Insertion of Mono- vs. Bi- vs. Trivalent Atoms in Prospective Active Electrode Materials for Electrochemical Batteries: An ab Initio Perspective



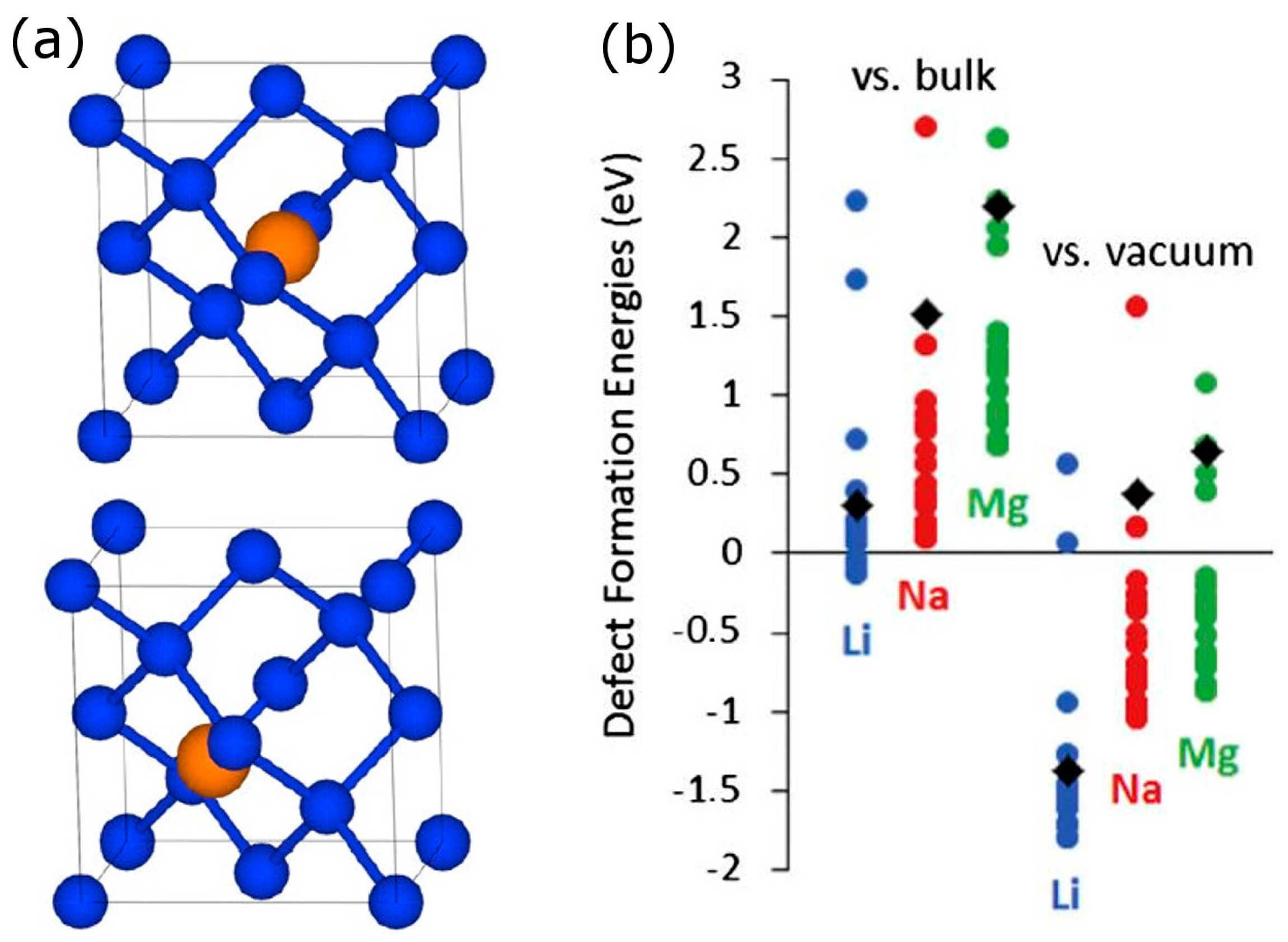{kind=link}
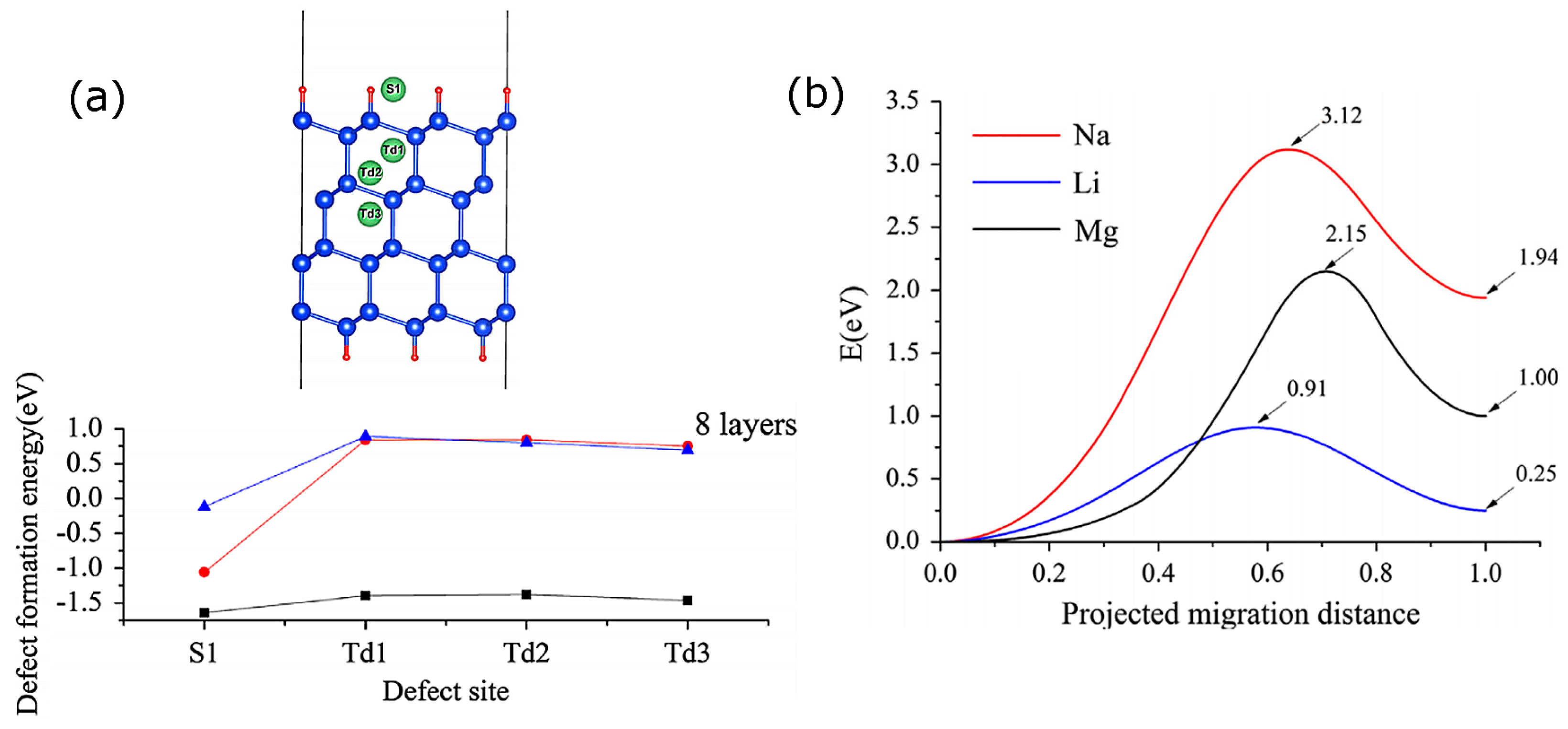{kind=link}
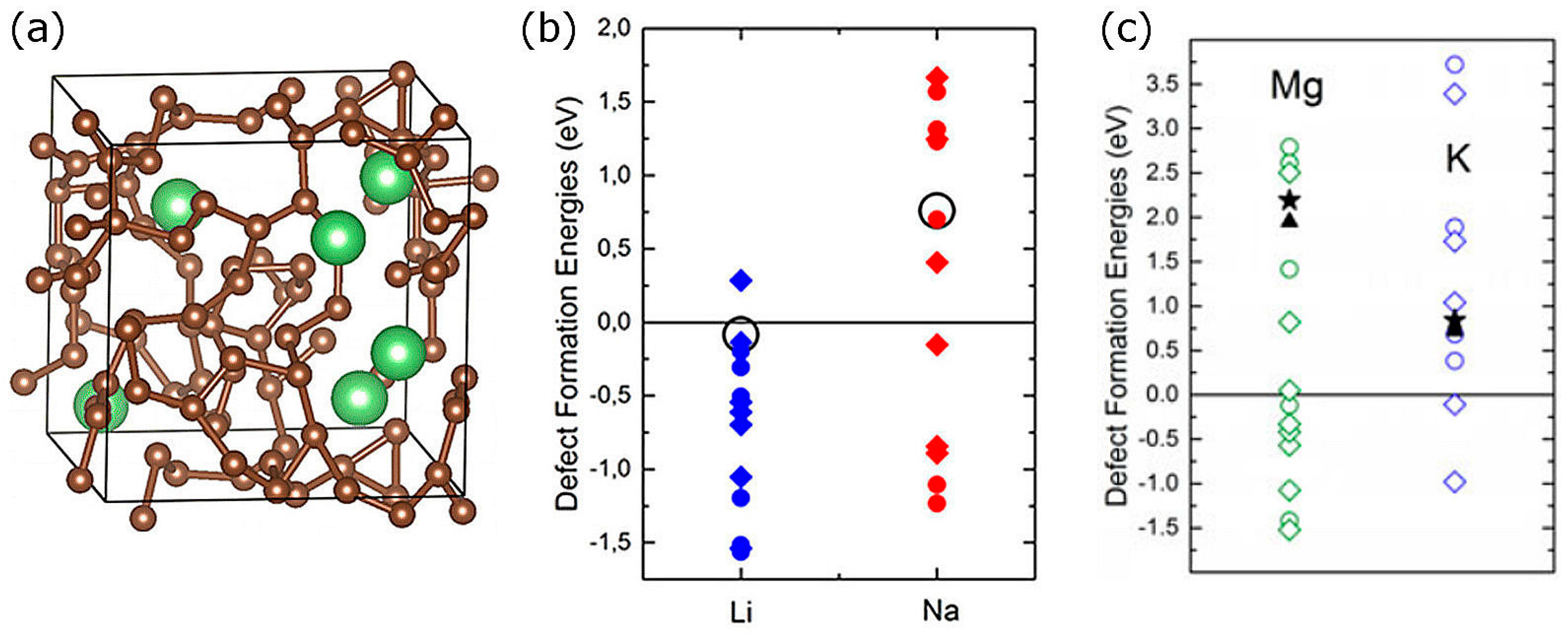{kind=link}
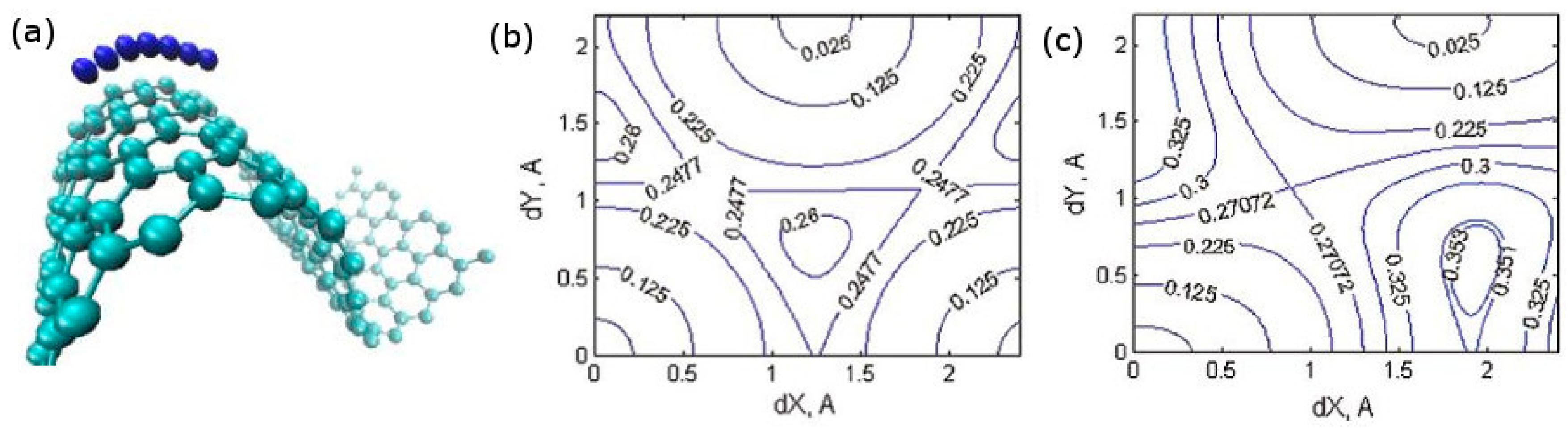{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Ab Initio Modeling Assists in Rational Design of Active Electrode Materials for Advanced Electrochemical Batteries
1.2. This Review
1.3. Key Computed Quantities
2. Results
2.1. Interactions of Li, Na, K, and Mg with Monoelemental Group IV Materials
2.1.1. Interaction of Li, Na, and Mg with Silicon: Effects Due to Ion Size and Valence, of Host Amorphization, and of Phonons, and How to Build Effective Localized Basis Sets
