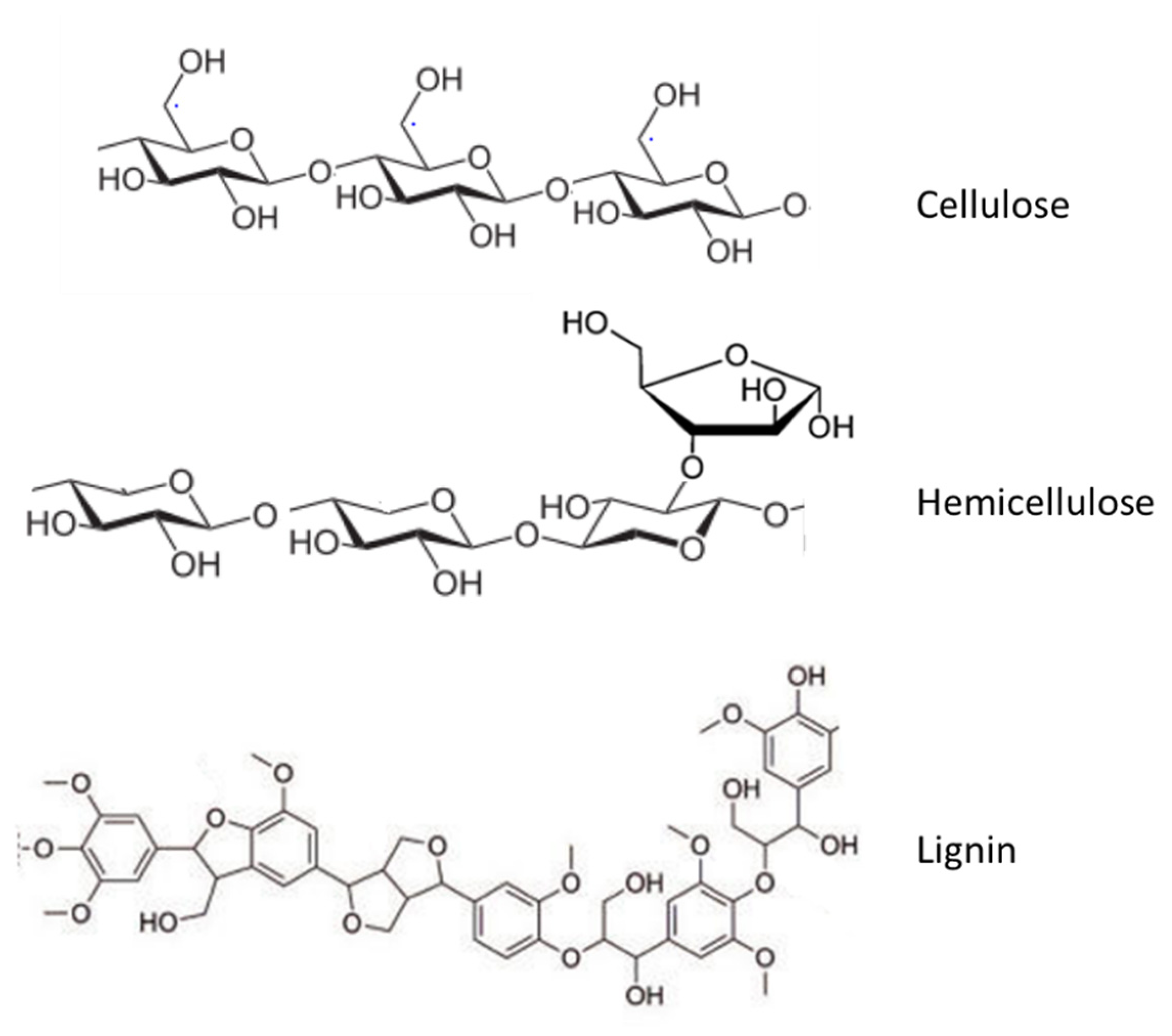
6.5. Lignin Conversion Processes
6.5.1. Lignin Thermal Pyrolysis
6.5.2. Lignin Microwave Pyrolysis
6.5.3. Lignin Catalytic Pyrolysis
6.5.4. Upgrading of Lignin Pyrolysis Oils
6.5.5. Lignin Hydropyrolysis
6.5.6. Lignin Liquefaction
6.6. Conversion of Polysaccharides to Hydrocarbons
6.7. Pyrolysis of Wood and Other Whole Biomass
6.8. Hydrocarbons from Algae
7. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hsu, C.S.; Robinson, P.R. Gasoline Production and Blending; Springer: Berlin, Germany, 2017; pp. 551–587. [Google Scholar] [CrossRef]
- Danielis, R.; Scorrano, M.; Giansoldati, M. Decarbonising transport in Europe: Trends, goals, policies and passenger car scenarios. Res. Transp. Econ. 2021, 101068. [Google Scholar] [CrossRef]
- Gray, N.; McDonagh, S.; O’Shea, R.; Smyth, B.; Murphy, J.D. Decarbonising ships, planes and trucks: An analysis of suitable low-carbon fuels for the maritime, aviation and haulage sectors. Adv. Appl. Energy 2021, 1, 100008. [Google Scholar] [CrossRef]
- Tibaquirá, J.E.; Huertas, J.I.; Ospina, S.; Quirama, L.F.; Niño, J.E. The Effect of Using Ethanol-Gasoline Blends on the Mechanical, Energy and Environmental Performance of In-Use Vehicles. Energies 2018, 11, 221. [Google Scholar] [CrossRef]
- Franck, H.-G.; Stadelhofer, J.W. Industrial Aromatic Chemistry; Springer: Berlin, Heidelberg, Germany, 1987. [Google Scholar]
- Speight, J.G. Handbook of Industrial Hydrocarbon Processes, 2nd ed.; Elesvier: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Available online: https://www.marketsandmarkets.com/Market-Reports/top-15-petrochemicals-market-193785497.html (accessed on 19 April 2021).
- Schmidt, V.A. Reactions for making widely used aniline compounds break norms of synthesis. Nat. Cell Biol. 2020, 584, 46–47. [Google Scholar] [CrossRef]
- Dos Santos, V.P.S.; Salgado, A.M.; Torres, A.G.; Pereira, K.S. Benzene as a Chemical Hazard in Processed Foods. Int. J. Food Sci. 2015, 2015, 1–7. [Google Scholar] [CrossRef]
- Talhout, R.; Schulz, T.; Florek, E.; Van Benthem, J.; Wester, P.; Opperhuizen, A. Hazardous Compounds in Tobacco Smoke. Int. J. Environ. Res. Public Health 2011, 8, 613–628. [Google Scholar] [CrossRef]
- Available online: https://www.statista.com/statistics/1099350/benzene-demand-globally/ (accessed on 23 April 2021).
- Smith, M.T. Advances in understanding benzene health effects and susceptibility. Annu. Rev. Public Health 2010, 31, 133–148. [Google Scholar] [CrossRef]
- Loomis, D.; Guyton, K.Z.; Grosse, Y.; El Ghissassi, F.; Bouvard, V.; Benbrahim-Tallaa, L.; Guha, N.; Vilahur, N.; Mattock, H.; Straif, K. Carcinogenicity of benzene. Lancet Oncol. 2017, 18, 1574–1575. [Google Scholar] [CrossRef]
- Altman, R. How the Benzene Tree Polluted the World. Available online: https://www.theatlantic.com/science/archive/2017/10/benzene-tree-organic-compounds/530655/ (accessed on 27 May 2021).
- Wittcoff, H.A.; Reuben, B.G.; Plotkin, J.S. Chemicals from Benzene; Wiley: Hoboken, NJ, USA, 2013; pp. 323–373. [Google Scholar] [CrossRef]
- Fiebelkorn, S.; Meredith, C. Estimation of the Leukemia Risk in Human Populations Exposed to Benzene from Tobacco Smoke Using Epidemiological Data. Risk Anal. 2017, 38, 1490–1501. [Google Scholar] [CrossRef]
- Available online: https://www.aiplastics.com/blog/applications-where-nylon-is-used/ (accessed on 27 May 2021).
- Available online: https://www.statista.com/statistics/1065877/global-toluene-production-capacity/#:~:text=Global%20production%20capacity%20of%20toluene%202018%20%26%202023&text=A%20five%2Dyear%20projection%20of,this%20chemical%20worldwide%20in%202018 (accessed on 27 May 2021).
