Mg6MnO8 as a Magnesium-Ion Battery Material: Defects, Dopants and Mg-Ion Transport




Abstract

1. Introduction
2. Computational Methods
3. Results
3.1. Crystal Structure of Mg6MnO8
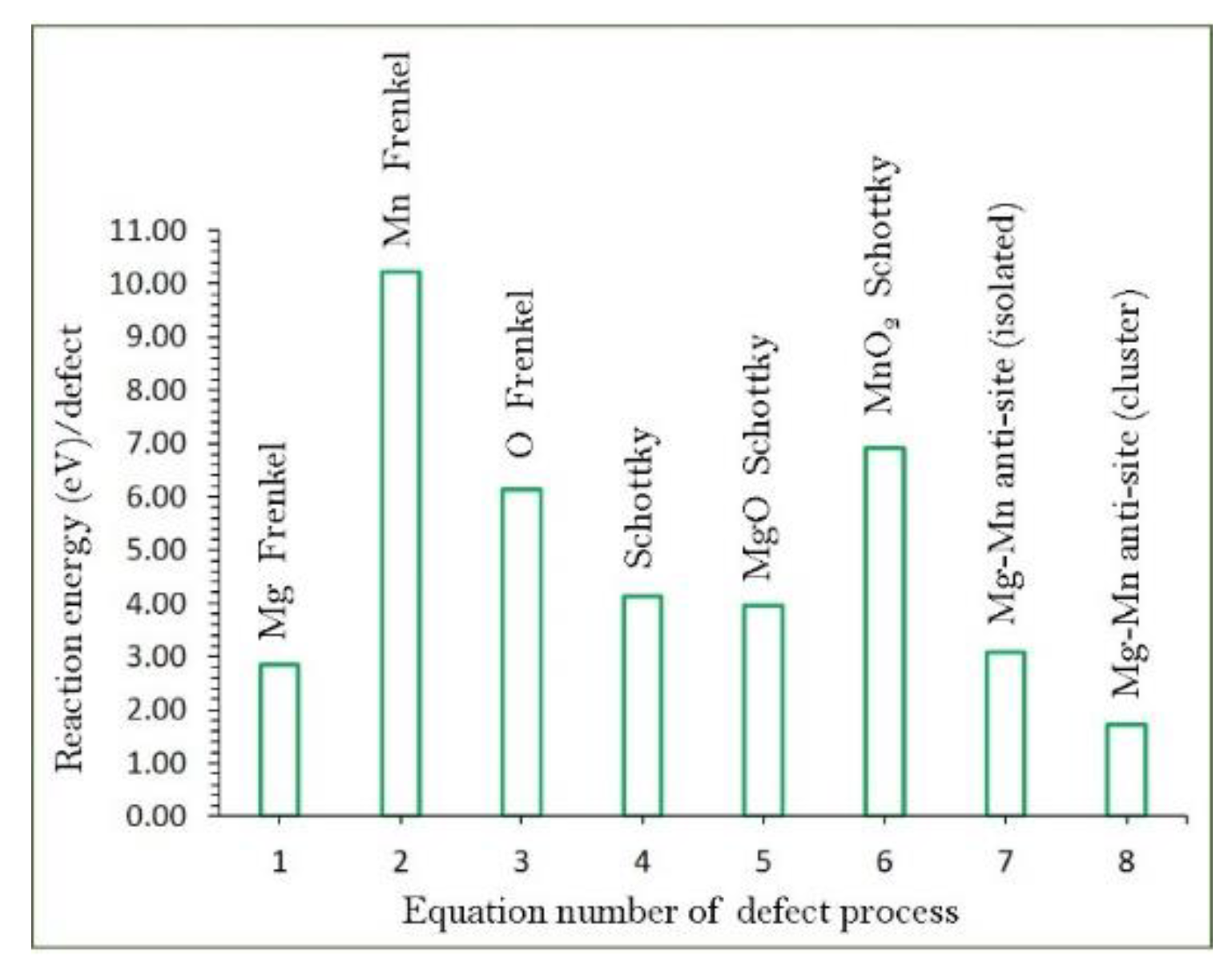
3.2. Intrinsic Defect Processes
3.3. Mg-Ion Diffusion
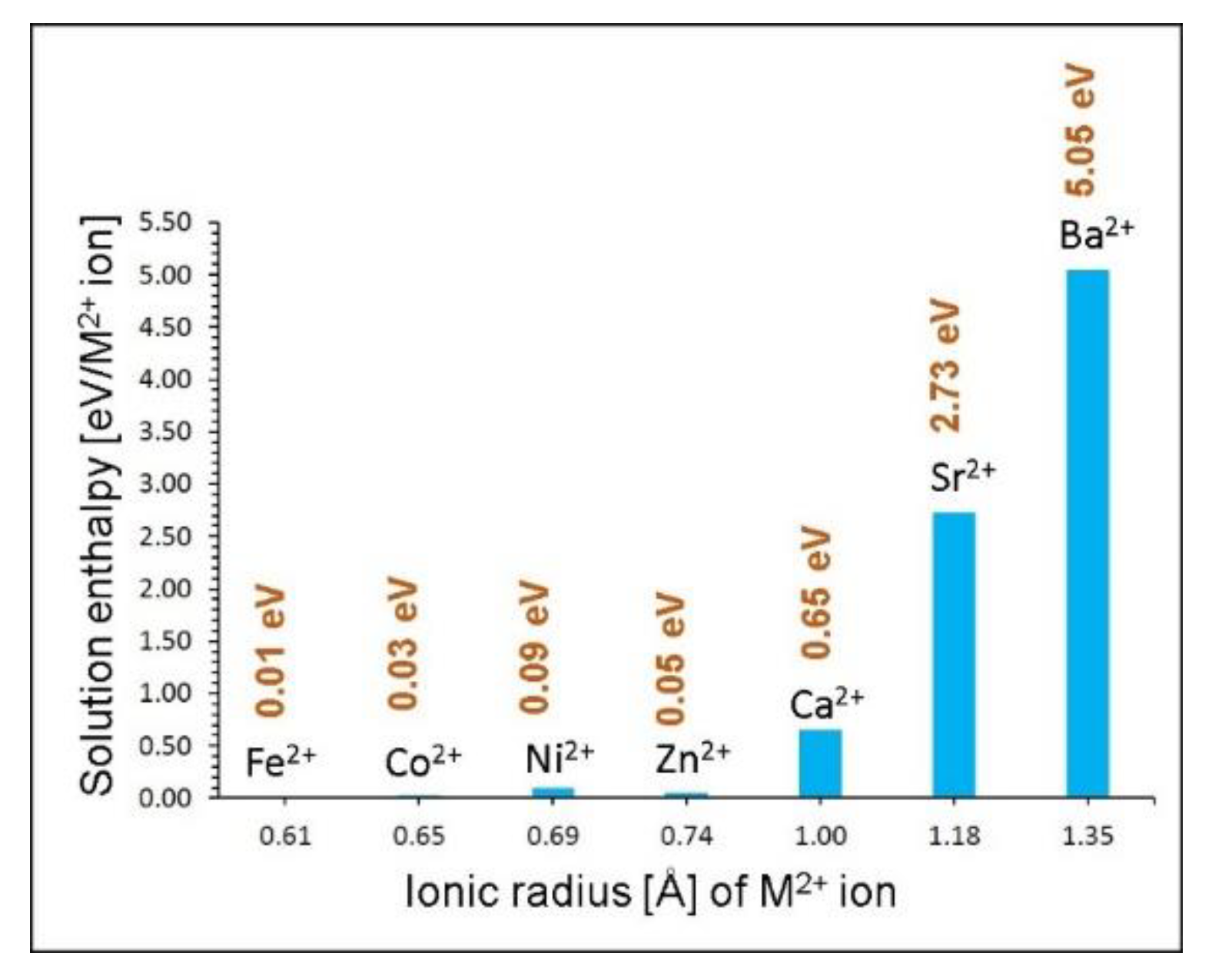
3.4. Dopant Substitution
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Xu, C.; Li, B.; Du, H.; Kang, F. Energetic Zinc Ion Chemistry: The Rechargeable Zinc Ion Battery. Angew. Chem. Int. Ed. 2012, 51, 933–935. [Google Scholar] [CrossRef]
- Muldoon, J.; Bucur, C.B.; Gregory, T. Quest for Nonaqueous Multivalent Secondary Batteries: Magnesium and Beyond. Chem. Rev. 2014, 114, 11683–11720. [Google Scholar] [CrossRef]
- Ponrouch, A.; Frontera, C.; Bardé, F.; Palacín, M.R. Towards a calcium-based rechargeable battery. Nat. Mater. 2015, 15, 169. [Google Scholar] [CrossRef] [PubMed]
- Novák, P.; Imhof, R.; Haas, O. Magnesium insertion electrodes for rechargeable nonaqueous batteries—A competitive alternative to lithium? Electrochim. Acta 1999, 45, 351–367. [Google Scholar] [CrossRef]
- Aurbach, D.; Gofer, Y.; Lu, Z.; Schechter, A.; Chusid, O.; Gizbar, H.; Cohen, Y.; Ashkenazi, V.; Moshkovich, M.; Turgeman, R.; et al. A short review on the comparison between Li battery systems and rechargeable magnesium battery technology. J. Power Sources 2001, 97–98, 28–32. [Google Scholar] [CrossRef]
- Huie, M.M.; Bock, D.C.; Takeuchi, E.S.; Marschilok, A.C.; Takeuchi, K.J. Cathode materials for magnesium and magnesium-ion based batteries. Coord. Chem. Rev. 2015, 287, 15–27. [Google Scholar] [CrossRef]
- Yoo, H.D.; Shterenberg, I.; Gofer, Y.; Gershinsky, G.; Pour, N.; Aurbach, D. Mg rechargeable batteries: An on-going challenge. Energy Environ. Sci. 2013, 6, 2265–2279. [Google Scholar] [CrossRef]
- Feng, Z.; Yang, J.; NuLi, Y.; Wang, J. Sol–gel synthesis of Mg1.03Mn0.97SiO4 and its electrochemical intercalation behavior. J. Power Sources 2008, 184, 604–609. [Google Scholar] [CrossRef]