2.1.2. Insertion of Li, Na, K, and Mg in Carbon: Effects Due to Ion Size and of Host Amorphization
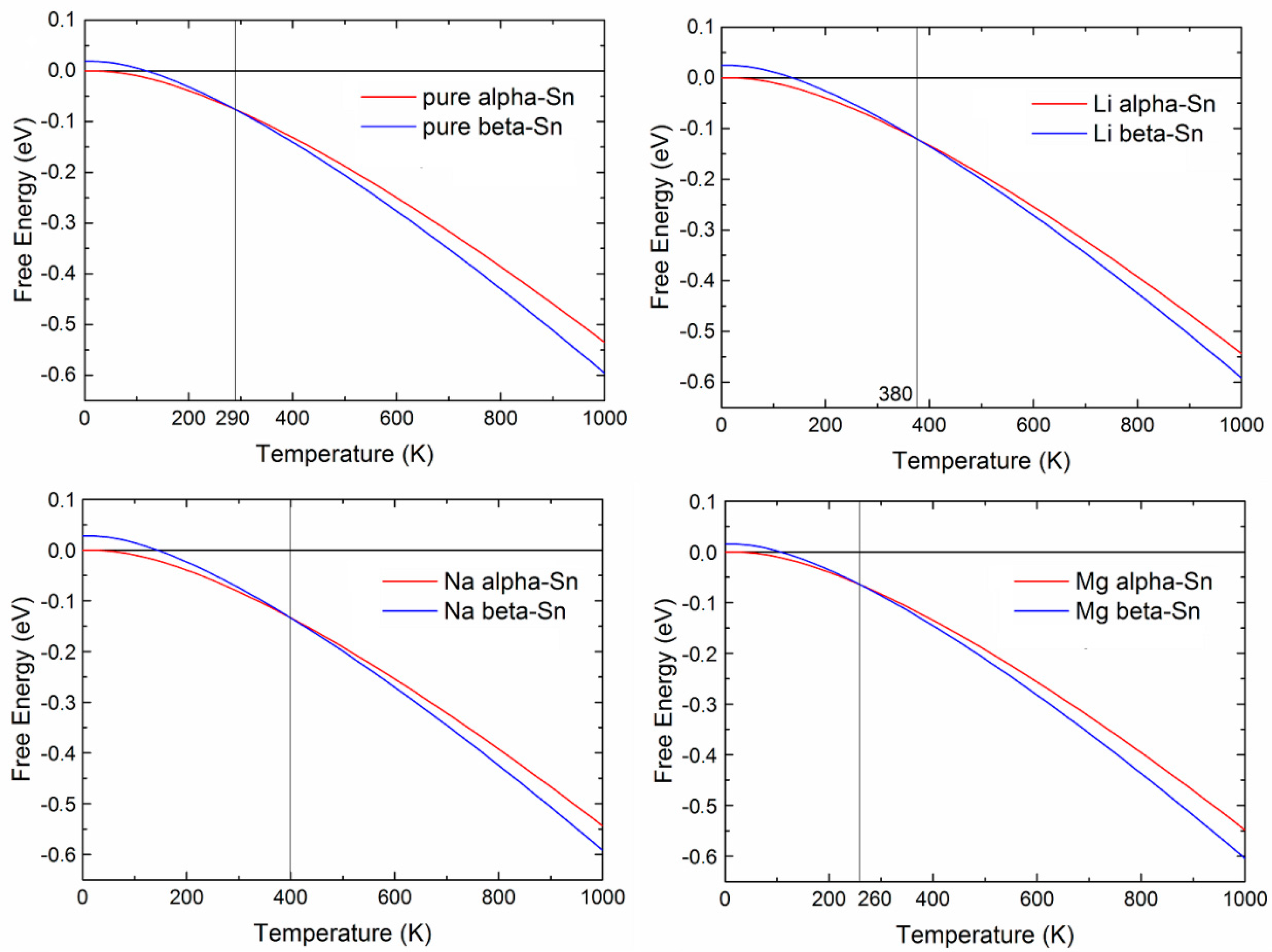
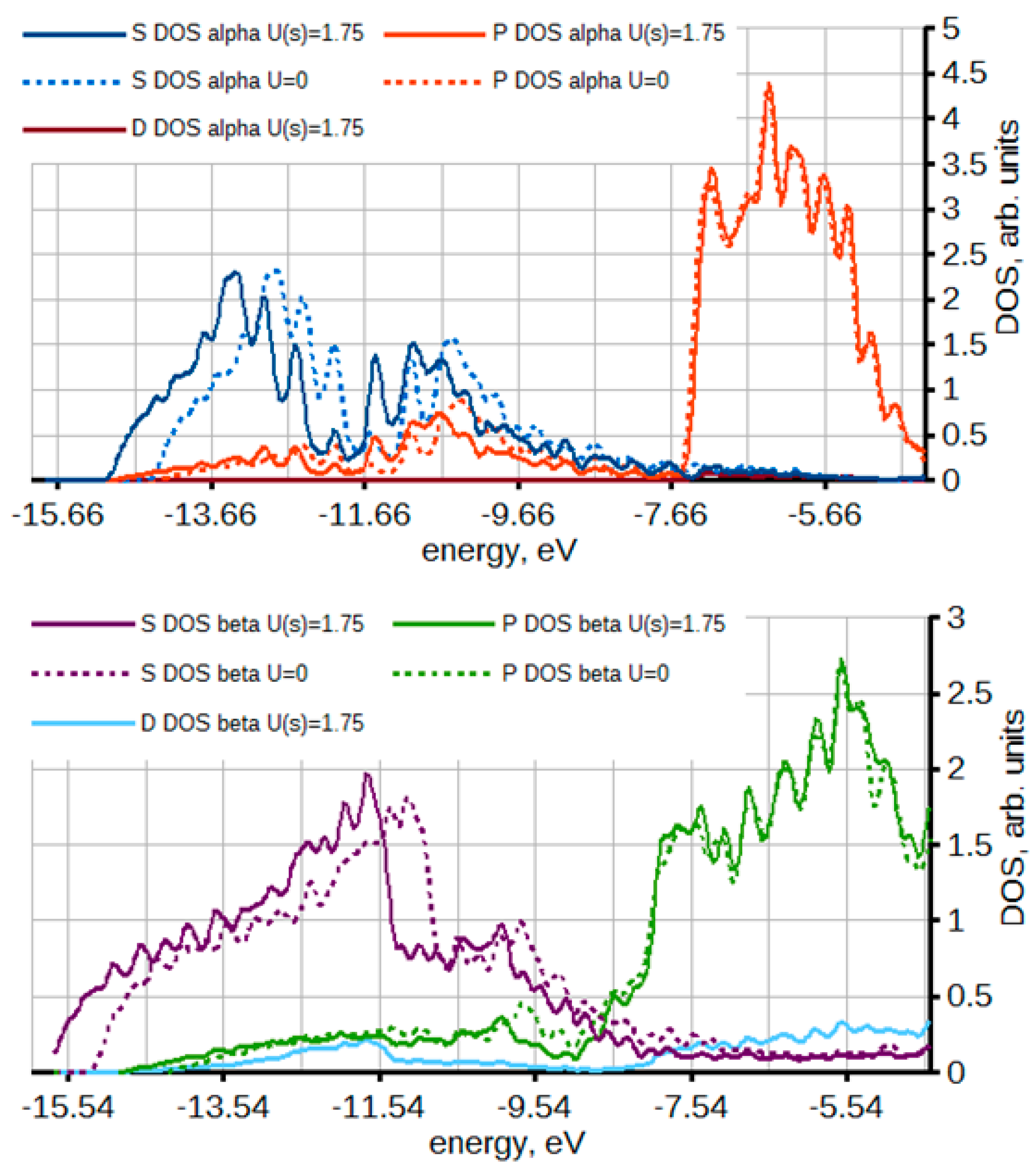
2.1.3. Insertion of Li, Na, and Mg in Tin: Effects Due to Ion Size and Valence, and Phase Competition
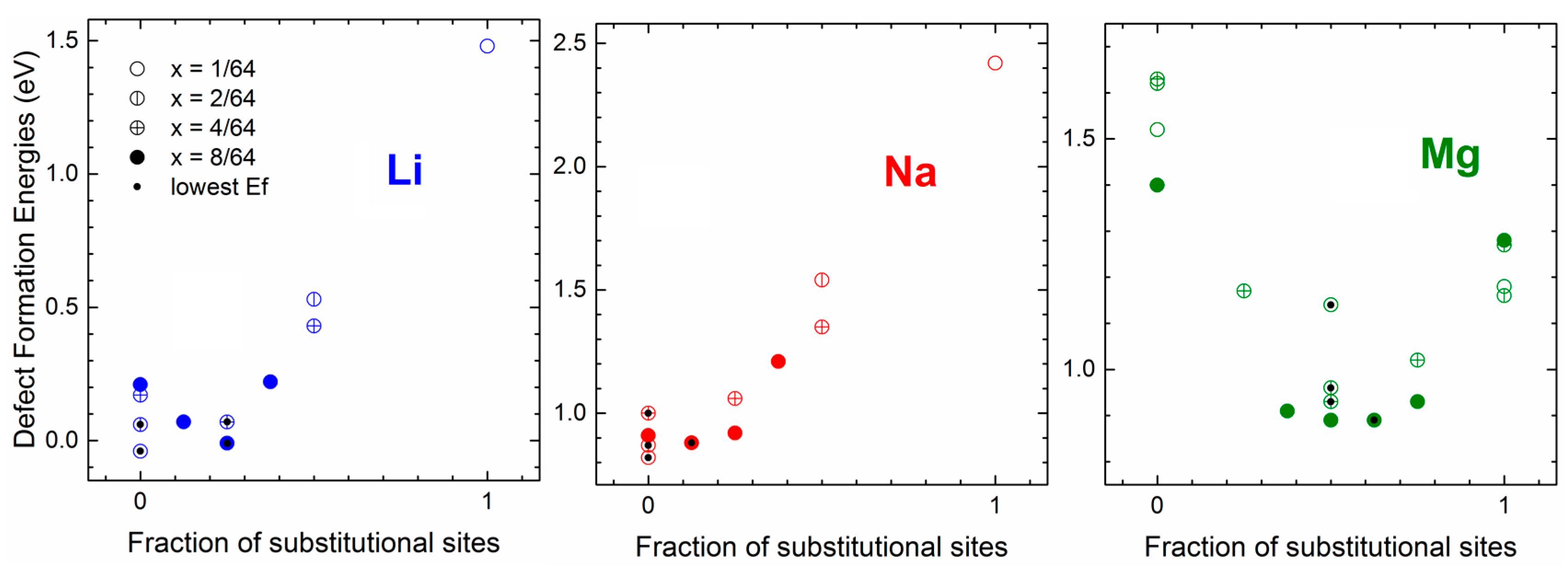
2.1.4. Interaction of Li, Na, and Mg with Germanium: Effects Due to Ion Size and Valence and Competition between Interstitial and Substitutional Sites
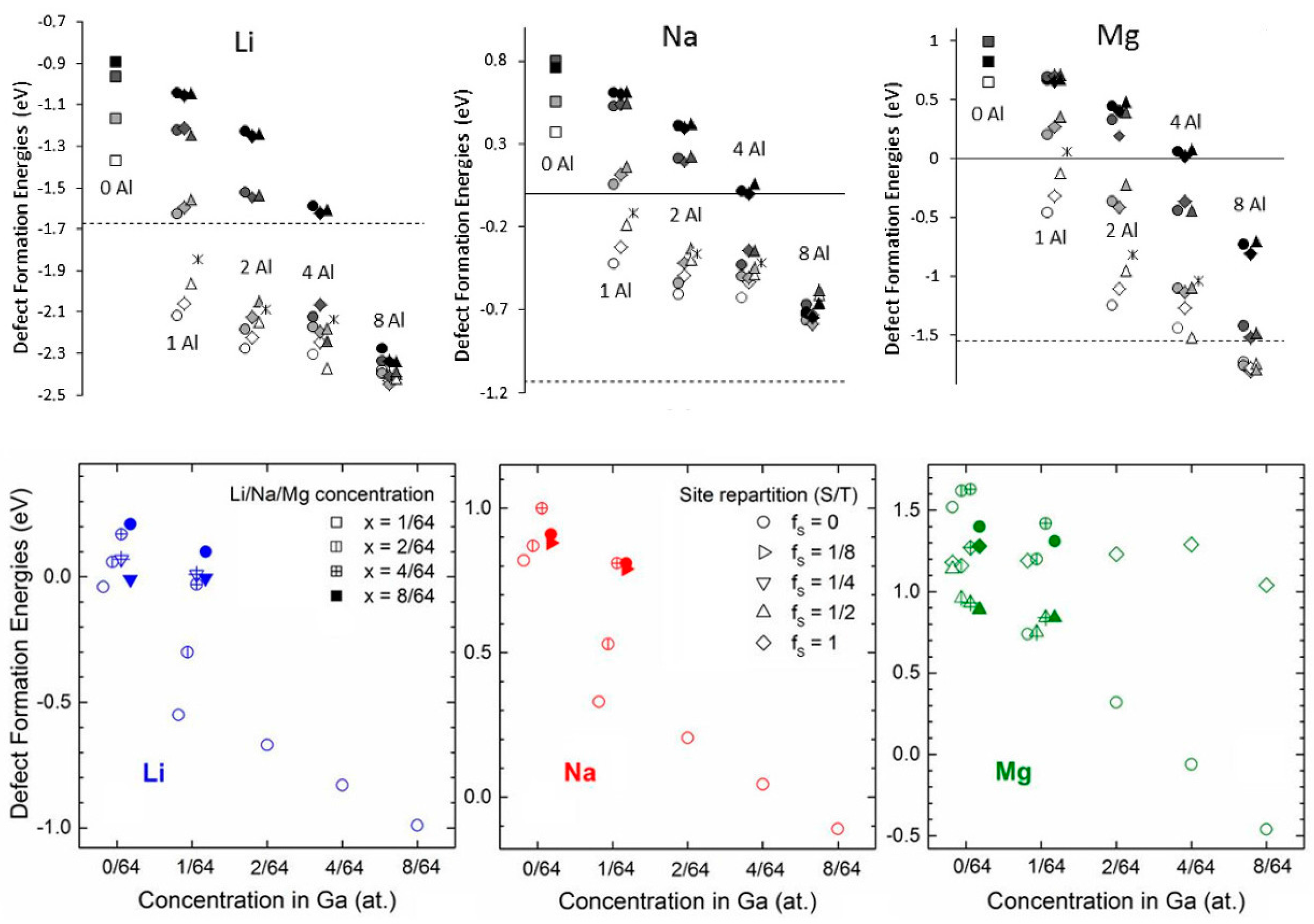
2.1.5. p-Doping of Si and Ge as a Way to Enhance Li, Na, and Mg Insertion Energetics
2.2. Interactions of Li, Na, and Mg with Different Phases of Titania
2.2.1. Mapping Li, Na, and Mg Insertion Energies among Different Phases Including Amorphous
2.2.2. Electronic Structure of Doped Titania
2.2.3. On the Charge and Oxidation State of Titanium in TiO2
2.3. Interactions of Li, Na, Na, Mg, and Al with Different Phases and Stoichiometries of Vanadium Oxides: Effects Due to Ion Size and Valency
2.3.1. Li, Na, K, Mg and Al Insertion Properties
2.3.2. On the Treatment of Dispersion Interactions
2.4. From Oxides to Sulfides: A Promising Direction for Multivalent Batteries
3. Discussion and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- British Petroleum. British Petroleum Company BP Statistical Review of World Energy, 66th ed.; British Petroleum Co.: London, UK, 2017; Available online: https://www.bp.com/content/dam/bp/en/corporate/pdf/energy-economics/statistical-review-2017/bp-statistical-review-of-world-energy-2017-full-report.pdf (accessed on 1 November 2017).