- Kim, S.; Park, E.; Song, S.-H.; Lee, C.-W.; Kwon, J.-T.; Park, E.Y.; Kim, B. Toluene concentrations in the blood and risk of thyroid cancer among residents living near national industrial complexes in South Korea: A population-based cohort study. Environ. Int. 2021, 146, 106304. [Google Scholar] [CrossRef]
- Wittcoff, H.A.; Reuben, B.G.; Plotkin, J.S. Chemicals from Toluene; Wiley: Hoboken, NJ, USA, 2013; pp. 375–382. [Google Scholar] [CrossRef]
- Ding, Z.; Pawar, P. GT-TolAlk: Toluene Methylation to Xylenes. In Handbook of Petrochemical Production Processes, 2nd ed.; Meyers, R.A., Ed.; McGraw Hill: New York, NY, USA, 2019; Chapter 1.11. [Google Scholar]
- Huff, J.; Chan, P.; Melnick, R. Clarifying carcinogenicity of ethylbenzene. Regul. Toxicol. Pharmacol. 2010, 58, 167–169. [Google Scholar] [CrossRef]
- Maerz, B.; Stein, L. Badger Ethylbenzene Technology. In Handbook of Petrochemical Production Processes, 2nd ed.; Meyers, R.A., Ed.; McGraw Hill: New York, NY, USA, 2019; Chapter 1.2. [Google Scholar]
- Donahoe, G.; Hubbell, D. Badger Styrene Technology. In Handbook of Petrochemical Production Processes; Meyers, R.A., Ed.; McGraw Hill: New York, NY, USA, 2019; Chapter 5.1. [Google Scholar]
- Available online: https://www.nevicolor.it/Apps/WebObjects/Nevicolor.woa/wa/viewFile?id=5858&lang=eng (accessed on 27 May 2021).
- Liu, Z.; Joshi, S.; Ma, H.; Zhang, R. Best practices for pygas-based styrene extraction, Hydrocarbon Process. March 2021. Available online: https://www.hydrocarbonprocessing.com/magazine/2021/march-2021/special-focus-petrochemical-technology/best-practices-for-pygas-based-styrene-extraction (accessed on 27 May 2021).
- Bao, Z.; Yang, L.; Cheng, Z.-M.; Zhou, Z. Selective Hydrogenation of the C8 Aromatic Fraction of Pyrolysis Gasoline over NiZn3/α-Al2O3: Experimental and Modeling Studies. Ind. Eng. Chem. Res. 2020, 59, 4322–4332. [Google Scholar] [CrossRef]
- Lynwood, C. (Ed.) Poystyrene, Synthesis, Characteristics and Applications; Nova Publishing: Hauppauge, NY, USA, 2014. [Google Scholar]
- Available online: https://www.repsol.com/en/products-and-services/chemicals/product-range/styrene/index.cshtml (accessed on 27 May 2021).
- Huff, J.; Infante, P.F. Styrene exposure and risk of cancer. Mutagen 2011, 26, 583–584. [Google Scholar] [CrossRef]
- Aransiola, E.F.; Daramola, M.O.; Ojumu, T.V. Xylenes: Production Technologies and Uses. Daramola, M.O., Ed.; Xylenes Nova Science Publishers, Inc.: New York, NY, USA, 2013; pp. 1–12. [Google Scholar]
- Wittcoff, H.A.; Reuben, B.G.; Plotkin, J.S. Chemicals from Xylenes; Wiley: Hoboken, NJ, USA, 2013; pp. 383–405. [Google Scholar] [CrossRef]
- Kandyala, R.; Raghavendra, S.P.C.; Rajasekharan, S.T. Xylene: An overview of its health hazards and preventive measures. J. Oral Maxillofac. Pathol. 2010, 14, 1–5. [Google Scholar] [CrossRef]
- Cavani, F.; Caldarelli, A.; Luciani, S.; Cortelli, C.; Cruzzolin, F. Selective oxidation of o-xylene to phthalic anhydride: From conventional catalysts and technologies toward innovative approaches. Catalysis 2012, 24, 204–222. [Google Scholar]
- Castillo-Welter, F. E PTA: The Lurgi-Eastman/SK Process. In Handbook of Petrochemical Production Processes, 1st ed.; Meyers, R.A., Ed.; Mc Graw Hill: New York, NY, USA, 2005. [Google Scholar]
- Available online: https://www.dupont.it/brands/kevlar.html (accessed on 27 May 2021).
- Long, X.-L.; Wang, Z.-H.; Wu, S.-Q.; Wu, S.-M.; Lv, H.-F.; Yuan, W.-K. Production of isophthalic acid from m-xylene oxidation under the catalysis of the H3PW12O40/carbon and cobalt catalytic system. J. Ind. Eng. Chem. 2014, 20, 100–107. [Google Scholar] [CrossRef]
- Khan, T.; Ram, S. Versalis/Lummus Cumene and Phenol Technology. In Handbook of Petrochemical Production Processes, 2nd ed.; Meyers, R.A., Ed.; McGraw Hill: New York, NY, USA, 2019. [Google Scholar]
- Available online: https://honeywelluop.chinacloudsites.cn/wp-content/uploads/2011/02/UOP-LAB-Complex-Data-Sheet.pdf (accessed on 28 May 2021).
- Cheng, L.S. UOP PAREX™ Process. In Handbook of Petrochemical Production Processes, 2nd ed.; Meyers, R.A., Ed.; McGraw Hill: New York, NY, USA, 2019; Chapter 1.15. [Google Scholar]
- Anastas, P.T.; Hammond, D.G. Inherent Safety at Chemical Sites; Elsevier: Amsterdam, The Netherlands, 2016; pp. 23–118. [Google Scholar] [CrossRef]
- Bennett, R.Q.; Goodwin, J.A.; Rich, J.D. Process for Synthesizing Diisopropylbenzene. U.S. Patent 7,102,043, 5 September 2006. [Google Scholar]
- He, J.; Zhang, J.; Li, J.H.; Zhu, Z.R. Shape-Selective Alkylation of Toluene with Propylene over Modified Zeolite. Adv. Mater. Res. 2011, 396–398, 739–744. [Google Scholar] [CrossRef]
- Gajbhiye, S.R.; Deshmukh, G.P.; Vinu, A.; Kantam, M.L.; Mannepalli, L. PRODUCTION OF p-CYMENE BY ALKYLATION OF TOLUENE WITH PROPAN-2-OL. Catal. Green Chem. Eng. 2019, 2, 121–131. [Google Scholar] [CrossRef]
- Marchese, A.; Arciola, C.R.; Barbieri, R.; Silva, A.S.; Nabavi, S.M.; Sokeng, A.J.T.; Izadi, M.; Jafari, N.J.; Suntar, I.; Daglia, M.; et al. Update on Monoterpenes as Antimicrobial Agents: A Particular Focus on p-Cymene. Materials 2017, 10, 947. [Google Scholar] [CrossRef]
- Schramm, R.M.; Langlois, G.E. The Alkali Metal Catalyzed Alkylation of Toluene with Propylene. J. Am. Chem. Soc. 1960, 82, 4912–4918. [Google Scholar] [CrossRef]
- Agee, B.M.; Mullins, G.; Swartling, D.J. Progress towards a more sustainable synthetic pathway to ibuprofen through the use of solar heating. Sustain. Chem. Process. 2016, 4, 8. [Google Scholar] [CrossRef]
- ASTM. Standard Specification for Aviation Gasolines. Available online: http://www.aviation-fuel.com/pdfs/avgas100llspecsastmd910_2011.pdf (accessed on 27 May 2021).
- Meyers, R.A. (Ed.) Handbook of Petroleum Refining Processes, 4th ed.; McGraw Hill: New York, NY, USA, 2016. [Google Scholar]
- Available online: https://www.enistation.com/instazione/prodotti (accessed on 26 May 2021).
- Available online: https://oilproducts.eni.com/it_IT/prodotti/aviazione (accessed on 26 May 2021).
- Antos, G.J.; Aitani, A.M. (Eds.) Catalytic Naphtha Reforming, 2nd ed.; Dekker: New York, NY, USA, 2004. [Google Scholar]
- Lapinski, M.; Baird, M.L.; James, R. UOP Platforming Process. In Handbook of Petroleum Refining Processes, 3rd ed.; Meyers, R.A., Ed.; Mc Graw-Hill: New York, NY, USA, 2004; pp. 4.3–4.31. [Google Scholar]
- Domergue, B.; le Goff, P.Y. Octanizing Reformer Options, Digital Refining. 2006. Available online: https://www.digitalrefining.com/article/1000276 (accessed on 27 May 2021).
- Lapinski, M.P.; Metro, S.; Pujadó, P.R.; Moser, M. Catalytic Reforming in Petroleum Processing. In Handbook of Petroleum Processing; Springer: Berlin, Germany, 2014; pp. 1–25. [Google Scholar] [CrossRef]
- Chevron Phillips Chem. Aromatics Technology. Available online: https://www.cpchem.com/what-we-do/licensing/aromatics-technology (accessed on 27 May 2021).
- Fukunaga, T.; Katsuno, H. Halogen-promoted Pt/KL Zeolite Catalyst for the Production of Aromatic Hydrocarbons from Light Naphtha. Catal. Surv. Asia 2010, 14, 96–102. [Google Scholar] [CrossRef]
- Kinnis, N.M.; Kuzma, P., Jr.; Quitmeier, W.D. Lummus Ethylene Process. In Handbook of Petrochemical Production Processes, 2nd ed.; Meyers, R.A., Ed.; McGraw Hill: New York, NY, USA, 2019; Chapter 2.4. [Google Scholar]
- Available online: https://www.ispatguru.com/crude-benzol-and-its-major-components/#:~:text=Benzol%20fraction%20produced%20during%20the,to%201.1%20%25%20of%20dry%20coal.&text=It%20is%20the%20series%20of,%2C%20and%20xylene%20(C8H10) (accessed on 27 May 2021).
