
The Synthesis and Preclinical Investigation of Lactosamine-Based Radiopharmaceuticals for the Detection of Galectin-3-Expressing Melanoma Cells
 , and
, and
Abstract
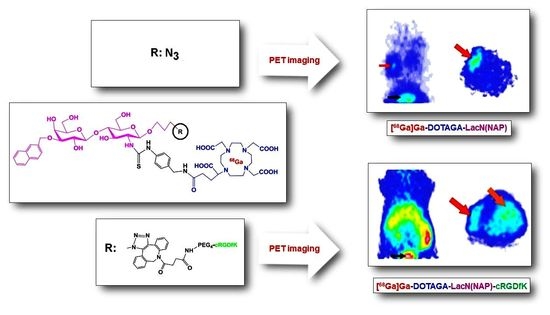
:
1. Introduction
2. Materials and Methods
2.1. General
2.2. Chemistry
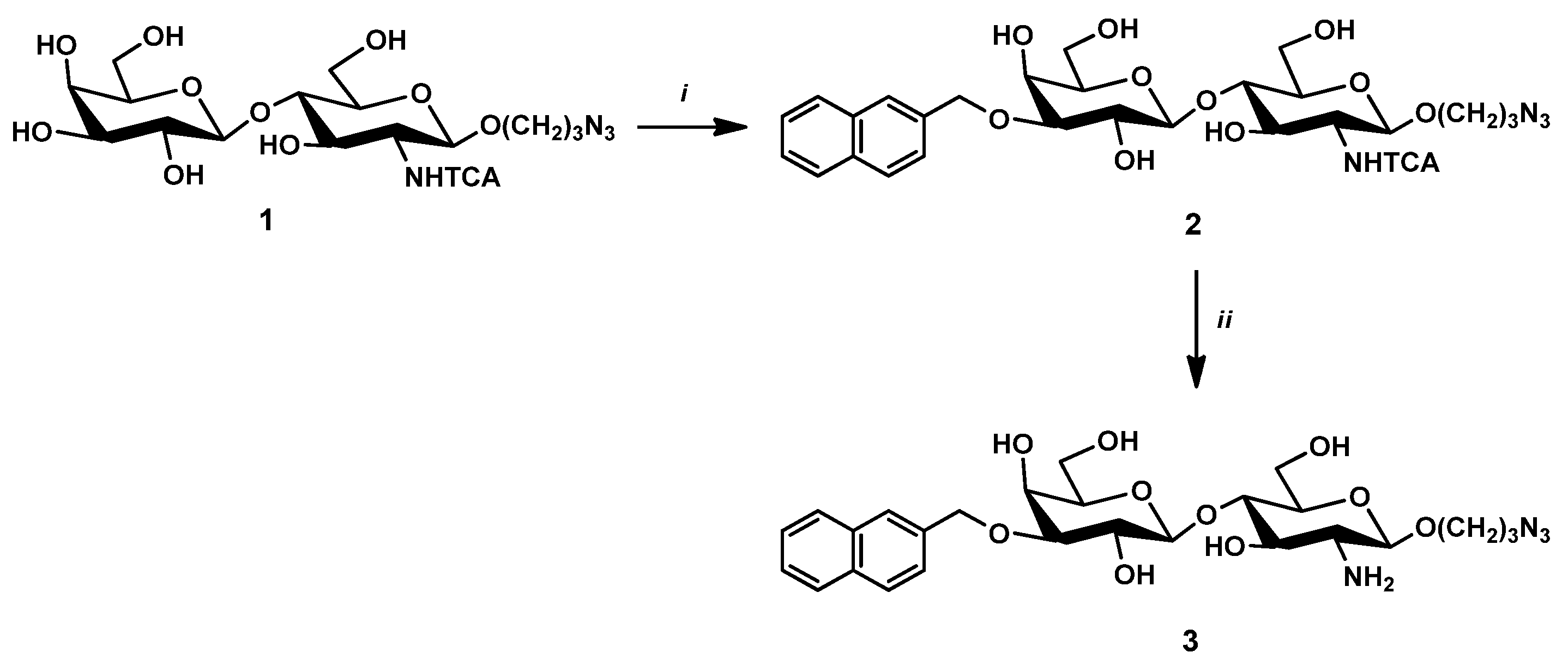