- Orikasa, Y.; Masese, T.; Koyama, Y.; Mori, T.; Hattori, M.; Yamamoto, K.; Okado, T.; Huang, Z.D.; Minato, T.; Tassel, C.; et al. High energy density rechargeable magnesium battery using earth-abundant and non-toxic elements. Sci. Rep. 2014, 4, 5622. [Google Scholar] [CrossRef] [PubMed]
- NuLi, Y.; Zheng, Y.; Wang, Y.; Yang, J.; Wang, J. Electrochemical intercalation of Mg2+ in 3D hierarchically porous magnesium cobalt silicate and its application as an advanced cathode material in rechargeable magnesium batteries. J. Mater. Chem. 2011, 21, 12437–12443. [Google Scholar] [CrossRef]
- Lee, S.H.; DiLeo, R.A.; Marschilok, A.C.; Takeuchi, K.J.; Takeuchi, E.S. Sol Gel Based Synthesis and Electrochemistry of Magnesium Vanadium Oxide: A Promising Cathode Material for Secondary Magnesium Ion Batteries. ECS Electrochem. Lett. 2014, 3, A87–A90. [Google Scholar] [CrossRef]
- Huang, Z.D.; Masese, T.; Orikasa, Y.; Mori, T.; Minato, T.; Tassel, C.; Kobayashi, Y.; Kageyama, H.; Uchimoto, Y. MgFePO4F as a feasible cathode material for magnesium batteries. J. Mater. Chem. A 2014, 2, 11578–11582. [Google Scholar] [CrossRef]
- Liu, M.; Jain, A.; Rong, Z.; Qu, X.; Canepa, P.; Malik, R.; Ceder, G.; Persson, K.A. Evaluation of sulfur spinel compounds for multivalent battery cathode applications. Energy Environ. Sci. 2016, 9, 3201–3209. [Google Scholar] [CrossRef]
- Wan, L.F.; Perdue, B.R.; Apblett, C.A.; Prendergast, D. Mg Desolvation and Intercalation Mechanism at the Mo6S8 Chevrel Phase Surface. Chem. Mater. 2015, 27, 5932–5940. [Google Scholar] [CrossRef]
- Aurbach, D.; Lu, Z.; Schechter, A.; Gofer, Y.; Gizbar, H.; Turgeman, R.; Cohen, Y.; Moshkovich, M.; Levi, E. Prototype systems for rechargeable magnesium batteries. Nature 2000, 407, 724–727. [Google Scholar] [CrossRef]
- Gu, S.; Hsieh, C.T.; Huq, M.M.; Hsu, J.P.; Gandomi, Y.A.; Li, J. Preparation of MgCo2O4/graphite composites as cathode materials for magnesium-ion batteries. J. Solid State Electrochem. 2019, 23, 1399–1407. [Google Scholar] [CrossRef]