- Available online: https://www.exxonmobilchemical.com/en/catalysts-and-technology-licensing/synthetic-fuels#:~:text=ExxonMobil’s%20methanol%20to%20gasoline%20(MTG,or%20with%20petroleum%20refinery%20stocks (accessed on 27 May 2021).
- Available online: https://www.globalsyngas.org/uploads/downloads/S6-2-ExxonMobil%20Catalysts-Mitch%20Hindman.pdf (accessed on 27 May 2021).
- Inoue, Y.; Nakashiro, K.; Ono, Y. Selective conversion of methanol into aromatic hydrocarbons over silver-exchanged ZSM-5 zeolites. Microporous Mater. 1995, 4, 379–383. [Google Scholar] [CrossRef]
- Niziolek, A.M.; Onel, O.; Floudas, C.A. Production of benzene, toluene, and xylenes from natural gas via methanol: Process synthesis and global optimization. AIChE J. 2016, 62, 1531–1556. [Google Scholar] [CrossRef]
- Xin, Y.; Qi, P.; Duan, X.; Lin, H.; Yuan, Y. Enhanced Performance of Zn–Sn/HZSM-5 Catalyst for the Conversion of Methanol to Aromatics. Catal. Lett. 2013, 143, 798–806. [Google Scholar] [CrossRef]
- Donath, E.E. History of catalysis in coal liquefaction. In Catalysis Science and Technology; Anderson, J.R., Boudart, M., Eds.; Springer: Berlin, Germany, 1982; Volume 3, pp. 1–38. [Google Scholar]
- Kong, Z.; Dong, X.; Xu, B.; Li, R.; Yin, Q.; Song, C. EROI Analysis for Direct Coal Liquefaction without and with CCS: The Case of the Shenhua DCL Project in China. Energies 2015, 8, 786–807. [Google Scholar] [CrossRef]
- Asaro, M.; Smith, R.M.; Davis, B.H. Coal to Liquids Technologies. In Fossil Energy; Malhotra, R., Ed.; Springer: Berlin, Germany, 2020; pp. 387–426. [Google Scholar] [CrossRef]
- Maloletnev, A.S.; Gyul’malieva, M.A. Manufacture of Aromatic Hydrocarbons from Coal Hydrogenation Products. Solid Fuel Chem. 2007, 41, 240–245. [Google Scholar] [CrossRef]
- E Dry, M. The Fischer–Tropsch process: 1950–2000. Catal. Today 2002, 71, 227–241. [Google Scholar] [CrossRef]
- Zhou, L. BP-UOP Cyclar Process. In Handbook of Petroleum Refining Processes, 3rd ed.; Meyers, R.A., Ed.; McGraw Hill: New York, NY, USA, 2004; pp. 2.29–2.37. [Google Scholar]
- Cox, H. UOP Cyclar Process. Available online: https://www.accessengineeringlibrary.com/binary/mheaeworks/2e9b5a9ed7781084/9f4b030862ce971be0b81448ade3f668edf1f4001fca5d29cd2d1aa5c356ca73/uop-cyclar-process.pdf?implicit-login=true&sigma-token=Et4NSeXiKVxl2aOxSCRY3fUCTsjlnae5npWmvSrUl30 (accessed on 27 May 2021).
- Bhan, A.; Delgass, W.N. Propane Aromatization over HZSM-5 and Ga/HZSM-5 Catalysts. Catal. Rev. 2008, 50, 19–151. [Google Scholar] [CrossRef]
- Xin, M.; Xing, E.; Gao, X.; Wang, Y.; Ouyang, Y.; Xu, G.; Luo, Y.; Shu, X. Ga Substitution during Modification of ZSM-5 and Its Influences on Catalytic Aromatization Performance. Ind. Eng. Chem. Res. 2019, 58, 6970–6981. [Google Scholar] [CrossRef]
- Available online: https://gtctech.com/technology-licensing/petrochemical-technology/aromatization-olefin-cracking/ (accessed on 6 May 2021).
- Pérez-Uresti, S.I.; Adrián-Mendiola, J.M.; El-Halwagi, M.M.; Jiménez-Gutiérrez, A. Techno-Economic Assessment of Benzene Production from Shale Gas. J. Process. 2017, 5, 33. [Google Scholar] [CrossRef]
- Chou, C. Find the best aromatics extraction system for industrial applications. Hydrocarbon Process. 2018. Available online: https://www.hydrocarbonprocessing.com/magazine/2018/september-2018/process-optimization/find-the-best-aromatics-extraction-system-for-industrial-applications (accessed on 27 May 2021).
- Available online: https://papers.gtctech.com/wp-content/uploads/GTC-BTX-Select.pdf (accessed on 5 May 2021).
- Gouzien, L.; Hombourger, T.; Mikitenko, P. Solvent Extractuion in the Oil Industry. In Petroleum Refining; Wauquier, J.-P., Ed.; Separation Processes, Editions Technip: Paris, France, 2000; Volume 2. [Google Scholar]
- Forte, P. Method for Aromatic Hydrocarbon Recovery. U.S. Patent 5073669, 17 December 1991. [Google Scholar]
- Emmrich, G.; Gehrke, H.; Ranke, U. Working with an extractive distillation process. Digit. Refin. 2001, 6, 125–129. [Google Scholar]
- ThyssenKrupp Industrial Solutions, Aromatics. Available online: https://ucpcdn.thyssenkrupp.com/_legacy/UCPthyssenkruppBAIS/assets.files/products___services/chemical_plants___processes/tkis_aromatics.pdf (accessed on 22 April 2021).
- Mittelstädt, S. Lurgi Distapex Extractive Distillation Process. In Handbook of Petrochemical Production Processes, 2nd ed.; Meyers, R.A., Ed.; McGraw Hill: New York, NY, USA, 2019; Chapter 1.13. [Google Scholar]
- Stoodt, T.J.; Negiz, A. UOP Sulfolane Process. In Handbook of Petroleum Refining Processes, 3rd ed.; Meyers, R.A., Ed.; McGraw Hill: New York, NY, USA, 2019. [Google Scholar]
- Cretoiu, L. GT-BTX Aromatics Extraction Process. In Handbook of Petrochemical Production Processes, 2nd ed.; Meyers, R.A., Ed.; McGraw Hill: New York, NY, USA, 2019; Chapter 1.8. [Google Scholar]
- Zhou, J.; Sui, H.; Jia, Z.; Yang, Z.; He, L.; Li, X. Recovery and purification of ionic liquids from solutions: A review. RSC Adv. 2018, 8, 32832–32864. [Google Scholar] [CrossRef]
- Skrollahza, S. Liquid Extraction of Aromatic Hydrocarbons by Tetrahydrofurfuryl Alcohol, An Environmentally Friendly Solvent. J. Appl. Sci. 2008, 8, 1320–1324. [Google Scholar] [CrossRef][Green Version]
- Liu, Z.; Wang, Y.; Zhao, L. Best practices for aromatics extractive distillation in integrated complexes. Hydrocarbon Process. 2020. Available online: https://www.hydrocarbonprocessing.com/magazine/2020/june-2020/special-focus-process-optimization/best-practices-for-aromatics-extractive-distillation-in-integrated-complexes (accessed on 27 May 2021).
- Johnson, J.A. Aromatics complexes. In Handbook of Petroleum Refining Processes, 3rd ed.; Meyers, R.A., Ed.; McGraw-Hill: New York, NY, USA, 2004; Available online: https://www.accessengineeringlibrary.com/content/book/9780071391092/chapter/chapter2 (accessed on 27 May 2021).
- Gentry, G.C. The Petrochemistry of Paraxylene. Digit. Refin. 2015, 1001045. [Google Scholar]
- Available online: https://www.exxonmobilchemical.com/en/catalysts-and-technology-licensing/xylenes-production/heavy-aromatics-alkylation (accessed on 28 May 2021).
- Available online: https://www.exxonmobilchemical.com/en/catalysts-and-technology-licensing/xylenes-production/selective-toluene-disproportionation (accessed on 28 May 2021).
- Bradley, T.W. ExxonMobil PxMaxTM Xylene isomerization. In Handbook of Petrochemical Production Processes, 1st ed; Meyers, R.A., Ed.; Mc Graw Hill: New York, NY, USA, 2005; Chapter 13.2. [Google Scholar]
- Available online: https://www.exxonmobilchemical.com/en/catalysts-and-technology-licensing/xylenes-production/liquid-phase-xylenes-isomerization (accessed on 28 May 2021).
- Lee, H.; Tyson, E. BP para-xylene process. In Handbook of Petrochemical Production Processes, 2nd ed.; Meyers, R.A., Ed.; McGraw Hill: New York, NY, USA, 2019; Chapter 1.3. [Google Scholar]
- Available online: https://www.lummustechnology.com/process-technologies/petrochemicals/aromatics/aromatics-production/DETOL-Hydrodealkylation-of-Toluene (accessed on 27 May 2021).
- Available online: https://www.exxonmobilchemical.com/en/catalysts-and-technology-licensing/xylenes-production/toluene-selective-alkylation-with-methanol (accessed on 28 May 2021).