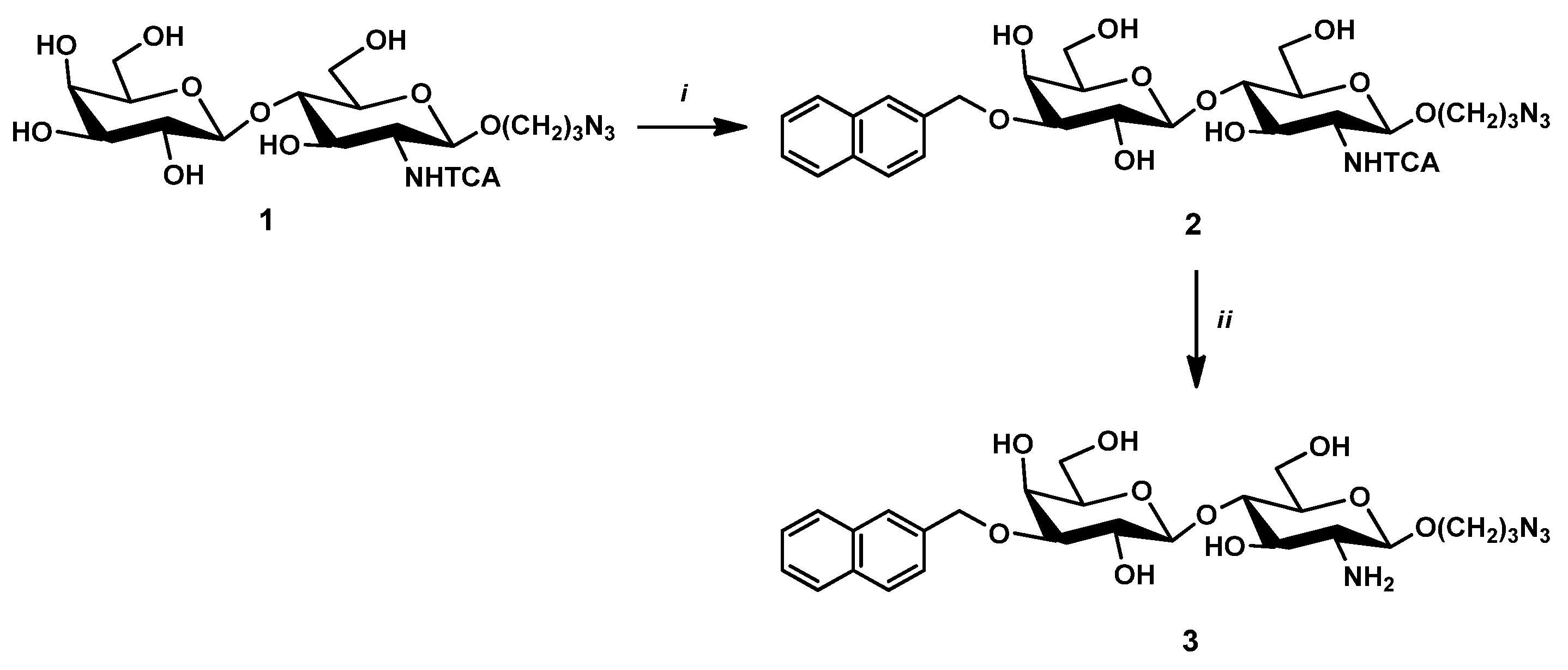
2.2.1. 3-Azidopropyl-(3-O-(2-naphtyl)methyl-β-D-galactopyranosyl)-(1→4)-(2-N-trichloroacetyl-2-deoxy-β-D-glucopyranoside) (2)
2.2.2. 3-Azidopropyl-(3-O-(2-naphtyl)methyl-β-D-galactopyranosyl)-(1→4)-(2-amino-2-deoxy-β-D-glucopyranoside) (3)
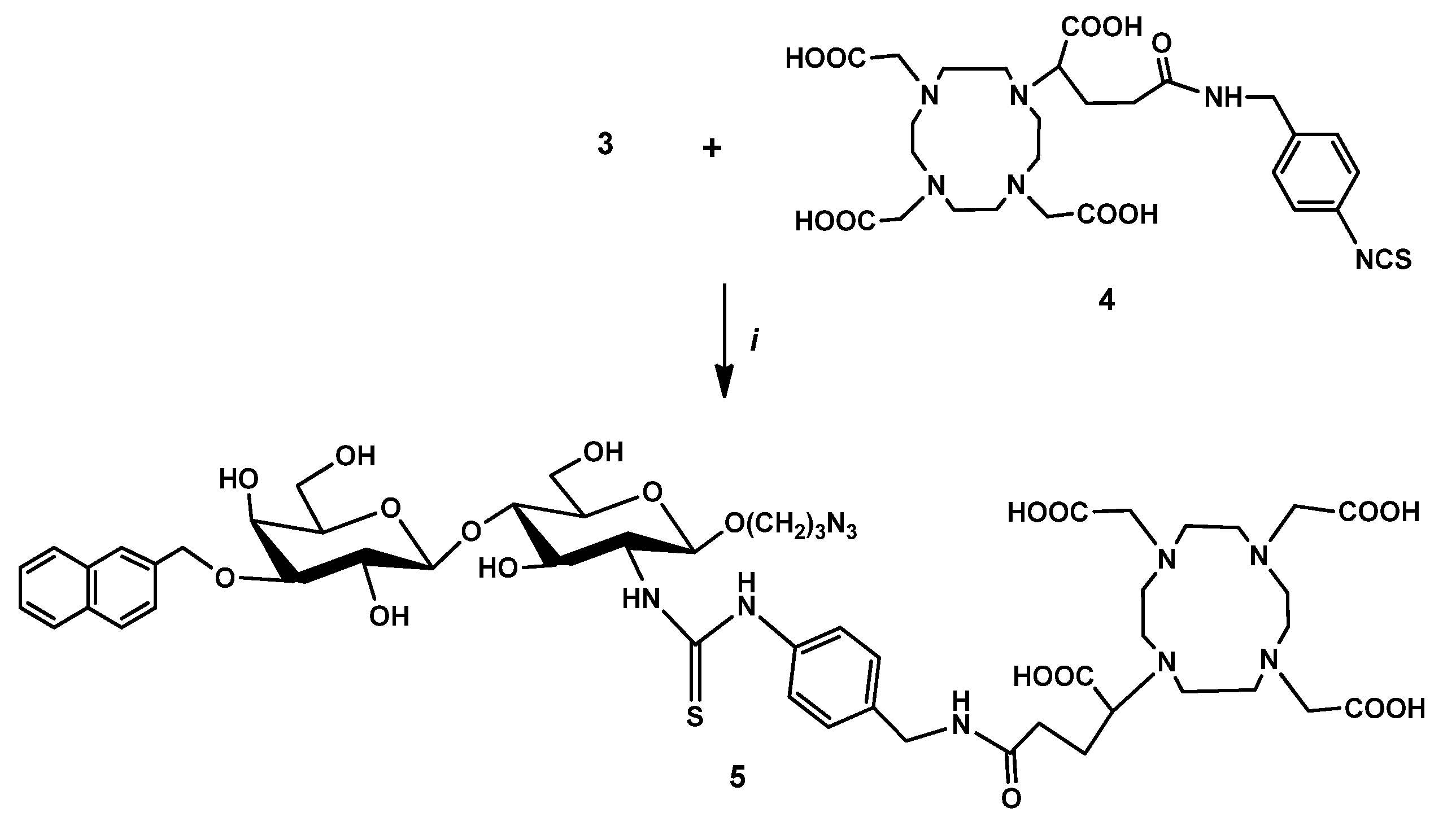
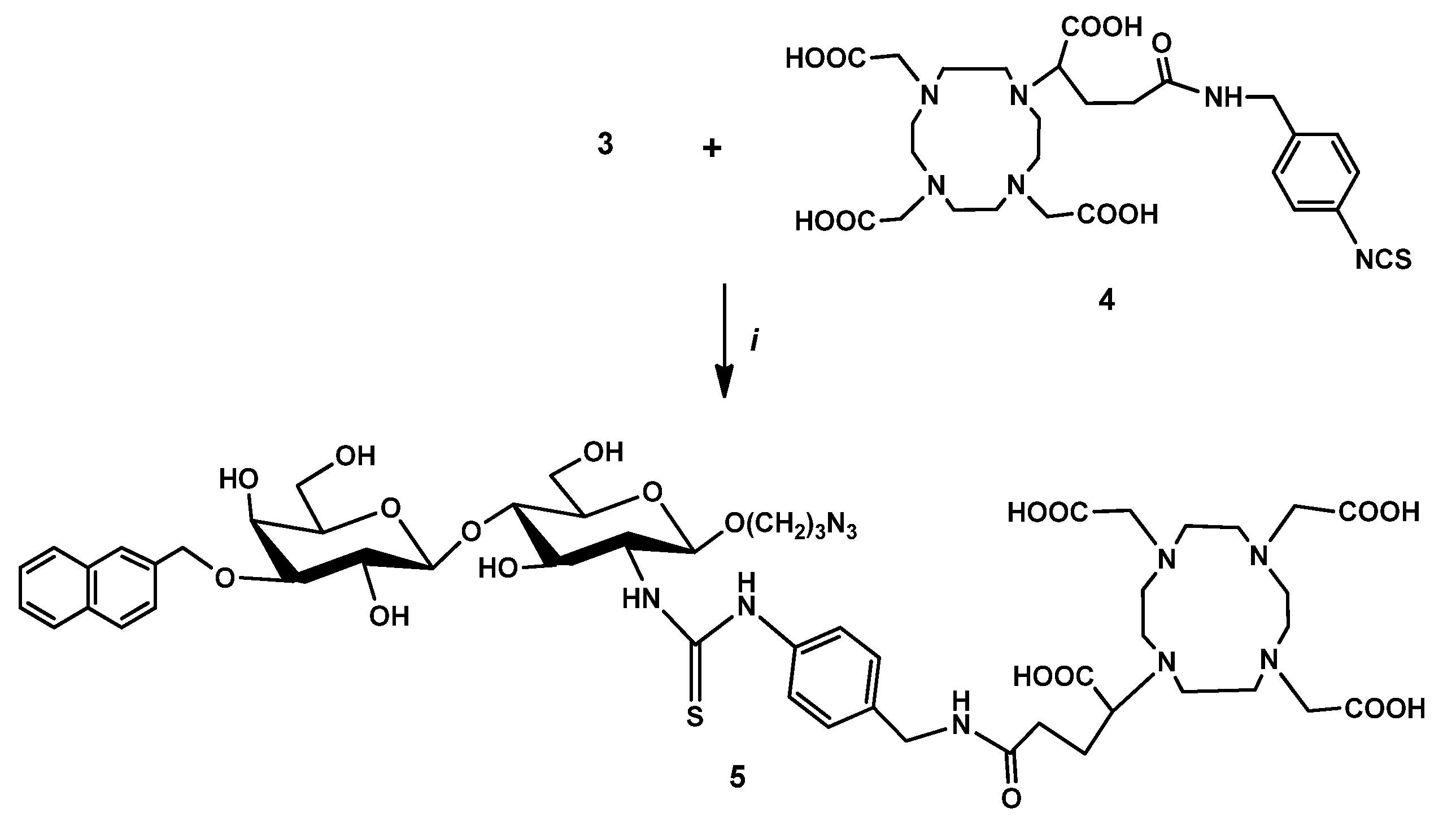