- Lee, J.; Seymour, I.D.; Pell, A.J.; Dutton, S.E.; Grey, C.P. A systematic study of 25Mg NMR in paramagnetic transition metal oxides: Applications to Mg-ion battery materials. Phys. Chem. Chem. Phys. 2017, 19, 613–625. [Google Scholar] [CrossRef]
- Armstrong, A.R.; Kuganathan, N.; Islam, M.S.; Bruce, P.G. Structure and Lithium Transport Pathways in Li2FeSiO4 Cathodes for Lithium Batteries. J. Am. Chem. Soc. 2011, 133, 13031–13035. [Google Scholar] [CrossRef]
- Kuganathan, N.; Iyngaran, P.; Chroneos, A. Lithium diffusion in Li5FeO4. Sci. Rep. 2018, 8, 5832. [Google Scholar] [CrossRef]
- Kuganathan, N.; Ganeshalingam, S.; Chroneos, A. Defects, Dopants and Lithium Mobility in Li9V3(P2O7)3 (PO4)2. Sci. Rep. 2018, 8, 8140. [Google Scholar] [CrossRef]
- Kuganathan, N.; Islam, M.S. Li2MnSiO4 Lithium Battery Material: Atomic-Scale Study of Defects, Lithium Mobility, and Trivalent Dopants. Chem. Mater. 2009, 21, 5196–5202. [Google Scholar] [CrossRef]
- Fisher, C.A.J.; Kuganathan, N.; Islam, M.S. Defect chemistry and lithium-ion migration in polymorphs of the cathode material Li2MnSiO4. J. Mater. Chem. A 2013, 1, 4207–4214. [Google Scholar] [CrossRef]
- Kuganathan, N.; Kordatos, A.; Chroneos, A. Li2SnO3 as a Cathode Material for Lithium-ion Batteries: Defects, Lithium Ion Diffusion and Dopants. Sci. Rep. 2018, 8, 12621. [Google Scholar] [CrossRef] [PubMed]
- Kuganathan, N.; Chroneos, A. Defects, Dopants and Sodium Mobility in Na2MnSiO4. Sci. Rep. 2018, 8, 14669. [Google Scholar] [CrossRef] [PubMed]
- Kuganathan, N.; Chroneos, A. Defects and dopant properties of Li3V2(PO4)3. Sci. Rep. 2019, 9, 333. [Google Scholar] [CrossRef] [PubMed]
- Kuganathan, N.; Kordatos, A.; Chroneos, A. Defect Chemistry and Li-ion Diffusion in Li2RuO3. Sci. Rep. 2019, 9, 550. [Google Scholar] [CrossRef] [PubMed]
- Kuganathan, N.; Kordatos, A.; Fitzpatrick, M.E.; Vovk, R.V.; Chroneos, A. Defect process and lithium diffusion in Li2TiO3. Solid State Ion. 2018, 327, 93–98. [Google Scholar] [CrossRef]