- US Geological Survey. U.S. Geological Survey Mineral Commodity Summaries; US Geological Survey: Reston, VA, USA, 2016.
- Wedepohl, K.H. The composition of the continental crust. Geochim. Cosmochim. Acta 1995, 59, 1217–1232. [Google Scholar] [CrossRef]
- Marcus, Y. Thermodynamic functions of transfer of single ions from water to nonaqueous and mixed solvents: Part 3—Standard potentials of selected electrodes. Pure Appl. Chem. 1985, 57, 1129–1132. [Google Scholar] [CrossRef]
- Komaba, S.; Hasegawa, T.; Dahbi, M.; Kubota, K. Potassium intercalation into graphite to realize high-voltage/high-power potassium-ion batteries and potassium-ion capacitors. Electrochem. Commun. 2015, 60, 172–175. [Google Scholar] [CrossRef]
- Haynes, W.M. CRC Handbook of Chemistry and Physics; CRC Press: Boca Raton, FL, USA, 2014. [Google Scholar]
- Lüder, J.; Legrain, F.; Chen, Y.; Manzhos, S. Doping of active electrode materials for electrochemical batteries: An electronic structure perspective. MRS Commun. 2017, 7, 523–540. [Google Scholar] [CrossRef]
- Meng, Y.S.; Arroyo-de Dompablo, M.E. First principles computational materials design for energy storage materials in lithium ion batteries. Energy Environ. Sci. 2009, 2, 589–609. [Google Scholar] [CrossRef]
- Urban, A.; Seo, D.-H.; Ceder, G. Computational understanding of Li-ion batteries. npj Comput. Mater. 2016, 2, 16002. [Google Scholar] [CrossRef]
- Lejaeghere, K.; Bihlmayer, G.; Björkman, T.; Blaha, P.; Blügel, S.; Blum, V.; Caliste, D.; Castelli, I.E.; Clark, S.J.; Dal Corso, A. Reproducibility in density functional theory calculations of solids. Science 2016, 351, aad3000. [Google Scholar] [CrossRef] [PubMed]
- Malyi, O.; Kulish, V.V.; Tan, T.L.; Manzhos, S. A computational study of the insertion of Li, Na, and Mg atoms into Si (111) nanosheets. Nano Energy 2013, 2, 1149–1157. [Google Scholar] [CrossRef]
- Malyi, O.I.; Tan, T.L.; Manzhos, S. A comparative computational study of structures, diffusion, and dopant interactions between Li and Na insertion into Si. Appl. Phys. Express 2013, 6, 027301. [Google Scholar] [CrossRef]
- Malyi, O.I.; Tan, T.L.; Manzhos, S. In search of high performance anode materials for Mg batteries: Computational studies of Mg in Ge, Si, and Sn. J. Power Sources 2013, 233, 341–345. [Google Scholar] [CrossRef]
- Legrain, F.; Malyi, O.I.; Manzhos, S. Comparative computational study of the diffusion of Li, Na, and Mg in silicon including the effect of vibrations. Solid State Ion. 2013, 253, 157–163. [Google Scholar] [CrossRef]
- Legrain, F.; Malyi, O.I.; Tan, T.L.; Manzhos, S. Computational study of Mg insertion into amorphous silicon: Advantageous energetics over crystalline silicon for Mg storage. MRS Online Proc. Libr. Arch. 2013, 1540. [Google Scholar] [CrossRef]
- Tan, T.L.; Malyi, O.I.; Legrain, F.; Manzhos, S. Role of Inter-Dopant Interactions on the Diffusion of Li and Na Atoms in Bulk Si Anodes. MRS Online Proc. Libr. Arch. 2013, 1541. [Google Scholar] [CrossRef]
- Carvalho, A.; Rayson, M.J.; Briddon, P.R.; Manzhos, S. Effect of the adsorption of ethylene carbonate on Si surfaces on the Li insertion behavior. Chem. Phys. Lett. 2013, 585, 157–161. [Google Scholar] [CrossRef]
- Koh, Y.W.; Manzhos, S. Curvature drastically changes diffusion properties of Li and Na on graphene. MRS Commun. 2013, 3, 171–175. [Google Scholar] [CrossRef]
- Kulish, V.V.; Malyi, O.I.; Ng, M.-F.; Chen, Z.; Manzhos, S.; Wu, P. Controlling Na diffusion by rational design of Si-based layered architectures. Phys. Chem. Chem. Phys. 2014, 16, 4260–4267. [Google Scholar] [CrossRef] [PubMed]
- Legrain, F.; Malyi, O.I.; Manzhos, S. Comparative computational study of the energetics of Li, Na, and Mg storage in amorphous and crystalline silicon. Comput. Mater. Sci. 2014, 94, 214–217. [Google Scholar] [CrossRef]
- Legrain, F.; Malyi, O.I.; Manzhos, S. A Comparative Computational Study of Li, Na, and Mg Insertion in α-Sn. MRS Online Proc. Libr. Arch. 2014, 1678. [Google Scholar] [CrossRef]
- Legrain, F.; Malyi, O.; Manzhos, S. Insertion energetics of lithium, sodium, and magnesium in crystalline and amorphous titanium dioxide: A comparative first-principles study. J. Power Sources 2015, 278, 197–202. [Google Scholar] [CrossRef]
- Legrain, F.; Malyi, O.I.; Persson, C.; Manzhos, S. Comparison of alpha and beta tin for lithium, sodium, and magnesium storage: An ab initio study including phonon contributions. J. Chem. Phys. 2015, 143, 204701. [Google Scholar] [CrossRef] [PubMed]
- Legrain, F.; Manzhos, S. Aluminum doping improves the energetics of lithium, sodium, and magnesium storage in silicon: A first-principles study. J. Power Sources 2015, 274, 65–70. [Google Scholar] [CrossRef]
- Legrain, F.; Sottmann, J.; Kotsis, K.; Gorantla, S.; Sartori, S.; Manzhos, S. Amorphous (Glassy) carbon, a promising material for sodium ion battery anodes: A combined first-principles and experimental study. J. Phys. Chem. C 2015, 119, 13496–13501. [Google Scholar] [CrossRef]
- Manzhos, S.; Giorgi, G.; Yamashita, K. A density functional tight binding study of acetic acid adsorption on crystalline and amorphous surfaces of titania. Molecules 2015, 20, 3371–3388. [Google Scholar] [CrossRef] [PubMed]
- Legrain, F.; Kotsis, K.; Manzhos, S. Mg and K Insertion in Glassy Amorphous Carbon vs Graphite as Potential Anode Materials: An Ab Initio Study. MRS Adv. 2016, 1, 3069–3074. [Google Scholar] [CrossRef]
- Legrain, F.; Manzhos, S. Understanding the difference in cohesive energies between alpha and beta tin in DFT calculations. AIP Adv. 2016, 6, 045116. [Google Scholar] [CrossRef]
- Sk, M.A.; Chen, Y.; Manzhos, S. Orbital order switching in molecular calculations using GGA functionals: Qualitative errors in materials modeling for electrochemical power sources and how to fix them. Chem. Phys. Lett. 2016, 659, 270–276. [Google Scholar] [CrossRef]
- Sk, M.A.; Manzhos, S. Exploring the sodium storage mechanism in disodium terephthalate as anode for organic battery using density-functional theory calculations. J. Power Sources 2016, 324, 572–581. [Google Scholar] [CrossRef]
- Koch, D.; Kulish, V.V.; Manzhos, S. A first-principles study of potassium insertion in crystalline vanadium oxide phases as possible potassium-ion battery cathode materials. MRS Commun. 2017, 1–7. [Google Scholar] [CrossRef]
- Koch, D.; Manzhos, S. Addition to “On the Charge State of Titanium in Titanium Dioxide”. J. Phys. Chem. Lett. 2017, 8, 3945–3946. [Google Scholar] [CrossRef] [PubMed]
- Koch, D.; Manzhos, S. On the Charge State of Titanium in Titanium Dioxide. J. Phys. Chem. Lett. 2017, 8, 1593–1598. [Google Scholar] [CrossRef] [PubMed]
- Kulish, V.V.; Koch, D.; Manzhos, S. Ab initio study of Li, Mg and Al insertion into rutile VO2: Fast diffusion and enhanced voltages for multivalent batteries. Phys. Chem. Chem. Phys. 2017, 19, 22538–22545. [Google Scholar] [CrossRef] [PubMed]
- Kulish, V.V.; Koch, D.; Manzhos, S. Aluminium and magnesium insertion in sulfur-based spinels: A first-principles study. Phys. Chem. Chem. Phys. 2017, 19, 6076–6081. [Google Scholar] [CrossRef] [PubMed]