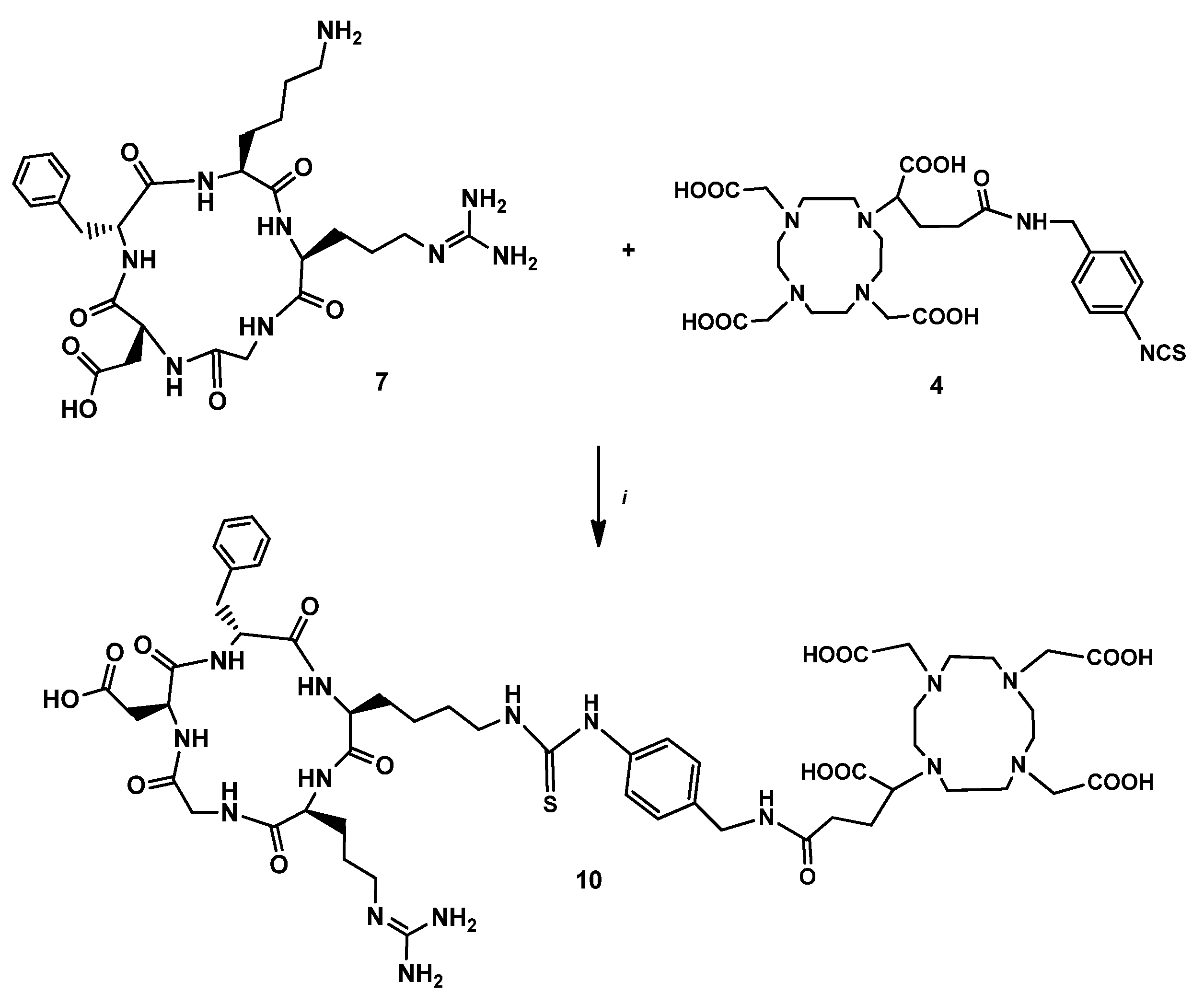
2.2.3. DOTAGA-LacN(NAP) (4)
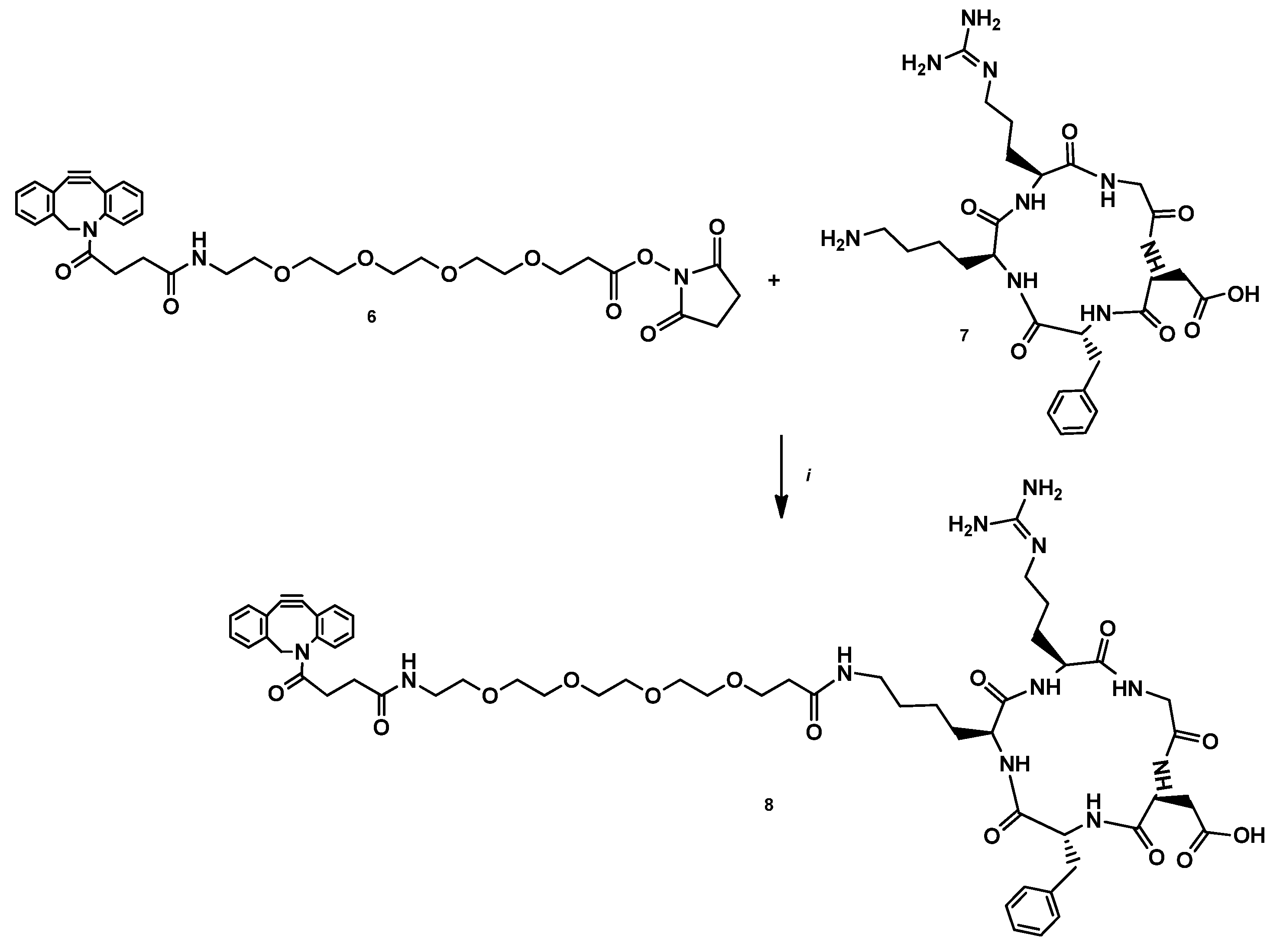