- Kuganathan, N.; Kordatos, A.; Anurakavan, S.; Iyngaran, P.; Chroneos, A. Li3SbO4 lithium-ion battery material: Defects, lithium ion diffusion and tetravalent dopants. Mater. Chem. Phys. 2019, 225, 34–41. [Google Scholar] [CrossRef]
- Kuganathan, N.; Chroneos, A. Na3V(PO4)2 cathode material for Na ion batteries: Defects, dopants and Na diffusion. Solid State Ion. 2019, 336, 75–79. [Google Scholar] [CrossRef]
- Kuganathan, N.; Iyngaran, P.; Vovk, R.; Chroneos, A. Defects, dopants and Mg diffusion in MgTiO3. Sci. Rep. 2019, 9, 4394. [Google Scholar] [CrossRef]
- Kuganathan, N.; Sgourou, E.N.; Panayiotatos, Y.; Chroneos, A. Defect Process, Dopant Behaviour and Li Ion Mobility in the Li2MnO3 Cathode Material. Energies 2019, 12, 1329. [Google Scholar] [CrossRef]
- Kordatos, A.; Kuganathan, N.; Kelaidis, N.; Iyngaran, P.; Chroneos, A. Defects and lithium migration in Li2CuO2. Sci. Rep. 2018, 8, 6754. [Google Scholar] [CrossRef] [PubMed]
- Kuganathan, N.; Kordatos, A.; Kelaidis, N.; Chroneos, A. Defects, Lithium Mobility and Tetravalent Dopants in the Li3NbO4 Cathode Material. Sci. Rep. 2019, 9, 2192. [Google Scholar] [CrossRef] [PubMed]
- Kuganathan, N.; Tsoukalas, L.H.; Chroneos, A. Defects, dopants and Li-ion diffusion in Li2SiO3. Solid State Ion. 2019, 335, 61–66. [Google Scholar] [CrossRef]
- Kuganathan, N.; Chroneos, A. Defect Chemistry and Na-Ion Diffusion in Na3Fe2(PO4)3 Cathode Material. Materials 2019, 12, 1348. [Google Scholar] [CrossRef] [PubMed]
- Kuganathan, N.; Dark, J.; Sgourou, E.N.; Panayiotatos, Y.; Chroneos, A. Atomistic Simulations of the Defect Chemistry and Self-Diffusion of Li-ion in LiAlO2. Energies 2019, 12, 2895. [Google Scholar] [CrossRef]
- Kaushalya, R.; Iyngaran, P.; Kuganathan, N.; Chroneos, A. Defect, Diffusion and Dopant Properties of NaNiO2: Atomistic Simulation Study. Energies 2019, 12, 3094. [Google Scholar] [CrossRef]
- Gale, J.D.; Rohl, A.L. The General Utility Lattice Program (GULP). Mol. Simul. 2003, 29, 291–341. [Google Scholar] [CrossRef]