- Kulish, V.V.; Manzhos, S. Comparison of Li, Na, Mg and Al-ion insertion in vanadium pentoxides and vanadium dioxides. RSC Adv. 2017, 7, 18643–18649. [Google Scholar] [CrossRef]
- Legrain, F.; Manzhos, S. A first-principles comparative study of lithium, sodium, and magnesium storage in pure and gallium-doped germanium: Competition between interstitial and substitutional sites. J. Chem. Phys. 2017, 146, 034706. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.-H.; Huang, T.-Y.; Shao, P.-S.; Chien, J.-H.; Weng, Y.-T.; Huang, H.-F.; Hwang, B.J.; Wu, N.-L. Experimental study on sodiation of amorphous silicon for use as sodium-ion battery anode. Electrochim. Acta 2016, 211, 265–272. [Google Scholar] [CrossRef]
- Zhang, M.; MacRae, A.C.; Liu, H.; Meng, Y.S. Communication—Investigation of Anatase-TiO2 as an Efficient Electrode Material for Magnesium-Ion Batteries. J. Electrochem. Soc. 2016, 163, A2368–A2370. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef]
- Aydinol, M.; Kohan, A.; Ceder, G.; Cho, K.; Joannopoulos, J. Ab initio study of lithium intercalation in metal oxides and metal dichalcogenides. Phys. Rev. B 1997, 56, 1354. [Google Scholar] [CrossRef]
- Morgan, D.; Van der Ven, A.; Ceder, G. Li conductivity in LixMPO4 (M = Mn, Fe, Co, Ni) olivine materials. Electrochem. Solid State Lett. 2004, 7, A30–A32. [Google Scholar] [CrossRef]
- Vineyard, G.H. Frequency factors and isotope effects in solid state rate processes. J. Phys. Chem. Solids 1957, 3, 121–127. [Google Scholar] [CrossRef]
- Park, M.; Zhang, X.; Chung, M.; Less, G.B.; Sastry, A.M. A review of conduction phenomena in Li-ion batteries. J. Power Sources 2010, 195, 7904–7929. [Google Scholar] [CrossRef]
- Lunell, S.; Stashans, A.; Ojamäe, L.; Lindström, H.; Hagfeldt, A. Li and Na Diffusion in TiO2 from Quantum Chemical Theory versus Electrochemical Experiment. J. Am. Chem. Soc. 1997, 119, 7374–7380. [Google Scholar] [CrossRef]
- Zhou, F.; Cococcioni, M.; Marianetti, C.A.; Morgan, D.; Ceder, G. First-principles prediction of redox potentials in transition-metal compounds with LDA + U. Phys. Rev. B 2004, 70, 235121. [Google Scholar] [CrossRef]
- Chan, C.K.; Peng, H.; Liu, G.; McIlwrath, K.; Zhang, X.F.; Huggins, R.A.; Cui, Y. High-performance lithium battery anodes using silicon nanowires. Nat. Nanotechnol. 2008, 3, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Park, C.-M.; Kim, J.-H.; Kim, H.; Sohn, H.-J. Li-alloy based anode materials for Li secondary batteries. Chem. Soc. Rev. 2010, 39, 3115–3141. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Adkins, E.R.; He, Y.; Zhong, L.; Luo, L.; Mao, S.X.; Wang, C.-M.; Korgel, B.A. Germanium as a Sodium Ion Battery Material: In Situ TEM Reveals Fast Sodiation Kinetics with High Capacity. Chem. Mater. 2016, 28, 1236–1242. [Google Scholar] [CrossRef]
- Li, Z.; Ding, J.; Mitlin, D. Tin and Tin Compounds for Sodium Ion Battery Anodes: Phase Transformations and Performance. Acc. Chem. Res. 2015, 48, 1657–1665. [Google Scholar] [CrossRef] [PubMed]
- Jung, I.-H.; Kim, J. Thermodynamic modeling of the Mg–Ge–Si, Mg–Ge–Sn, Mg–Pb–Si and Mg–Pb–Sn systems. J. Alloys Compd. 2010, 494, 137–147. [Google Scholar] [CrossRef]
- Shenoy, V.; Johari, P.; Qi, Y. Elastic softening of amorphous and crystalline Li–Si phases with increasing Li concentration: A first-principles study. J. Power Sources 2010, 195, 6825–6830. [Google Scholar] [CrossRef]
- Mortazavi, M.; Deng, J.; Shenoy, V.B.; Medhekar, N.V. Elastic softening of alloy negative electrodes for Na-ion batteries. J. Power Sources 2013, 225, 207–214. [Google Scholar] [CrossRef]
- Ge, P.; Fouletier, M. Electrochemical intercalation of sodium in graphite. Solid State Ion. 1988, 28, 1172–1175. [Google Scholar] [CrossRef]
- Li, Y.; Mu, L.; Hu, Y.-S.; Li, H.; Chen, L.; Huang, X. Pitch-derived amorphous carbon as high performance anode for sodium-ion batteries. Energy Storage Mater. 2016, 2, 139–145. [Google Scholar] [CrossRef]
- Li, Y.; Hu, Y.-S.; Li, H.; Chen, L.; Huang, X. A superior low-cost amorphous carbon anode made from pitch and lignin for sodium-ion batteries. J. Mater. Chem. A 2016, 4, 96–104. [Google Scholar] [CrossRef]
- Chan, T.-L.; Chelikowsky, J.R. Controlling diffusion of lithium in silicon nanostructures. Nano Lett. 2010, 10, 821–825. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kweon, K.E.; Chou, C.-Y.; Ekerdt, J.G.; Hwang, G.S. On the nature and behavior of Li atoms in Si: A first principles study. J. Phys. Chem. C 2010, 114, 17942–17946. [Google Scholar] [CrossRef]
- Wan, W.; Zhang, Q.; Cui, Y.; Wang, E. First principles study of lithium insertion in bulk silicon. J. Phys. Condens. Matter 2010, 22, 415501. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, W.; Wan, W.; Cui, Y.; Wang, E. Lithium insertion in silicon nanowires: An ab initio study. Nano Lett. 2010, 10, 3243–3249. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y. Density functional theory calculations of alkali metal (Li, Na, and K) graphite intercalation compounds. J. Phys. Chem. C 2013, 118, 16–19. [Google Scholar] [CrossRef]
- Wang, D.; Liu, L.-M.; Zhao, S.-J.; Li, B.-H.; Liu, H.; Lang, X.-F. β-MnO2 as a cathode material for lithium ion batteries from first principles calculations. Phys. Chem. Chem. Phys. 2013, 15, 9075–9083. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.C.; Jung, D.S.; Choi, J.W.; Han, Y.-K. Atom-level understanding of the sodiation process in silicon anode material. J. Phys. Chem. Lett. 2014, 5, 1283–1288. [Google Scholar] [CrossRef] [PubMed]
- Arrouvel, C.; Parker, S.C.; Islam, M.S. Lithium insertion and transport in the TiO2-B anode material: A computational study. Chem. Mater. 2009, 21, 4778–4783. [Google Scholar] [CrossRef]
- Panduwinata, D.; Gale, J.D. A first principles investigation of lithium intercalation in TiO2-B. J. Mater. Chem. 2009, 19, 3931–3940. [Google Scholar] [CrossRef]
- Tritsaris, G.A.; Zhao, K.; Okeke, O.U.; Kaxiras, E. Diffusion of Lithium in Bulk Amorphous Silicon: A Theoretical Study. J. Phys. Chem. C 2012, 116, 22212–22216. [Google Scholar] [CrossRef]
- Liu, X.H.; Zheng, H.; Zhong, L.; Huang, S.; Karki, K.; Zhang, L.Q.; Liu, Y.; Kushima, A.; Liang, W.T.; Wang, J.W. Anisotropic swelling and fracture of silicon nanowires during lithiation. Nano Lett. 2011, 11, 3312–3318. [Google Scholar] [CrossRef] [PubMed]
- McDowell, M.T.; Lee, S.W.; Harris, J.T.; Korgel, B.A.; Wang, C.; Nix, W.D.; Cui, Y. In situ TEM of two-phase lithiation of amorphous silicon nanospheres. Nano Lett. 2013, 13, 758–764. [Google Scholar] [CrossRef] [PubMed]
- Bucur, C.B.; Gregory, T.; Oliver, A.G.; Muldoon, J. Confession of a magnesium battery. J. Phys. Chem. Lett. 2015, 6, 3578–3591. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Sahadeo, E.; Noked, M.; Lee, S.B. Mapping the challenges of magnesium battery. J. Phys. Chem. Lett. 2016, 7, 1736–1749. [Google Scholar] [CrossRef] [PubMed]
- Stevens, D.; Dahn, J. The mechanisms of lithium and sodium insertion in carbon materials. J. Electrochem. Soc. 2001, 148, A803–A811. [Google Scholar] [CrossRef]