- Jin, W. CrystPX: Modern Crystallization Technology for Para-Xylene Production. In Handbook of Petrochemical Production Processes, 2nd ed.; Meyers, R.A., Ed.; McGraw Hill: New York, NY, USA, 2019; Chapter 1.5. [Google Scholar]
- Stepanski, M. Economic recovery of meta-xylene. Sulzer Tech. Rev. 2000, 3, 8–9. [Google Scholar]
- Tao, G.; Geladi, P.; Lestander, T.A.; Xiong, S. Biomass properties in association with plant species and assortments. II: A synthesis based on literature data for ash elements. Renew. Sustain. Energy Rev. 2012, 16, 3507–3522. [Google Scholar] [CrossRef]
- Vassilev, S.V.; Baxter, D.; Andersen, L.K.; Vassileva, C.G. An overview of the chemical composition of biomass. Fuel 2010, 89, 913–933. [Google Scholar] [CrossRef]
- Vassilev, S.V.; Baxter, D.; Andersen, L.K.; Vassileva, C.G.; Morgan, T.J. An overview of the organic and inorganic phase composition of biomass. Fuel 2012, 94, 1–33. [Google Scholar] [CrossRef]
- Rabemanolontsoa, H.; Saka, S. Comparative study on chemical composition of various biomass species. RSC Adv. 2013, 3, 3946–3956. [Google Scholar] [CrossRef]
- Vassilev, S.V.; Vassileva, C.G. Composition, properties and challenges of algae biomass for biofuel application: An overview. Fuel 2016, 181, 1–33. [Google Scholar] [CrossRef]
- Niziolek, A.M.; Onel, O.; Guzman, Y.A.; Floudas, C.A. Biomass-Based Production of Benzene, Toluene, and Xylenes via Methanol: Process Synthesis and Deterministic Global Optimization. Energy Fuels 2016, 30, 4970–4998. [Google Scholar] [CrossRef]
- Olah, G.A. Towards Oil Independence Through Renewable Methanol Chemistry. Angew. Chem. Int. Ed. 2013, 52, 104–107. [Google Scholar] [CrossRef]
- Roode-Gutzmer, Q.I.; Kaiser, D.; Bertau, M. Renewable Methanol Synthesis. ChemBioEng Rev. 2019, 6, 209–236. [Google Scholar] [CrossRef]
- Berenger, E. Hardwood Distillation Industry; Report Nr. 738; US Department of Agriculture: Washington, DC, USA, 1956. [Google Scholar]
- Goldstein, I.S. Organic Chemicals from Biomass; CRC Press: Boca Raton, FL, USA, 1981. [Google Scholar]
- Available online: https://arpa-e.energy.gov/technologies/projects/methanol-fermentation-clostridium-bacteria (accessed on 5 May 2021).
- Sheldon, D. Methanol Production—A Technical History. Johns. Matthey Technol. Rev. 2017, 61, 172–182. [Google Scholar] [CrossRef]
- Bozzano, G.; Manenti, F. Efficient methanol synthesis: Perspectives, technologies and optimization strategies. Prog. Energy Combust. Sci. 2016, 56, 71–105. [Google Scholar] [CrossRef]
- Bowker, M. Methanol Synthesis from CO2 Hydrogenation. ChemCatChem 2019, 11, 4238–4264. [Google Scholar] [CrossRef]
- Topsoe, towards Sustainable Future with MK-317 SUSTAIN™. Available online: https://info.topsoe.com/hubfs/DOWNLOADS/DOWNLOADS%20-%20Leaflets/MK-317%20SUSTAIN%E2%84%A2.pdf?hsCtaTracking=2ede2d0f-1230-4f63-b2ee-e30c3811f806%7C741a3c3a-a506-4d0a-a2dd-0f7c0ce74a6a (accessed on 5 May 2021).
- Molino, A.; LaRocca, V.; Chianese, S.; Musmarra, D. Biofuels Production by Biomass Gasification: A Review. Energies 2018, 11, 811. [Google Scholar] [CrossRef]
- Available online: https://www.carbonrecycling.is/products (accessed on 30 April 2021).
- Available online: https://www.thyssenkrupp-industrial-solutions.com/en/products-and-services/chemical-plants-and-processes/methanol-plants/small-scale-methanol (accessed on 30 April 2021).
- Montanari, T.; Finocchio, E.; Salvatore, E.; Garuti, G.; Giordano, A.; Pistarino, C.; Busca, G. CO2 separation and landfill biogas upgrading: A comparison of 4A and 13X zeolite adsorbents. Energy 2011, 36, 314–319. [Google Scholar] [CrossRef]
- Available online: https://www.methanol.org/wp-content/uploads/2019/01/MethanolReport.pdf (accessed on 30 May 2021).
- Beenackers, A.A.C.M.; Van Swaaij, W.P.M. Methanol from wood I. Process principles and technologies for producing methanol from biomass. Int. J. Sol. Energy. 1984, 2, 349–367. [Google Scholar] [CrossRef]
- Rauch, R.; Hrbek, J.; Hofbauer, H. Biomass Gasification for Synthesis Gas Production and Applications of the Syngas. In Advances in Bioenergy: The Sustainability Challenge; Lund, P.D., Byrne, J., Berndes, G., Vasalos, I.A., Eds.; Wiley: Hoboken, NJ, USA, 2013; pp. 73–91. [Google Scholar]
- Molino, A.; Chianese, S.; Musmarra, D. Biomass gasification technology: The state of the art overview. J. Energy Chem. 2016, 25, 10–25. [Google Scholar] [CrossRef]
- Sansaniwal, S.K.; Pal, K.; Rosen, M.; Tyagi, S. Recent advances in the development of biomass gasification technology: A comprehensive review. Renew. Sustain. Energy Rev. 2017, 72, 363–384. [Google Scholar] [CrossRef]
- Mai, T.P.; Nguyen, D.Q. Gasification of Biomass, Intechopen. 2020. Available online: https://www.intechopen.com/online-first/gasification-of-biomass (accessed on 27 May 2021).
- Andersson, K.J.; Rasmussen, M.S.-S.; Nielsen, P.E.H. Industrial-scale gas conditioning including Topsoe tar reforming and purification downstream biomass gasifiers: An overview and recent examples. Fuel 2017, 203, 1026–1030. [Google Scholar] [CrossRef]
- Available online: https://www.energy.gov/sites/prod/files/2015/04/f22/demonstration_market_transformation_knight_3417.pdf (accessed on 27 May 2021).
- Available online: https://www.varmlandsmetanol.se/About%20bioMethanol.htm (accessed on 4 May 2021).
- Saleh, A.R.; Sudarmanta, B.; Fansuri, H.; Muraza, O. Improved Municipal Solid Waste Gasification Efficiency Using a Modified Downdraft Gasifier with Variations of Air Input and Preheated Air Temperature. Energy Fuels 2019, 33, 11049–11056. [Google Scholar] [CrossRef]
- Enerkem Biofuels and Chemicals from Mixed Waste. Available online: https://www.etipbioenergy.eu/images/SPM9_Presentations/Day1/5_%20ETIP%20B%20SPM9_R.%20Vierhout_Enerkem.pdf (accessed on 3 May 2021).
- Checa, M.; Nogales-Delgado, S.; Montes, V.; Encinar, J.M. Recent Advances in Glycerol Catalytic Valorization: A Review. Catalyst 2020, 10, 1279. [Google Scholar] [CrossRef]
- Adeniyi, A.G.; Ighalo, J.O. A review of steam reforming of glycerol. Chem. Pap. 2019, 73, 2619–2635. [Google Scholar] [CrossRef]
- Fasolini, A.; Cespi, D.; Tabanelli, T.; Cucciniello, R.; Cavani, F. Hydrogen from Renewables: A Case Study of Glycerol Reforming. Catalysts 2019, 9, 722. [Google Scholar] [CrossRef]
- van Bennekom, J.G.; Venderbosch, R.H.; Heeres, H.J. Biomethanol from Glycerol. In BiodieselFeedstocks, Production and Applications, Chapter XX; Fang, Z., Ed.; Intechopen: London, UK, 2012; pp. 331–360. [Google Scholar]
- Available online: https://brintbranchen.dk/wp-content/uploads/2017/10/Paul-Compagne_BioMCN.pdf (accessed on 30 April 2021).
- Achinas, S.; Achinas, V.; Euverink, G.J.W. A Technological Overview of Biogas Production from Biowaste. Engineering 2017, 3, 299–307. [Google Scholar] [CrossRef]
- Khan, I.U.; Othman, M.H.D.; Hashim, H.; Matsuura, T.; Ismail, A.; Rezaei-DashtArzhandi, M.; Azelee, I.W. Biogas as a renewable energy fuel—A review of biogas upgrading, utilisation and storage. Energy Convers. Manag. 2017, 150, 277–294. [Google Scholar] [CrossRef]
- García-Diéguez, M.; Finocchio, E.; Larrubia, M.Á.; Alemany, L.J.; Busca, G. Characterization of alumina-supported Pt, Ni and PtNi alloy catalysts for the dry reforming of methane. J. Catal. 2010, 274, 11–20. [Google Scholar] [CrossRef]
- Zhao, X.; Joseph, B.; Kuhn, J.; Ozcan, S. Biogas Reforming to Syngas: A Review. iScience 2020, 23, 101082. [Google Scholar] [CrossRef] [PubMed]
- Hernández, B.; Martín, M. Optimal Process Operation for Biogas Reforming to Methanol: Effects of Dry Reforming and Biogas Composition. Ind. Eng. Chem. Res. 2016, 55, 6677–6685. [Google Scholar] [CrossRef]
- Available online: https://oberonfuels.com/technology/oberon-process/ (accessed on 4 May 2021).