- Gale, J.D. GULP: A computer program for the symmetry-adapted simulation of solids. J. Chem. Soc. Faraday Trans. 1997, 93, 629–637. [Google Scholar] [CrossRef]
- Mott, N.F.; Littleton, M.J. Conduction in polar crystals. I. Electrolytic conduction in solid salts. Trans. Faraday Soc. 1938, 34, 485–499. [Google Scholar] [CrossRef]
- Taguchi, H.; Ohta, A.; Nagao, M.; Kido, H.; Ando, H.; Tabata, K. Crystal Structure and Magnetic Properties of (Mg6−xLix)MnO8. J. Solid State Chem. 1996, 124, 220–223. [Google Scholar] [CrossRef]
- Kröger, F.A.; Vink, H.J. Relations between the Concentrations of Imperfections in Crystalline Solids. In Solid State Physics; Seitz, F., Turnbull, D., Eds.; Academic Press: Cambridge, MA, USA, 1956; Volume 3, pp. 307–435. [Google Scholar]
- Nytén, A.; Kamali, S.; Häggström, L.; Gustafsson, T.; Thomas, J.O. The lithium extraction/insertion mechanism in Li2FeSiO4. J. Mater. Chem. 2006, 16, 2266–2272. [Google Scholar] [CrossRef]
- Ensling, D.; Stjerndahl, M.; Nytén, A.; Gustafsson, T.; Thomas, J.O. A comparative XPS surface study of Li2FeSiO4/C cycled with LiTFSI- and LiPF6-based electrolytes. J. Mater. Chem. 2009, 19, 82–88. [Google Scholar] [CrossRef]
- Liu, H.; Choe, M.J.; Enrique, R.A.; Orvañanos, B.; Zhou, L.; Liu, T.; Thornton, K.; Grey, C.P. Effects of Antisite Defects on Li Diffusion in LiFePO4 Revealed by Li Isotope Exchange. J. Phys. Chem. C 2017, 121, 12025–12036. [Google Scholar] [CrossRef]
- Devaraju, M.K.; Truong, Q.D.; Hyodo, H.; Sasaki, Y.; Honma, I. Synthesis, characterization and observation of antisite defects in LiNiPO4 nanomaterials. Sci. Rep. 2015, 5, 11041. [Google Scholar] [CrossRef]
- Politaev, V.V.; Petrenko, A.A.; Nalbandyan, V.B.; Medvedev, B.S.; Shvetsova, E.S. Crystal structure, phase relations and electrochemical properties of monoclinic Li2MnSiO4. J. Solid State Chem. 2007, 180, 1045–1050. [Google Scholar] [CrossRef]