- Komaba, S.; Murata, W.; Ishikawa, T.; Yabuuchi, N.; Ozeki, T.; Nakayama, T.; Ogata, A.; Gotoh, K.; Fujiwara, K. Electrochemical Na insertion and solid electrolyte interphase for hard-carbon electrodes and application to Na-Ion batteries. Adv. Funct. Mater. 2011, 21, 3859–3867. [Google Scholar] [CrossRef]
- Jiang, X.; Århammar, C.; Liu, P.; Zhao, J.; Ahuja, R. The R3-carbon allotrope: A pathway towards glassy carbon under high pressure. Sci. Rep. 2013, 3, 1877. [Google Scholar] [CrossRef] [PubMed]
- Jian, Z.; Luo, W.; Ji, X. Carbon Electrodes for K-Ion Batteries. J. Am. Chem. Soc. 2015, 137, 11566–11569. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.-S.; Ren, W.; Wen, L.; Gao, L.; Zhao, J.; Chen, Z.; Zhou, G.; Li, F.; Cheng, H.-M. Graphene anchored with Co3O4 nanoparticles as anode of lithium ion batteries with enhanced reversible capacity and cyclic performance. ACS Nano 2010, 4, 3187–3194. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Cui, L.-F.; Yang, Y.; Sanchez Casalongue, H.; Robinson, J.T.; Liang, Y.; Cui, Y.; Dai, H. Mn3O4-Graphene Hybrid as a High-Capacity Anode Material for Lithium Ion Batteries. J. Am. Chem. Soc. 2010, 132, 13978–13980. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Smith, K.B.; Hayner, C.M.; Kung, H.H. Silicon nanoparticles–graphene paper composites for Li ion battery anodes. Chem. Commun. 2010, 46, 2025–2027. [Google Scholar] [CrossRef] [PubMed]
- Pollak, E.; Geng, B.; Jeon, K.-J.; Lucas, I.T.; Richardson, T.J.; Wang, F.; Kostecki, R. The interaction of Li+ with single-layer and few-layer graphene. Nano Lett. 2010, 10, 3386–3388. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Kutana, A.; Liu, Y.; Yakobson, B.I. First-principles studies of Li nucleation on graphene. J. Phys. Chem. Lett. 2014, 5, 1225–1229. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Zheng, W.; Kuo, J.-L.; Singh, D.J. Adsorption of single Li and the formation of small Li clusters on graphene for the anode of lithium-ion batteries. ACS Appl. Mater. Interfaces 2013, 5, 7793–7797. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Ren, Z.; Guo, P.; Fang, L.; Fan, J. Diffusion of Li+ ion on graphene: A DFT study. Appl. Surf. Sci. 2011, 258, 1651–1655. [Google Scholar] [CrossRef]
- Hardikar, R.P.; Das, D.; Han, S.S.; Lee, K.-R.; Singh, A.K. Boron doped defective graphene as a potential anode material for Li-ion batteries. Phys. Chem. Chem. Phys. 2014, 16, 16502–16508. [Google Scholar] [CrossRef] [PubMed]
- Malyi, O.I.; Sopiha, K.; Kulish, V.V.; Tan, T.L.; Manzhos, S.; Persson, C. A computational study of Na behavior on graphene. Appl. Surf. Sci. 2015, 333, 235–243. [Google Scholar] [CrossRef]
- Khantha, M.; Cordero, N.; Molina, L.; Alonso, J.; Girifalco, L. Interaction of lithium with graphene: An ab initio study. Phys. Rev. B 2004, 70, 125422. [Google Scholar] [CrossRef]
- Singh, N.; Arthur, T.S.; Ling, C.; Matsui, M.; Mizuno, F. A high energy-density tin anode for rechargeable magnesium-ion batteries. Chem. Commun. 2013, 49, 149–151. [Google Scholar] [CrossRef] [PubMed]
- Hirai, K.; Ichitsubo, T.; Uda, T.; Miyazaki, A.; Yagi, S.; Matsubara, E. Effects of volume strain due to Li–Sn compound formation on electrode potential in lithium-ion batteries. Acta Mater. 2008, 56, 1539–1545. [Google Scholar] [CrossRef]
- Im, H.S.; Cho, Y.J.; Lim, Y.R.; Jung, C.S.; Jang, D.M.; Park, J.; Shojaei, F.; Kang, H.S. Phase Evolution of Tin Nanocrystals in Lithium Ion Batteries. ACS Nano 2013, 7, 11103–11111. [Google Scholar] [CrossRef] [PubMed]
- Soler, J.M.; Artacho, E.; Gale, J.D.; García, A.; Junquera, J.; Ordejón, P.; Sánchez-Portal, D. The SIESTA method for ab initio order-N materials simulation. J. Phys. Condens. Matter 2002, 14, 2745. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Kittel, C. Introduction to Solid State Physics; Wiley: Hoboken, GA, USA, 2005. [Google Scholar]
- Blum, V.; Gehrke, R.; Hanke, F.; Havu, P.; Havu, V.; Ren, X.; Reuter, K.; Scheffler, M. Ab initio molecular simulations with numeric atom-centered orbitals. Comput. Phys. Commun. 2009, 180, 2175–2196. [Google Scholar] [CrossRef]
- Havu, V.; Blum, V.; Havu, P.; Scheffler, M. Efficient O(N) integration for all-electron electronic structure calculation using numeric basis functions. J. Comput. Phys. 2009, 228, 8367–8379. [Google Scholar] [CrossRef]
- The Elk FP-LAPW Code. Available online: http://elk.sourceforge.net/ (accessed on 1 November 2017).
- Gani, T.Z.; Kulik, H.J. Where does the density localize? Convergent behavior for global hybrids, range separation, and DFT + U. J. Chem. Theory Comput. 2016, 12, 5931–5945. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.-Y.; Lee, M.; Hwang, G.S. A Comparative First-Principles Study on Sodiation of Silicon, Germanium, and Tin for Sodium-Ion Batteries. J. Phys. Chem. C 2015, 119, 14843–14850. [Google Scholar] [CrossRef]
- Moutanabbir, O.; Isheim, D.; Blumtritt, H.; Senz, S.; Pippel, E.; Seidman, D.N. Colossal injection of catalyst atoms into silicon nanowires. Nature 2013, 496, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Deng, D.; Kim, M.G.; Lee, J.Y.; Cho, J. Green energy storage materials: Nanostructured TiO2 and Sn-based anodes for lithium-ion batteries. Energy Environ. Sci. 2009, 2, 818–837. [Google Scholar] [CrossRef]
- Huang, J.P.; Yuan, D.D.; Zhang, H.Z.; Cao, Y.L.; Li, G.R.; Yang, H.X.; Gao, X.P. Electrochemical sodium storage of TiO2(B) nanotubes for sodium ion batteries. RSC Adv. 2013, 3, 12593–12597. [Google Scholar] [CrossRef]
- Frank, A.J.; Kopidakis, N.; van de Lagemaat, J. Electrons in nanostructured TiO2 solar cells: Transport, recombination and photovoltaic properties. Coord. Chem. Rev. 2004, 248, 1165–1179. [Google Scholar] [CrossRef]
- Liu, J.; Wang, J.; Ku, Z.; Wang, H.; Chen, S.; Zhang, L.; Lin, J.; Shen, Z.X. Aqueous Rechargeable Alkaline CoxNi2−xS2/TiO2 Battery. ACS Nano 2016, 10, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Nakata, K.; Fujishima, A. TiO2 photocatalysis: Design and applications. J. Photochem. Photobiol. C Photochem. Rev. 2012, 13, 169–189. [Google Scholar] [CrossRef]
- Oh, S.-M.; Hwang, J.-Y.; Yoon, C.S.; Lu, J.; Amine, K.; Belharouak, I.; Sun, Y.-K. High Electrochemical Performances of Microsphere C-TiO2 Anode for Sodium-Ion Battery. ACS Appl. Mater. Interfaces 2014, 6, 11295–11301. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.; Matsuoka, M.; Takeuchi, M.; Zhang, J.; Horiuchi, Y.; Anpo, M.; Bahnemann, D.W. Understanding TiO2 Photocatalysis: Mechanisms and Materials. Chem. Rev. 2014, 114, 9919–9986. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, V.; Karki, A.; Gnanasekar, K.I.; Eddy, F.P.; Rambabu, B. Nanocrystalline TiO2 (anatase) for Li-ion batteries. J. Power Sources 2006, 159, 186–192. [Google Scholar] [CrossRef]
- Usui, H.; Domi, Y.; Yoshioka, S.; Kojima, K.; Sakaguchi, H. Electrochemical Lithiation and Sodiation of Nb-Doped Rutile TiO2. ACS Sustain. Chem. Eng. 2016, 4, 6695–6702. [Google Scholar] [CrossRef]