- Available online: https://www.ieabioenergy.com/wp-content/uploads/2013/09/5083.pdf (accessed on 4 May 2021).
- van Kasteren, J.M.N. Production of Bioalcohols via Gasification. In Handbook of Biofuels Production, 2nd ed.; Luque, R., Sze, C., Lin, K., Wilson, K., Clark, J., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 495–507. [Google Scholar] [CrossRef]
- Nahar, G.; Dupont, V. Hydrogen via steam reforming of liquid biofeedstock. Biofuels 2012, 3, 167–191. [Google Scholar] [CrossRef]
- Bion, N.; Duprez, D.; Epron, F. Design of Nanocatalysts for Green Hydrogen Production from Bioethanol. ChemSusChem 2012, 5, 76–84. [Google Scholar] [CrossRef]
- Garbarino, G.; Cavattoni, T.; Riani, P.; Brescia, R.; Canepa, F.; Busca, G. On the Role of Support in Metallic Heterogeneous Catalysis: A Study of Unsupported Nickel–Cobalt Alloy Nanoparticles in Ethanol Steam Reforming. Catal. Lett. 2019, 149, 929–941. [Google Scholar] [CrossRef]
- Available online: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/866383/Phase_1_-_Wood_-_Novel_Renewable_Ethanol_Steam_Reformer.pdf (accessed on 14 May 2021).
- Oakley, J.H.; Hoadley, A.F. Industrial scale steam reforming of bioethanol: A conceptual study. Int. J. Hydrogen Energy 2010, 35, 8472–8485. [Google Scholar] [CrossRef]
- Garbarino, G.; Pugliese, F.; Cavattoni, T.; Busca, G.; Costamagna, P. A Study on CO2 Methanation and Steam Methane Reforming over Commercial Ni/Calcium Aluminate Catalysts. Energies 2020, 13, 2792. [Google Scholar] [CrossRef]
- Sanchez, N.; Ruiz, R.; Hacker, V.; Cobo, M. Impact of bioethanol impurities on steam reforming for hydrogen production: A review. Int. J. Hydrogen Energy 2020, 45, 11923–11942. [Google Scholar] [CrossRef]
- Riani, P.; Garbarino, G.; Canepa, F.; Busca, G. Cobalt nanoparticles mechanically deposited on α-Al2O3: A competitive catalyst for the production of hydrogen through ethanol steam reforming. J. Chem. Technol. Biotechnol. 2019, 94, 538–546. [Google Scholar] [CrossRef]
- Awatmongkhon, B.; Theinnoi, K.; Wongchang, T.; Haoharn, C.; Wongkhorsub, C.; Tsolakis, A. Hydrogen Production via the Catalytic Partial Oxidation of Ethanol on a Platinum–Rhodium Catalyst: Effect of the Oxygen-to-Ethanol Molar Ratio and the Addition of Steam. Energy Fuels 2019, 33, 6742–6753. [Google Scholar] [CrossRef]
- Baruah, R.; Dixit, M.; Basarkar, P.; Parikh, D.; Bhargav, A. Advances in ethanol autothermal reforming. Renew. Sustain. Energy Rev. 2015, 51, 1345–1353. [Google Scholar] [CrossRef]
- Nanda, S.; Rana, R.; Hunter, H.N.; Fang, Z.; Dalai, A.K.; Kozinski, J.A. Hydrothermal catalytic processing of waste cooking oil for hydrogen-rich syngas production. Chem. Eng. Sci. 2019, 195, 935–945. [Google Scholar] [CrossRef]
- Nahar, G.; Dupont, V.; Twigg, M.V.; Dvininov, E. Feasibility of hydrogen production from steam reforming of biodiesel (FAME) feedstock on Ni-supported catalysts. Appl. Catal. B Environ. 2015, 168–169, 228–242. [Google Scholar] [CrossRef]
- Lin, K.-W.; Wu, H.-W. Thermodynamic analysis and experimental study of partial oxidation reforming of biodiesel and hydrotreated vegetable oil for hydrogen-rich syngas production. Fuel 2019, 236, 1146–1155. [Google Scholar] [CrossRef]
- Najafpour, G.D.; Shahavi, M.H.; A Neshat, S. Assessment of biological Hydrogen production processes: A review. IOP Conf. Series Earth Environ. Sci. 2016, 36, 012068. [Google Scholar] [CrossRef]
- Hernández, B.; Blázquez, C.G.; Aristizábal-Marulanda, V.; Martín, M. Production of H2 and Methanol via Dark Fermentation: A Process Optimization Study. Ind. Eng. Chem. Res. 2020, 59, 16720–16729. [Google Scholar] [CrossRef]
- Haider, M.H.; Dummer, N.F.; Knight, D.W.; Jenkins, R.L.; Howard, M.; Moulijn, J.; Taylor, S.H.; Hutchings, G.J. Efficient green methanol synthesis from glycerol. Nat. Chem. 2015, 7, 1028–1032. [Google Scholar] [CrossRef]
- Available online: https://www.topsoe.com/processes/gasoline-synthesis/tigas#:~:text=Gasoline%20from%20synthesis%20gas%20(STG,gasoline%20in%20the%20gasoline%20reactors (accessed on 3 May 2021).
- Soccol, C.R.; Faraco, V.; Karp, S.G.; Vandenberghe, L.P.; Thomaz-Soccol, V.; Woiciechowski, A.L.; Pandey, A. Lignocellulosic Bioethanol: Current Status and Future Perspectives. In Biofuels: Alternative Feedstocks and Conversion Processes for the Production of Liquid and Gaseous Biofuels; Elsevier: Amsterdam, The Netherlands, 2019; pp. 331–354. [Google Scholar]
- Jambo, S.A.; Abdulla, R.; Azhar, S.H.M.; Marbawi, H.; Gansau, J.A.; Ravindra, P. A review on third generation bioethanol feedstock. Renew. Sustain. Energy Rev. 2016, 65, 756–769. [Google Scholar] [CrossRef]
- Sun, J.; Wang, Y. Recent Advances in Catalytic Conversion of Ethanol to Chemicals. ACS Catal. 2014, 4, 1078–1090. [Google Scholar] [CrossRef]
- Phung, T.K.; Busca, G. Selective Bioethanol Conversion to Chemicals and Fuels via Advanced Catalytic Approaches. In Biorefinery of Alternative Resources: Targeting Green Fuels and Platform Chemicals; Springer: Berlin, Germany, 2020; pp. 75–103. [Google Scholar]
- Mohsenzadeh, A.; Zamani, A.; Taherzadeh, M.J. Bioethylene Production from Ethanol: A Review and Techno-economical Evaluation. ChemBioEng Rev. 2017, 4, 75–91. [Google Scholar] [CrossRef]
- Makshina, E.V.; Dusselier, M.; Janssens, W.; Degrève, J.; Jacobs, P.A.; Sels, B.F. Review of old chemistry and new catalytic advances in the on-purpose synthesis of butadiene. Chem. Soc. Rev. 2014, 43, 7917–7953. [Google Scholar] [CrossRef] [PubMed]
- Costa, E.; Uguina, A.; Aguado, J.; Hernandez, P.J. Ethanol to gasoline process: Effect of variables, mechanism, and kinetics. Ind. Eng. Chem. Process. Des. Dev. 1985, 24, 239–244. [Google Scholar] [CrossRef]
- Eagan, N.M.; Kumbhalkar, M.D.; Buchanan, J.S.; Dumesic, J.A.; Huber, G.W. Chemistries and processes for the conversion of ethanol into middle-distillate fuels. Nat. Rev. Chem. 2019, 3, 223–249. [Google Scholar] [CrossRef]
- Inaba, M.; Murata, K.; Saito, M.; Takahara, I. Ethanol conversion to aromatic hydrocarbons over several zeolite catalysts. React. Kinet. Catal. Lett. 2006, 88, 135–141. [Google Scholar] [CrossRef]
- Phung, T.K.; Radikapratama, R.; Garbarino, G.; Lagazzo, A.; Riani, P.; Busca, G. Tuning of product selectivity in the conversion of ethanol to hydrocarbons over H-ZSM-5 based zeolite catalysts. Fuel Process. Technol. 2015, 137, 290–297. [Google Scholar] [CrossRef]
- Sudiyarmanto; Kristiani, A.; Andreas, W.; Abimanyu, H. Catalytic conversion of ethanol to aromatic compounds using metal/zeolite catalysts. AIP Conf. Proc. 2016, 1755, 080007. [Google Scholar] [CrossRef]
- Li, Z.; Lepore, A.W.; Salazar, M.F.; Foo, G.S.; Davison, B.H.; Wu, Z.; Narula, C.K. Selective conversion of bio-derived ethanol to renewable BTX over Ga-ZSM-5. Green Chem. 2017, 19, 4344–4352. [Google Scholar] [CrossRef]
- Douvartzides, S.L.; Charisiou, N.; Papageridis, K.N.; Goula, M.A. Green Diesel: Biomass Feedstocks, Production Technologies, Catalytic Research, Fuel Properties and Performance in Compression Ignition Internal Combustion Engines. Energies 2019, 12, 809. [Google Scholar] [CrossRef]
- Available online: https://www.eni.com/en-IT/circular-economy/ecofind-biofuel.html (accessed on 30 May 2021).