- Fisher, C.A.J.; Hart Prieto, V.M.; Islam, M.S. Lithium Battery Materials LiMPO4 (M = Mn, Fe, Co, and Ni): Insights into Defect Association, Transport Mechanisms, and Doping Behavior. Chem. Mater. 2008, 20, 5907–5915. [Google Scholar] [CrossRef]
- Nishimura, S.I.; Kobayashi, G.; Ohoyama, K.; Kanno, R.; Yashima, M.; Yamada, A. Experimental visualization of lithium diffusion in LixFePO4. Nat. Mater. 2008, 7, 707. [Google Scholar] [CrossRef]
- Peng, B.; Liang, J.; Tao, Z.; Chen, J. Magnesium nanostructures for energy storage and conversion. J. Mater. Chem. 2009, 19, 2877–2883. [Google Scholar] [CrossRef]
- Novák, P.; Desilvestro, J. Electrochemical Insertion of Magnesium in Metal Oxides and Sulfides from Aprotic Electrolytes. J. Electrochem. Soc. 1993, 140, 140–144. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Calc | Expt41 | |∆| (%) |
|---|---|---|---|
| a = b = c (Å) | 8.4259 | 8.3818 | 0.53 |
| α = β = γ (°) | 90.0000 | 90.0000 | 0.00 |
| V (Å3) | 598.21 | 588.86 | 1.59 |
| Migration Path | Mg–Mg Separation/Å | Activation Energy/eV |
|---|---|---|
| A | 2.98 | 0.82 |
| B | 5.96 | 3.72 |
| Long-Range Path | Overall Activation Energy/eV |
|---|---|
| A→A→A→A | 0.82 |
| B→B→B→B | 3.72 |
| A→B→B→A | 3.72 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuganathan, N.; Gkanas, E.I.; Chroneos, A. Mg6MnO8 as a Magnesium-Ion Battery Material: Defects, Dopants and Mg-Ion Transport. Energies 2019, 12, 3213. https://doi.org/10.3390/en12173213
Kuganathan N, Gkanas EI, Chroneos A. Mg6MnO8 as a Magnesium-Ion Battery Material: Defects, Dopants and Mg-Ion Transport. Energies. 2019; 12(17):3213. https://doi.org/10.3390/en12173213
Chicago/Turabian StyleKuganathan, Navaratnarajah, Evangelos I. Gkanas, and Alexander Chroneos. 2019. "Mg6MnO8 as a Magnesium-Ion Battery Material: Defects, Dopants and Mg-Ion Transport" Energies 12, no. 17: 3213. https://doi.org/10.3390/en12173213
APA StyleKuganathan, N., Gkanas, E. I., & Chroneos, A. (2019). Mg6MnO8 as a Magnesium-Ion Battery Material: Defects, Dopants and Mg-Ion Transport. Energies, 12(17), 3213. https://doi.org/10.3390/en12173213