- Wang, B.; Zhao, F.; Du, G.; Porter, S.; Liu, Y.; Zhang, P.; Cheng, Z.; Liu, H.K.; Huang, Z. Boron-Doped Anatase TiO2 as a High-Performance Anode Material for Sodium-Ion Batteries. ACS Appl. Mater. Interfaces 2016, 8, 16009–16015. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, C.; Yang, Y.; Zhang, Y.; Jia, X.; Chen, J.; Ji, X. Anatase TiO2 nanocubes for fast and durable sodium ion battery anodes. J. Mater. Chem. A 2015, 3, 8800–8807. [Google Scholar] [CrossRef]
- Zhang, Y.; Pu, X.; Yang, Y.; Zhu, Y.; Hou, H.; Jing, M.; Yang, X.; Chen, J.; Ji, X. An electrochemical investigation of rutile TiO2 microspheres anchored by nanoneedle clusters for sodium storage. Phys. Chem. Chem. Phys. 2015, 17, 15764–15770. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Liu, L.; Ji, G.; Yang, Q.; Zheng, L.; Zhang, J. Hydrogenated Anatase TiO2 as Lithium-Ion Battery Anode: Size–Reactivity Correlation. ACS Appl. Mater. Interfaces 2016, 8, 20074–20081. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Huang, Z.; NuLi, Y.; Tuerxun, F.; Yang, J.; Wang, J. A novel rechargeable battery with a magnesium anode, a titanium dioxide cathode, and a magnesium borohydride/tetraglyme electrolyte. Chem. Commun. 2015, 51, 2641–2644. [Google Scholar] [CrossRef] [PubMed]
- Morgan, B.J.; Watson, G.W. GGA + U description of lithium intercalation into anatase TiO2. Phys. Rev. B 2010, 82, 144119. [Google Scholar] [CrossRef]
- Morgan, B.J.; Madden, P.A. Lithium intercalation into TiO2(B): A comparison of LDA, GGA, and GGA + U density functional calculations. Phys. Rev. B 2012, 86, 035147. [Google Scholar] [CrossRef]
- Raebiger, H.; Lany, S.; Zunger, A. Charge self-regulation upon changing the oxidation state of transition metals in insulators. Nature 2008, 453, 763–766. [Google Scholar] [CrossRef] [PubMed]
- Wolczanski, P.T. Flipping the Oxidation State Formalism: Charge Distribution in Organometallic Complexes as Reported by Carbon Monoxide. Organometallics 2017, 36, 622–631. [Google Scholar] [CrossRef]
- West, K.; Zachau-Christiansen, B.; Østergård, M.; Jacobsen, T. Vanadium oxides as electrode materials for rechargeable lithium cells. J. Power Sources 1987, 20, 165–172. [Google Scholar] [CrossRef]
- Murphy, D.; Christian, P.; DiSalvo, F.; Waszczak, J. Lithium incorporation by vanadium pentoxide. Inorg. Chem. 1979, 18, 2800–2803. [Google Scholar] [CrossRef]
- Pereira-Ramos, J.; Messina, R.; Piolet, C.; Devynck, J. A thermodynamic study of electrochemical lithium insertion into vanadium pentoxide. Electrochim. Acta 1988, 33, 1003–1008. [Google Scholar] [CrossRef]
- Lee, S.; Sun, X.-G.; Lubimtsev, A.A.; Gao, X.; Ganesh, P.; Ward, T.Z.; Eres, G.; Chisholm, M.F.; Dai, S.; Lee, H.N. Persistent electrochemical performance in epitaxial VO2(B). Nano Lett. 2017, 17, 2229–2233. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Li, X.; Bai, Z.; Lin, L.; Chen, G.; Song, X.; Xiong, D.; Li, D.; Sun, X. Superior sodium storage of novel VO2 nano-microspheres encapsulated into crumpled reduced graphene oxide. J. Mater. Chem. A 2017, 5, 4850–4860. [Google Scholar] [CrossRef]
- Pei, C.; Xiong, F.; Sheng, J.; Yin, Y.; Tan, S.; Wang, D.; Han, C.; An, Q.; Mai, L. VO2 Nanoflakes as the Cathode Material of Hybrid Magnesium-Lithium-Ion Batteries with High Energy Density. ACS Appl. Mater. Interfaces 2017, 9, 17060–17066. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Lee, C.W.; Kim, J.-C.; Song, H.J.; Shim, H.-W.; Lee, S.; Kim, D.-W. Heteroepitaxy-Induced Rutile VO2 with Abundantly Exposed (002) Facets for High Lithium Electroactivity. ACS Energy Lett. 2016, 1, 216–224. [Google Scholar] [CrossRef]
- Moretti, A.; Maroni, F.; Nobili, F.; Passerini, S. V2O5 electrodes with extended cycling ability and improved rate performance using polyacrylic acid as binder. J. Power Sources 2015, 293, 1068–1072. [Google Scholar] [CrossRef]
- Chiku, M.; Takeda, H.; Matsumura, S.; Higuchi, E.; Inoue, H. Amorphous Vanadium Oxide/Carbon Composite Positive Electrode for Rechargeable Aluminum Battery. ACS Appl. Mater. Interfaces 2015, 7, 24385–24389. [Google Scholar] [CrossRef] [PubMed]
- Raju, V.; Rains, J.; Gates, C.; Luo, W.; Wang, X.; Stickle, W.F.; Stucky, G.D.; Ji, X. Superior Cathode of Sodium-Ion Batteries: Orthorhombic V2O5 Nanoparticles Generated in Nanoporous Carbon by Ambient Hydrolysis Deposition. Nano Lett. 2014, 14, 4119–4124. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Gong, Y.; Liu, Z.; Zhan, L.; Hashim, D.P.; Ma, L.; Vajtai, R.; Ajayan, P.M. Bottom-up approach toward single-crystalline VO2-graphene ribbons as cathodes for ultrafast lithium storage. Nano Lett. 2013, 13, 1596–1601. [Google Scholar] [CrossRef] [PubMed]
- Sasidharan, M.; Gunawardhana, N.; Yoshio, M.; Nakashima, K. V2O5 hollow nanospheres: A lithium intercalation host with good rate capability and capacity retention. J. Electrochem. Soc. 2012, 159, A618–A621. [Google Scholar] [CrossRef]
- Shklover, V.; Haibach, T.; Ried, F.; Nesper, R.; Novak, P. Crystal Structure of the Product of Mg2+ Insertion into V2O5 Single Crystals. J. Solid State Chem. 1996, 123, 317–323. [Google Scholar] [CrossRef]
- Jain, A.; Ong, S.P.; Hautier, G.; Chen, W.; Richards, W.D.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G. Commentary: The Materials Project: A materials genome approach to accelerating materials innovation. APL Mater. 2013, 1, 011002. [Google Scholar] [CrossRef]
- Morin, F. Oxides which show a metal-to-insulator transition at the Neel temperature. Phys. Rev. Lett. 1959, 3, 34. [Google Scholar] [CrossRef]
- Yoon, H.; Choi, M.; Lim, T.-W.; Kwon, H.; Ihm, K.; Kim, J.K.; Choi, S.-Y.; Son, J. Reversible phase modulation and hydrogen storage in multivalent VO2 epitaxial thin films. Nat. Mater. 2016, 15, 1113–1119. [Google Scholar] [CrossRef] [PubMed]
- Alivio, T.E.; Sellers, D.G.; Asayesh-Ardakani, H.; Braham, E.J.; Horrocks, G.A.; Pelcher, K.E.; Villareal, R.; Zuin, L.; Shamberger, P.J.; Arroyave, R. A Post-Synthetic Route for Modifying the Metal—Insulator Transition of VO2 by Interstitial Dopant Incorporation. Chem. Mater. 2017, 29, 5401–5412. [Google Scholar] [CrossRef]
- Tang, Y.; Rui, X.; Zhang, Y.; Lim, T.M.; Dong, Z.; Hng, H.H.; Chen, X.; Yan, Q.; Chen, Z. Vanadium pentoxide cathode materials for high-performance lithium-ion batteries enabled by a hierarchical nanoflower structure via an electrochemical process. J. Mater. Chem. A 2013, 1, 82–88. [Google Scholar] [CrossRef]
- Tepavcevic, S.; Xiong, H.; Stamenkovic, V.R.; Zuo, X.; Balasubramanian, M.; Prakapenka, V.B.; Johnson, C.S.; Rajh, T. Nanostructured bilayered vanadium oxide electrodes for rechargeable sodium-ion batteries. ACS Nano 2011, 6, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Shi, H.; Cao, R.; Zhang, X.; Jiang, Z. Theoretical study on the initial stage of a magnesium battery based on a V2O5 cathode. Phys. Chem. Chem. Phys. 2014, 16, 18578–18585. [Google Scholar] [CrossRef] [PubMed]
- Delmas, C.; Cognac-Auradou, H.; Cocciantelli, J.; Menetrier, M.; Doumerc, J. The LixV2O5 system: An overview of the structure modifications induced by the lithium intercalation. Solid State Ion. 1994, 69, 257–264. [Google Scholar] [CrossRef]