- Available online: https://www.neste.com/sites/default/files/attachments/neste_renewable_diesel_handbook.pdf (accessed on 30 May 2021).
- Available online: https://services.totalenergies.fr/pro/produits-services/carburants/carburants-adaptes-professionnels/total-hvo (accessed on 30 May 2021).
- Available online: https://www.regi.com/products/transportation-fuels/renewable-diesel (accessed on 30 May 2021).
- Sannita, E.; Aliakbarian, B.; A Casazza, A.; Perego, P.; Busca, G. Medium-temperature conversion of biomass and wastes into liquid products, a review. Renew. Sustain. Energy Rev. 2012, 16, 6455–6475. [Google Scholar] [CrossRef]
- Chang, C.-C.; Wan, S.-W. China’s Motor Fuels from Tung Oil. Ind. Eng. Chem. 1947, 39, 1543–1548. [Google Scholar] [CrossRef]
- Weisz, P.B.; Haag, W.O.; Rodewald, P.G. Catalytic Production of High-Grade Fuel (Gasoline) from Biomass Compounds by Shape-Selective Catalysis. Science 1979, 206, 57–58. [Google Scholar] [CrossRef]
- Romero, M.; Pizzi, A.; Toscano, G.; Busca, G.; Bosio, B.; Arato, E. Deoxygenation of waste cooking oil and non-edible oil for the production of liquid hydrocarbon biofuels. Waste Manag. 2016, 47, 62–68. [Google Scholar] [CrossRef]
- Trabelsi, A.B.H.; Zaafouri, K.; Baghdadi, W.; Naoui, S.; Ouerghi, A. Second generation biofuels production from waste cooking oil via pyrolysis process. Renew. Energy 2018, 126, 888–896. [Google Scholar] [CrossRef]
- Phung, T.K.; A Casazza, A.; Perego, P.; Capranica, P.; Busca, G. Catalytic pyrolysis of vegetable oils to biofuels: Catalyst functionalities and the role of ketonization on the oxygenate paths. Fuel Process. Technol. 2015, 140, 119–124. [Google Scholar] [CrossRef]
- Yeletsky, P.; Kukushkin, R.; Yakovlev, V.; Chen, B. Recent advances in one-stage conversion of lipid-based biomass-derived oils into fuel components—aromatics and isomerized alkanes. Fuel 2020, 278, 118255. [Google Scholar] [CrossRef]
- Arapova, O.V.; Chistyakov, A.V.; Tsodikov, M.V.; Moiseev, I.I. Lignin as a Renewable Resource of Hydrocarbon Products and Energy Carriers (A Review). Pet. Chem. 2020, 60, 227–243. [Google Scholar] [CrossRef]
- Tarasov, D.; Leitch, M.; Fatehi, P. Lignin–carbohydrate complexes: Properties, applications, analyses, and methods of extraction: A review. Biotechnol. Biofuels 2018, 11, 1–28. [Google Scholar] [CrossRef]
- Kumar, A.; Kumar, J.; Bhaskar, T. Utilization of lignin: A sustainable and eco-friendly approach. J. Energy Inst. 2020, 93, 235–271. [Google Scholar] [CrossRef]
- Bajwa, D.; Pourhashem, G.; Ullah, A.; Bajwa, S. A concise review of current lignin production, applications, products and their environmental impact. Ind. Crop. Prod. 2019, 139, 111526. [Google Scholar] [CrossRef]
- Gordobil, O.; Moriana, R.; Zhang, L.; Labidi, J.; Sevastyanova, O. Assesment of technical lignins for uses in biofuels and biomaterials: Structure-related properties, proximate analysis and chemical modification. Ind. Crop. Prod. 2016, 83, 155–165. [Google Scholar] [CrossRef]
- Tribot, A.; Amer, G.; Alio, M.A.; de Baynast, H.; Delattre, C.; Pons, A.; Mathias, J.-D.; Callois, J.-M.; Vial, C.; Michaud, P.; et al. Wood-lignin: Supply, extraction processes and use as bio-based material. Eur. Polym. J. 2019, 112, 228–240. [Google Scholar] [CrossRef]
- Shen, D.; Gu, S.; Luo, K.; Wang, S.; Fang, M. The pyrolytic degradation of wood-derived lignin from pulping process. Bioresour. Technol. 2010, 101, 6136–6146. [Google Scholar] [CrossRef]
- Mu, W.; Ben, H.; Ragauskas, A.; Deng, Y. Lignin Pyrolysis Components and Upgrading—Technology Review. BioEnergy Res. 2013, 6, 1183–1204. [Google Scholar] [CrossRef]
- Ansari, K.B.; Arora, J.S.; Chew, J.W.; Dauenhauer, P.J.; Mushrif, S.H. Fast Pyrolysis of Cellulose, Hemicellulose, and Lignin: Effect of Operating Temperature on Bio-oil Yield and Composition and Insights into the Intrinsic Pyrolysis Chemistry. Ind. Eng. Chem. Res. 2019, 58, 15838–15852. [Google Scholar] [CrossRef]
- Ferdous, D.; Dalai, A.; Bej, S.; Thring, R.; Bakhshi, N. Production of H2 and medium Btu gas via pyrolysis of lignins in a fixed-bed reactor. Fuel Process. Technol. 2001, 70, 9–26. [Google Scholar] [CrossRef]
- Zhao, C.; Jiang, E.; Chen, A. Volatile production from pyrolysis of cellulose, hemicellulose and lignin. J. Energy Inst. 2017, 90, 902–913. [Google Scholar] [CrossRef]
- Fan, L.; Zhang, Y.; Liu, S.; Zhou, N.; Chen, P.; Cheng, Y.; Addy, M.; Lu, Q.; Omar, M.M.; Liu, Y.; et al. Bio-oil from fast pyrolysis of lignin: Effects of process and upgrading parameters. Bioresour. Technol. 2017, 241, 1118–1126. [Google Scholar] [CrossRef]
- Sharma, R.K.; Wooten, J.B.; Baliga, V.L.; Lin, X.; Chan, W.G.; Hajaligol, M.R. Characterization of chars from pyrolysis of lignin. Fuel 2004, 83, 1469–1482. [Google Scholar] [CrossRef]
- Gooty, A.T.; Li, D.; Berruti, F.; Briens, C. Kraft-lignin pyrolysis and fractional condensation of its bio-oil vapors. J. Anal. Appl. Pyrolysis 2014, 106, 33–40. [Google Scholar] [CrossRef]
- Mullen, C.A.; Boateng, A.A. Characterization of water insoluble solids isolated from various biomass fast pyrolysis oils. J. Anal. Appl. Pyrolysis 2011, 90, 197–203. [Google Scholar] [CrossRef]
- Yin, C. Microwave-assisted pyrolysis of biomass for liquid biofuels production. Bioresour. Technol. 2012, 120, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Yang, H.; Chen, P.; Liu, Z.; Chen, Y.; Wang, L.; Wang, X.; Chen, H. Lignin Characterization and Catalytic Pyrolysis for Phenol-Rich Oil with TiO2-Based Catalysts. Energy Fuels 2019, 33, 9934–9941. [Google Scholar] [CrossRef]
- Han, T.; Ding, S.; Yang, W.; Jönsson, P. Catalytic pyrolysis of lignin using low-cost materials with different acidities and textural properties as catalysts. Chem. Eng. J. 2019, 373, 846–856. [Google Scholar] [CrossRef]
- Thring, R.W.; Katikaneni, S.P.; Bakhshi, N.N. The production of gasoline range hydrocarbons from Alcell® lignin using HZSM-5 catalyst. Fuel Process. Technol. 2000, 62, 17–30. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Lee, J.H.; Park, J.; Kim, J.K.; An, D.; Song, I.K.; Choi, J.W. Catalytic pyrolysis of lignin over HZSM-5 catalysts: Effect of various parameters on the production of aromatic hydrocarbon. J. Anal. Appl. Pyrolysis 2015, 114, 273–280. [Google Scholar] [CrossRef]
- Lazaridis, P.A.; Fotopoulos, A.P.; Karakoulia, S.A.; Triantafyllidis, K. Catalytic Fast Pyrolysis of Kraft Lignin with Conventional, Mesoporous and Nanosized ZSM-5 Zeolite for the Production of Alkyl-Phenols and Aromatics. Front. Chem. 2018, 6, 295. [Google Scholar] [CrossRef]
- Luna-Murillo, B.; Pala, M.; Paioni, A.L.; Baldus, M.; Ronsse, F.; Prins, W.; Bruijnincx, P.C.A.; Weckhuysen, B.M. Catalytic Fast Pyrolysis of Biomass: Catalyst Characterization Reveals the Feed-Dependent Deactivation of a Technical ZSM-5-Based Catalyst. ACS Sustain. Chem. Eng. 2021, 9, 291–304. [Google Scholar] [CrossRef]
- Bu, Q.; Lei, H.; Zacher, A.H.; Wang, L.; Ren, S.; Liang, J.; Wei, Y.; Liu, Y.; Tang, J.; Zhang, Q.; et al. A review of catalytic hydrodeoxygenation of lignin-derived phenols from biomass pyrolysis. Bioresour. Technol. 2012, 124, 470–477. [Google Scholar] [CrossRef]
- Shu, R.; Li, R.; Lin, B.; Wang, C.; Cheng, Z.; Chen, Y. A review on the catalytic hydrodeoxygenation of lignin-derived phenolic compounds and the conversion of raw lignin to hydrocarbon liquid fuels. Biomass Bioenergy 2020, 132, 105432. [Google Scholar] [CrossRef]