- Shao, J.; Li, X.; Wan, Z.; Zhang, L.; Ding, Y.; Zhang, L.; Qu, Q.; Zheng, H. Low-cost synthesis of hierarchical V2O5 microspheres as high-performance cathode for lithium-ion batteries. ACS Appl. Mater. Interfaces 2013, 5, 7671–7675. [Google Scholar] [CrossRef] [PubMed]
- An, Q.; Li, Y.; Yoo, H.D.; Chen, S.; Ru, Q.; Mai, L.; Yao, Y. Graphene decorated vanadium oxide nanowire aerogel for long-cycle-life magnesium battery cathodes. Nano Energy 2015, 18, 265–272. [Google Scholar] [CrossRef]
- Chan, C.K.; Peng, H.; Twesten, R.D.; Jarausch, K.; Zhang, X.F.; Cui, Y. Fast, completely reversible Li insertion in vanadium pentoxide nanoribbons. Nano Lett. 2007, 7, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Smolinski, H.; Gros, C.; Weber, W.; Peuchert, U.; Roth, G.; Weiden, M.; Geibel, C. NaV2O5 as a quarter-filled ladder compound. Phys. Rev. Lett. 1998, 80, 5164. [Google Scholar] [CrossRef]
- Scanlon, D.O.; Walsh, A.; Morgan, B.J.; Watson, G.W. An ab initio Study of Reduction of V2O5 through the Formation of Oxygen Vacancies and Li Intercalation. J. Phys. Chem. C 2008, 112, 9903–9911. [Google Scholar] [CrossRef]
- Sun, X.; Bonnick, P.; Duffort, V.; Liu, M.; Rong, Z.; Persson, K.A.; Ceder, G.; Nazar, L.F. A high capacity thiospinel cathode for Mg batteries. Energy Environ. Sci. 2016, 9, 2273–2277. [Google Scholar] [CrossRef]
- Zhang, R.; Yu, X.; Nam, K.-W.; Ling, C.; Arthur, T.S.; Song, W.; Knapp, A.M.; Ehrlich, S.N.; Yang, X.-Q.; Matsui, M. α-MnO2 as a cathode material for rechargeable Mg batteries. Electrochem. Commun. 2012, 23, 110–113. [Google Scholar] [CrossRef]
- Miao, X.; Chen, Z.; Wang, N.; Nuli, Y.; Wang, J.; Yang, J.; Hirano, S.-I. Electrospun V2MoO8 as a cathode material for rechargeable batteries with Mg metal anode. Nano Energy 2017, 34, 26–35. [Google Scholar] [CrossRef]
- Hu, Y.; Luo, B.; Ye, D.; Zhu, X.; Lyu, M.; Wang, L. An Innovative Freeze-Dried Reduced Graphene Oxide Supported SnS2 Cathode Active Material for Aluminum-Ion Batteries. Adv. Mater. 2017. [Google Scholar] [CrossRef]
- Wang, S.; Jiao, S.; Wang, J.; Chen, H.-S.; Tian, D.; Lei, H.; Fang, D.-N. High-Performance Aluminum-Ion Battery with CuS@C Microsphere Composite Cathode. ACS Nano 2016, 11, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, Y.; Du, S.; Gao, H.-J.; Zhang, S. Organic salts as super-high rate capability materials for lithium-ion batteries. Appl. Phys. Lett. 2012, 100, 091905. [Google Scholar] [CrossRef]
- Sun, X.; Bonnick, P.; Nazar, L.F. Layered TiS2 positive electrode for Mg batteries. ACS Energy Lett. 2016, 1, 297–301. [Google Scholar] [CrossRef]
- Liu, M.; Jain, A.; Rong, Z.; Qu, X.; Canepa, P.; Malik, R.; Ceder, G.; Persson, K.A. Evaluation of sulfur spinel compounds for multivalent battery cathode applications. Energy Environ. Sci. 2016, 9, 3201–3209. [Google Scholar] [CrossRef]
- Liu, M.; Rong, Z.; Malik, R.; Canepa, P.; Jain, A.; Ceder, G.; Persson, K.A. Spinel compounds as multivalent battery cathodes: A systematic evaluation based on ab initio calculations. Energy Environ. Sci. 2015, 8, 964–974. [Google Scholar] [CrossRef]
- Wang, Q.; Yu, H.-T.; Xie, Y.; Li, M.-X.; Yi, T.-F.; Guo, C.-F.; Song, Q.-S.; Lou, M.; Fan, S.-S. Structural stabilities, surface morphologies and electronic properties of spinel LiTi2O4 as anode materials for lithium-ion battery: A first-principles investigation. J. Power Sources 2016, 319, 185–194. [Google Scholar] [CrossRef]
- Ling, C.; Mizuno, F. Phase stability of post-spinel compound AMn2O4 (A = Li, Na, or Mg) and its application as a rechargeable battery cathode. Chem. Mater. 2013, 25, 3062–3071. [Google Scholar] [CrossRef]
- Song, T.; Xia, J.; Lee, J.-H.; Lee, D.H.; Kwon, M.-S.; Choi, J.-M.; Wu, J.; Doo, S.K.; Chang, H.; Park, W.I. Arrays of sealed silicon nanotubes as anodes for lithium ion batteries. Nano Lett. 2010, 10, 1710–1716. [Google Scholar] [CrossRef] [PubMed]
- Park, M.-H.; Kim, M.G.; Joo, J.; Kim, K.; Kim, J.; Ahn, S.; Cui, Y.; Cho, J. Silicon nanotube battery anodes. Nano Lett. 2009, 9, 3844–3847. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.; Yuan, H.; Wang, Y.; Cao, J.; Wang, Y. Mg intercalation properties into open-ended vanadium oxide nanotubes. Electrochem. Commun. 2005, 7, 431–436. [Google Scholar] [CrossRef]
- Liu, S.; Jia, H.; Han, L.; Wang, J.; Gao, P.; Xu, D.; Yang, J.; Che, S. Nanosheet-constructed porous TiO2-B for advanced lithium ion batteries. Adv. Mater. 2012, 24, 3201–3204. [Google Scholar] [CrossRef] [PubMed]
- Kulish, V.V.; Ng, M.F.; Malyi, O.I.; Wu, P.; Chen, Z. Enhanced Li Adsorption and Diffusion in Single-Walled Silicon Nanotubes: An ab Initio Study. ChemPhysChem 2013, 14, 1161–1167. [Google Scholar] [CrossRef] [PubMed]
- Seh, Z.W.; Li, W.; Cha, J.J.; Zheng, G.; Yang, Y.; McDowell, M.T.; Hsu, P.-C.; Cui, Y. Sulphur-TiO2 yolk-shell nanoarchitecture with internal void space for long-cycle lithium-sulphur batteries. Nat. Commun. 2013, 4, 1331. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Slater, M.D.; Balasubramanian, M.; Johnson, C.S.; Rajh, T. Amorphous TiO2 nanotube anode for rechargeable sodium ion batteries. J. Phys. Chem. Lett. 2011, 2, 2560–2565. [Google Scholar] [CrossRef]
- Chao, D.; Zhu, C.; Yang, P.; Xia, X.; Liu, J.; Wang, J.; Fan, X.; Savilov, S.V.; Lin, J.; Fan, H.J. Array of nanosheets render ultrafast and high-capacity Na-ion storage by tunable pseudocapacitance. Nat. Commun. 2016, 7, 12122. [Google Scholar] [CrossRef] [PubMed]
- Kulish, V.V.; Malyi, O.I.; Ng, M.-F.; Wu, P.; Chen, Z. Enhanced Li adsorption and diffusion in silicon nanosheets based on first principles calculations. RSC Adv. 2013, 3, 4231–4236. [Google Scholar] [CrossRef]
- Hu, Z.; Liu, Q.; Chou, S.L.; Dou, S.X. Advances and Challenges in Metal Sulfides/Selenides for Next-Generation Rechargeable Sodium-Ion Batteries. Adv. Mater. 2017. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Dall’Agnese, Y.; Naguib, M.; Gogotsi, Y.; Barsoum, M.W.; Zhuang, H.L.; Kent, P.R. Prediction and characterization of MXene nanosheet anodes for non-lithium-ion batteries. ACS Nano 2014, 8, 9606–9615. [Google Scholar] [CrossRef] [PubMed]













© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulish, V.V.; Koch, D.; Manzhos, S. Insertion of Mono- vs. Bi- vs. Trivalent Atoms in Prospective Active Electrode Materials for Electrochemical Batteries: An ab Initio Perspective. Energies 2017, 10, 2061. https://doi.org/10.3390/en10122061
Kulish VV, Koch D, Manzhos S. Insertion of Mono- vs. Bi- vs. Trivalent Atoms in Prospective Active Electrode Materials for Electrochemical Batteries: An ab Initio Perspective. Energies. 2017; 10(12):2061. https://doi.org/10.3390/en10122061
Chicago/Turabian StyleKulish, Vadym V., Daniel Koch, and Sergei Manzhos. 2017. "Insertion of Mono- vs. Bi- vs. Trivalent Atoms in Prospective Active Electrode Materials for Electrochemical Batteries: An ab Initio Perspective" Energies 10, no. 12: 2061. https://doi.org/10.3390/en10122061
APA StyleKulish, V. V., Koch, D., & Manzhos, S. (2017). Insertion of Mono- vs. Bi- vs. Trivalent Atoms in Prospective Active Electrode Materials for Electrochemical Batteries: An ab Initio Perspective. Energies, 10(12), 2061. https://doi.org/10.3390/en10122061