- Stummann, M.Z.; Høj, M.; Gabrielsen, J.; Clausen, L.R.; Jensen, P.A.; Jensen, A.D. A perspective on catalytic hydropyrolysis of biomass. Renew. Sustain. Energy Rev. 2021, 143, 110960. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Lee, H.W.; Lee, S.M.; Jae, J.; Park, Y.-K. Overview of the recent advances in lignocellulose liquefaction for producing biofuels, bio-based materials and chemicals. Bioresour. Technol. 2019, 279, 373–384. [Google Scholar] [CrossRef]
- Molina, M.J.C.; Granados, M.L.; Gervasini, A.; Carniti, P. Exploitment of niobium oxide effective acidity for xylose dehydration to furfural. Catal. Today 2015, 254, 90–98. [Google Scholar] [CrossRef]
- Bernal, H.G.; Galletti, A.M.R.; Garbarino, G.; Busca, G.; Finocchio, E. NbP catalyst for furfural production: FT IR studies of surface properties. Appl. Catal. A Gen. 2015, 502, 388–398. [Google Scholar] [CrossRef]
- Marzo, M.; Gervasini, A.; Carniti, P. Improving stability of Nb2O5 catalyst in fructose dehydration reaction in water solvent by ion-doping. Catal. Today 2012, 192, 89–95. [Google Scholar] [CrossRef]
- Khemthong, P.; Yimsukanan, C.; Narkkun, T.; Srifa, A.; Witoon, T.; Pongchaiphol, S.; Kiatphuengporn, S.; Faungnawakij, K. Advances in catalytic production of value-added biochemicals and biofuels via furfural platform derived lignocellulosic biomass. Biomass Bioenergy 2021, 148, 106033. [Google Scholar] [CrossRef]
- Yabushita, M.; Kobayashi, H.; Fukuoka, A. Catalytic transformation of cellulose into platform chemicals. Appl. Catal. B Environ. 2014, 145, 1–9. [Google Scholar] [CrossRef]
- Takkellapati, S.; Li, T.; Gonzalez, M.A. An overview of biorefinery-derived platform chemicals from a cellulose and hemicellulose biorefinery. Clean Technol. Environ. Policy 2018, 20, 1615–1630. [Google Scholar] [CrossRef]
- Genuino, H.C.; Muizenbelt, I.; Heeres, A.; Schenk, N.J.; Winkelman, J.G.M.; Heeres, H.J. An improved catalytic pyrolysis concept for renewable aromatics from biomass involving a recycling strategy for co-produced polycyclic aromatic hydrocarbons. Green Chem. 2019, 21, 3802–3806. [Google Scholar] [CrossRef]
- Zhao, Y.; Pan, T.; Zuo, Y.; Guo, Q.-X.; Fu, Y. Production of aromatic hydrocarbons through catalytic pyrolysis of 5-Hydroxymethylfurfural from biomass. Bioresour. Technol. 2013, 147, 37–42. [Google Scholar] [CrossRef]
- Yu, J.; Paterson, N.; Blamey, J.; Millan, M. Cellulose, xylan and lignin interactions during pyrolysis of lignocellulosic biomass. Fuel 2017, 191, 140–149. [Google Scholar] [CrossRef]
- Lédé, J. Cellulose pyrolysis kinetics: An historical review on the existence and role of intermediate active cellulose. J. Anal. Appl. Pyrolysis 2012, 94, 17–32. [Google Scholar] [CrossRef]
- Yu, J.; Paterson, N.; Millan, M. The primary products of cellulose pyrolysis in the absence of extraparticle reactions. Fuel 2019, 237, 911–915. [Google Scholar] [CrossRef]
- Xin, S.; Yang, H.; Chen, Y.; Yang, M.; Chen, L.; Wang, X.; Chen, H. Chemical structure evolution of char during the pyrolysis of cellulose. J. Anal. Appl. Pyrolysis 2015, 116, 263–271. [Google Scholar] [CrossRef]
- Chen, L.; Li, Y.; Zhang, X.; Liu, Y.; Zhang, Q.; Wang, C.; Ma, L. One-pot conversion of cellulose to liquid hydrocarbon efficiently catalyzed by Ru/C and boron phosphate in aqueous medium. Energy Procedia 2019, 158, 160–166. [Google Scholar] [CrossRef]
- Industrial Charcoal Making; FAO: Rome, Italy, 1985. Available online: http://www.fao.org/3/x5555e/x5555e.pdf (accessed on 25 May 2021).
- Antal, M.J.; Grønli, M.G. The Art, Science, and Technology of Charcoal Production. Ind. Eng. Chem. Res. 2003, 42, 1619–1640. [Google Scholar] [CrossRef]
- Kajina, W.; Junpen, A.; Garivait, S.; Kamnoet, O.; Keeratiisariyakul, P.; Rousset, P. Charcoal production processes: An overview. J. Sustain. Energy Environ. 2019, 10, 19–25. [Google Scholar]
- Available online: http://www.fao.org/3/ca7967en/ca7967en.pdf (accessed on 17 May 2021).
- Oasmaa, A.; Solantausta, Y.; Arpiainen, V.; Kuoppala, E.; Sipilä, K. Fast Pyrolysis Bio-Oils from Wood and Agricultural Residues. Energy Fuels 2010, 24, 1380–1388. [Google Scholar] [CrossRef]
- Available online: https://uop.honeywell.com/en/industry-solutions/renewable-fuels/rtp-biomass-conversion (accessed on 7 May 2021).
- Available online: https://bioenergitidningen.se/app/uploads/2018/09/5.Dan_Szeezil.pdf (accessed on 7 May 2021).
- Available online: https://corporate.exxonmobil.com/Energy-and-innovation/Advanced-biofuels/Advanced-biofuels-and-algae-research#Algaeforbiofuelsproduction. (accessed on 27 May 2021).
- Singh, R.; Upadhyay, A.K.; Chandra, P.; Singh, D.P. Biotechnological Application of Algae in Pharmaceuticals Industries with Special Reference to Omega-3 Fatty Acid and Human Health. In Algae and Sustainable Technologies: Bioenergy, Nanotechnology and Green; Upadhyay, A.K., Singh, D.P., Eds.; CRC Press: Abington, UK, 2021; pp. 29–42. [Google Scholar]
- Aratboni, H.A.; Rafiei, N.; Garcia-Granados, R.; Alemzadeh, A.; Morones-Ramírez, J.R. Biomass and lipid induction strategies in microalgae for biofuel production and other applications. Microb. Cell Factories 2019, 18, 1–17. [Google Scholar] [CrossRef]
- Khan, M.I.; Shin, J.H.; Kim, J.D. The promising future of microalgae: Current status, challenges, and optimization of a sustainable and renewable industry for biofuels, feed, and other products. Microb. Cell Factories 2018, 17, 1–21. [Google Scholar] [CrossRef]
- Saad, M.G.; Dosoky, N.S.; Zoromba, M.S.; Shafik, H.M. Algal Biofuels: Current Status and Key Challenges. Energies 2019, 12, 1920. [Google Scholar] [CrossRef]
- Kumar, G.; Shobana, S.; Chen, W.-H.; Bach, Q.-V.; Kim, S.-H.; Atabani, A.; Chang, J.-S. A review of thermochemical conversion of microalgal biomass for biofuels: Chemistry and processes. Green Chem. 2017, 19, 44–67. [Google Scholar] [CrossRef]
- Yang, C.; Li, R.; Zhang, B.; Qiu, Q.; Wang, B.; Yang, H.; Ding, Y.; Wang, C. Pyrolysis of microalgae: A critical review. Fuel Process. Technol. 2019, 186, 53–72. [Google Scholar] [CrossRef]
- Aliyu, A.; Lee, J.; Harvey, A. Microalgae for biofuels: A review of thermochemical conversion processes and associated opportunities and challenges. Bioresour. Technol. Rep. 2021, 15, 100694. [Google Scholar] [CrossRef]
- Casazza, A.A.; Spennati, E.; Converti, A.; Busca, G. Production of carbon-based biofuels by pyrolysis of exhausted Arthrospira platensis biomass after protein or lipid recovery. Fuel Process. Technol. 2020, 201, 106336. [Google Scholar] [CrossRef]
- Borges, F.C.; Xie, Q.; Min, M.; Muniz, L.A.R.; Farenzena, M.; Trierweiler, J.O.; Chen, P.; Ruan, R. Fast microwave-assisted pyrolysis of microalgae using microwave absorbent and HZSM-5 catalyst. Bioresour. Technol. 2014, 166, 518–526. [Google Scholar] [CrossRef]
- Thangalazhy-Gopakumar, S.; Adhikari, S.; Chattanathan, S.A.; Gupta, R.B. Catalytic pyrolysis of green algae for hy-drocarbon production using H +ZSM-5 catalyst. Bioresour. Technol. 2012, 118, 150–157. [Google Scholar]
- Spennati, E.; Casazza, A.A.; Converti, A.; Busca, G. Thermocatalytic Pyrolysis of Exhausted Arthrospira platensis Biomass after Protein or Lipid Recovery. Energies 2020, 13, 5246. [Google Scholar] [CrossRef]
- Zabeti, M.; Nguyen, T.S.; Lefferts, L.; Heeres, H.J.; Seshan, K. In situ catalytic pyrolysis of lignocellulose using alka-li-modified amorphous silica alumina. Bioresour. Technol. 2012, 118, 374–381. [Google Scholar]
- Ueckerdt, F.; Bauer, C.; Dirnaichner, A.; Everall, J.; Sacchi, R.; Luderer, G. Potential and risks of hydrogen-based e-fuels in climate change mitigation. Nat. Clim. Chang. 2021, 11, 384–393. [Google Scholar] [CrossRef]
- Available online: https://ec.europa.eu/energy/sites/ener/files/documents/iea-the_future_of_petrochemicals.pdf (accessed on 27 May 2021).











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Monocyclic Aromatic Molecule | Teb °C | Tm °C | Vp kPa | d g/mL | Øc nm | RON | MON | Fp °C | IARC Classification |
|---|---|---|---|---|---|---|---|---|---|
| Benzene | 80.1 | +5.5 | 12.70 | 0.879 | 0.67 | 101.0 | 93.0 | −11 | Group 1 |
| Toluene | 110.6 | −59.4 | 3.79 | 0.867 | 0.67 | 121.0 | 103.0 | 4 | Group 3 |
| Ethylbenzene | 136.2 | −95.0 | 1.28 | 0.867 | 0.67 | 108.3 | 97.9 | 15 | Group 2b |
| Styrene | 145.2 | −30.0 | 0.81 | 0.906 | 0.67 | 103.0 | 100.2 | 31 | Group 2a |
| Para-xylene | 138.3 | +13.3 | 1.18 | 0.861 | 0.67 | 146.0 | 101.2 | 25 | Group 3 |
| Meta-xylene | 139.1 | −47.9 | 1.12 | 0.864 | 0.71 | 145.0 | 102.8 | 23 | Group 3 |
| Ortho-xylene | 144.4 | −25.2 | 0.89 | 0.880 | 0.74 | 120.0 | 100.0 | 17 | Group 3 |
| Component | Crude Oil | Full Straight Run Gasoline | Reformate Gasoline | Light Reformate Gasoline | Pyrolysis Gasoline | Coke Oven Light Oil |
|---|---|---|---|---|---|---|
| Benzene | 0.1–0.3 | 1–3 | 3–12 | 20–30 | 25–34 | 60–80 |
| Toluene | 0.3–2 | 2–8 | 12–25 | 40–50 | 15–22 | 9–18 |
| Xylenes | 0.5–3 | 2–8 | 15–30 | <0.5 | 5–15 | 1–6 |
| Ethylbenzene | 2–8 | <0.5 | 2–5 | 1–2 | ||
| Styrene | 0 | 0 | tr | tr | 2–6 | 1–2 |
| C9+ aromatics | 0.3–2 | 2–8 | 10–20 | 0 | 2–5 | 5–10 |
| Total monocyclic aromatics | 1–7 | 7–20 | 35–65 | 60–80 | 45–65 | 95–98 |
| Mogas [50] | Avgas100LL [51] | ||
|---|---|---|---|
| Sulfur | ppm wt max | 10 | 500 |
| Benzene | % v/v max | 1 | - |
| Olefins | % v/v max | 18 | - |
| Aromatics | % v/v max | 35 | - |
| Oxygen | % w/w max | 3.7 | - |
| Lead | mg/L max | 5 | 560 |
| Manganese | mg/L max | 2 | - |
| Biofuel (Bioethanol) | % w/w min | 1 | - |
| Density | kg/m3 range | 720–775 | 720 * |
| Vapor pressure ** | kPa range | 45–60 | 38–49 *** |
| RON | min | 95 | 99.5 |
| MON | min | 85 | - |
| Freezing point | °C max | - | −58 |
| Distillation end point | °C | 210 | 170 |
| Solvent | Acronym | Tb °C | d g/cm3 | Rv | Process Name | Companies | |
|---|---|---|---|---|---|---|---|
| LLE | ED | ||||||
| Diethylene Glycol | DEG | 244 | 1.12 | - | Udex [78] | - | Dow, UOP |
| Triethylene Glycol | TEG | 285 | 1.12 | 1.44 | - | - | |
| Tetraethylene Glycol | Tetra | 327 | 1.12 | 1.39 | Tetra [78] | - | Union Carbide/UOP |
| Mixed Glycol Ethers | CAROM | - | - | 1.35 | Carom [79] | - | UOP |
| N-formyl-morpholine | NFM | 244 | 1.15 | 1,89 | Morphylex [80] | Morphylane [80,81] | Uhde |
| N-methyl-pyrrolidone | NMP | 206 | 1.03 | 1.95 | Aerosolvan [78] | Distapex [82] | Lurgi |
| Dimethylsulfoxide | DMSO | 189 | 1.10 | DMSO [78] | - | IFP | |
| Sulfolane | - | 287 | 1.26 | 2.00 | Sulfolane [83] | ED sulfolane [83] | UOP/Shell |
| Techtiv 100 | - | 280–290 | 1.24−1.27 | 2.44 | - | GT-BTX [84] | GTC-Sulzer |
| Techtiv 500 | - | - | - | 2.83 | - | GT-BTX Select [84] | GTC-Sulzer |
| No solvent | - | - | - | 0.57 | - | - | - |
| Biomass Type | Cellulose | Hemicellulose | Lignin | Proteins | Lipids | Ash |
|---|---|---|---|---|---|---|
| Woody | 42–64 | 7–33 | 10–36 | 0–0.5 | - | 0.8–10 |
| Barks | 19–25 | 30–60 | 20–45 | - | - | 2–7 |
| Grasses | 31–50 | 25–53 | 6–34 | - | - | 0.8–10 |
| Straws | 29–55 | 23–35 | 15–35 | - | - | 4–20 |
| Algae | 0–28 | 21–66 | 0–15 | 7–50 | 1–9 | 8–49 |
| Vegetable oils * | - | - | - | - | >95 | <2 |
| Biomass Type | C | O | H | N | S | Cl |
|---|---|---|---|---|---|---|
| Woody biomass | 42–58 | 34–49 | 3–9 | 0.1–3.4 | 0.01–0.6 | 0.01–0.8 |
| Grasses | 46–52 | 42–44 | 5–6.5 | 0.3–2.6 | 0.04–0.27 | 0.04–0.83 |
| Straws | 48–51 | 40–45 | 5.5–6.5 | 0.5–2.8 | 0.08–0.28 | 0.03–0.64 |
| Industrial lignins | 55–70 | 23–40 | 4.5–6.0 | 0–0.2 | 0.01–6 | - |
| Cellulose | 40–46 | 45–55 | 6–6.5 | 0.1–1.5 | - | - |
| Triglyceride oils * | 70–78 | 10–15 | 10–13 | 0.05–0.1 | 0.01–0.1 | 0.003 |
| Algae | 38–54 | 26–53 | 4.5–13 | 1.1–12.5 | 0.5–3.3 | 0.2–2 |
| Coal (mean) | 63–87 | 4–30 | 3.5–6.3 | 0.5–2–9 | 0.2–9.8 | 0.05–0.11 |
| Crude oil | 83–87 | 0.05–1.5 | 10–14 | 0.1–2.0 | 0.05–6.0 | 0–0.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Busca, G. Production of Gasolines and Monocyclic Aromatic Hydrocarbons: From Fossil Raw Materials to Green Processes. Energies 2021, 14, 4061. https://doi.org/10.3390/en14134061
Busca G. Production of Gasolines and Monocyclic Aromatic Hydrocarbons: From Fossil Raw Materials to Green Processes. Energies. 2021; 14(13):4061. https://doi.org/10.3390/en14134061
Chicago/Turabian StyleBusca, Guido. 2021. "Production of Gasolines and Monocyclic Aromatic Hydrocarbons: From Fossil Raw Materials to Green Processes" Energies 14, no. 13: 4061. https://doi.org/10.3390/en14134061
APA StyleBusca, G. (2021). Production of Gasolines and Monocyclic Aromatic Hydrocarbons: From Fossil Raw Materials to Green Processes. Energies, 14(13), 4061. https://doi.org/10.3390/en14134061