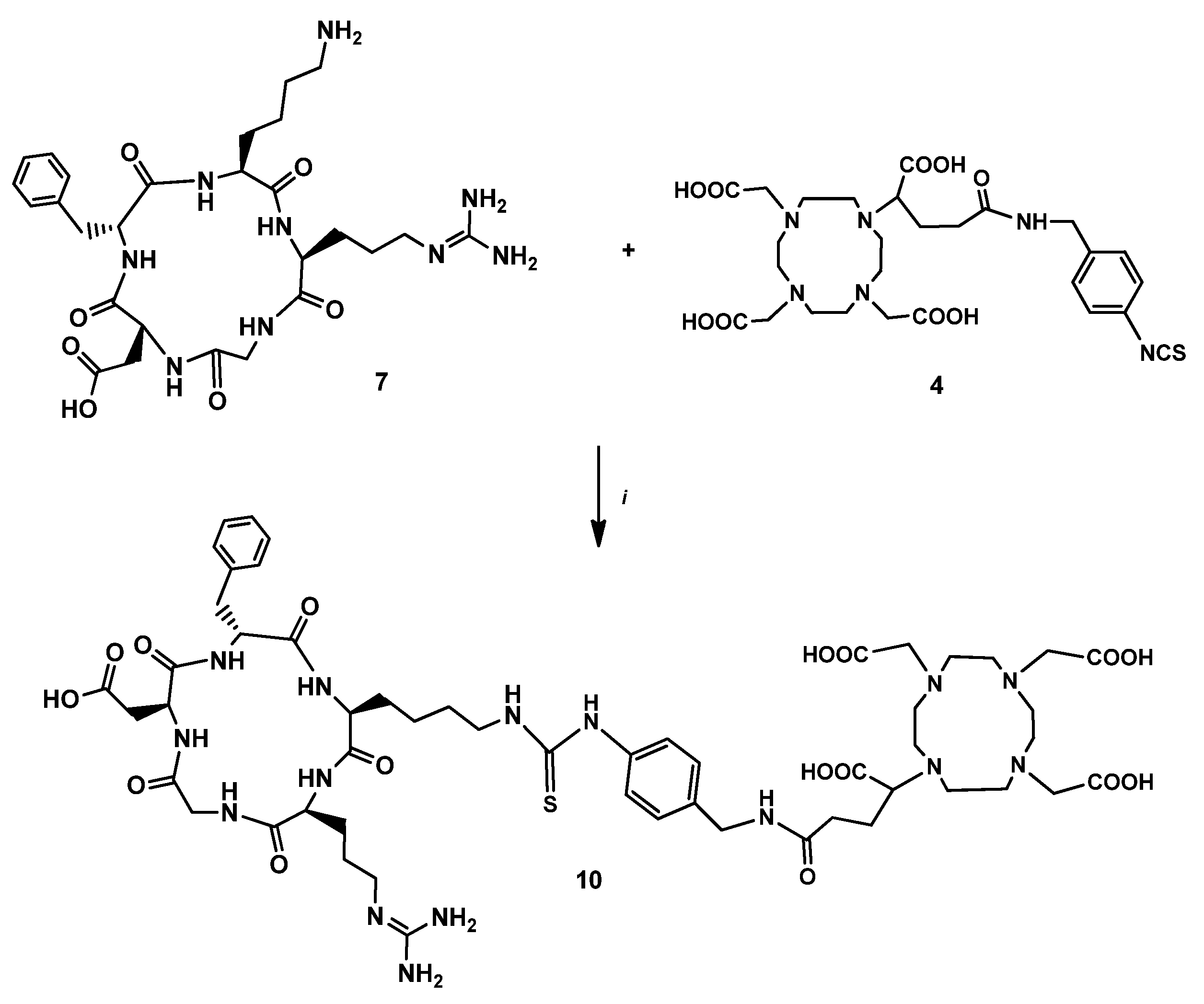
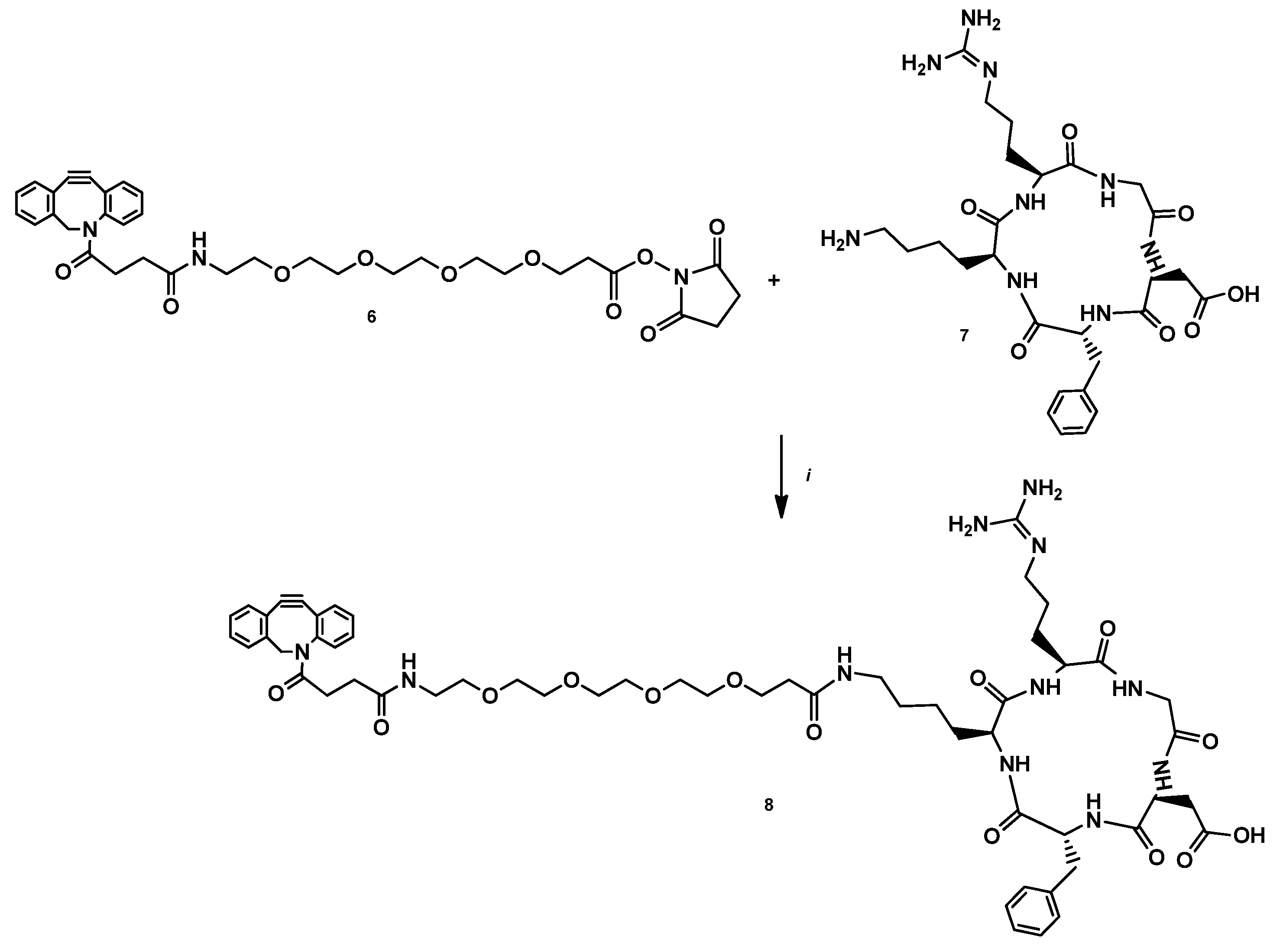
2.2.4. DBCO-PEG4-cRGDfK (7)
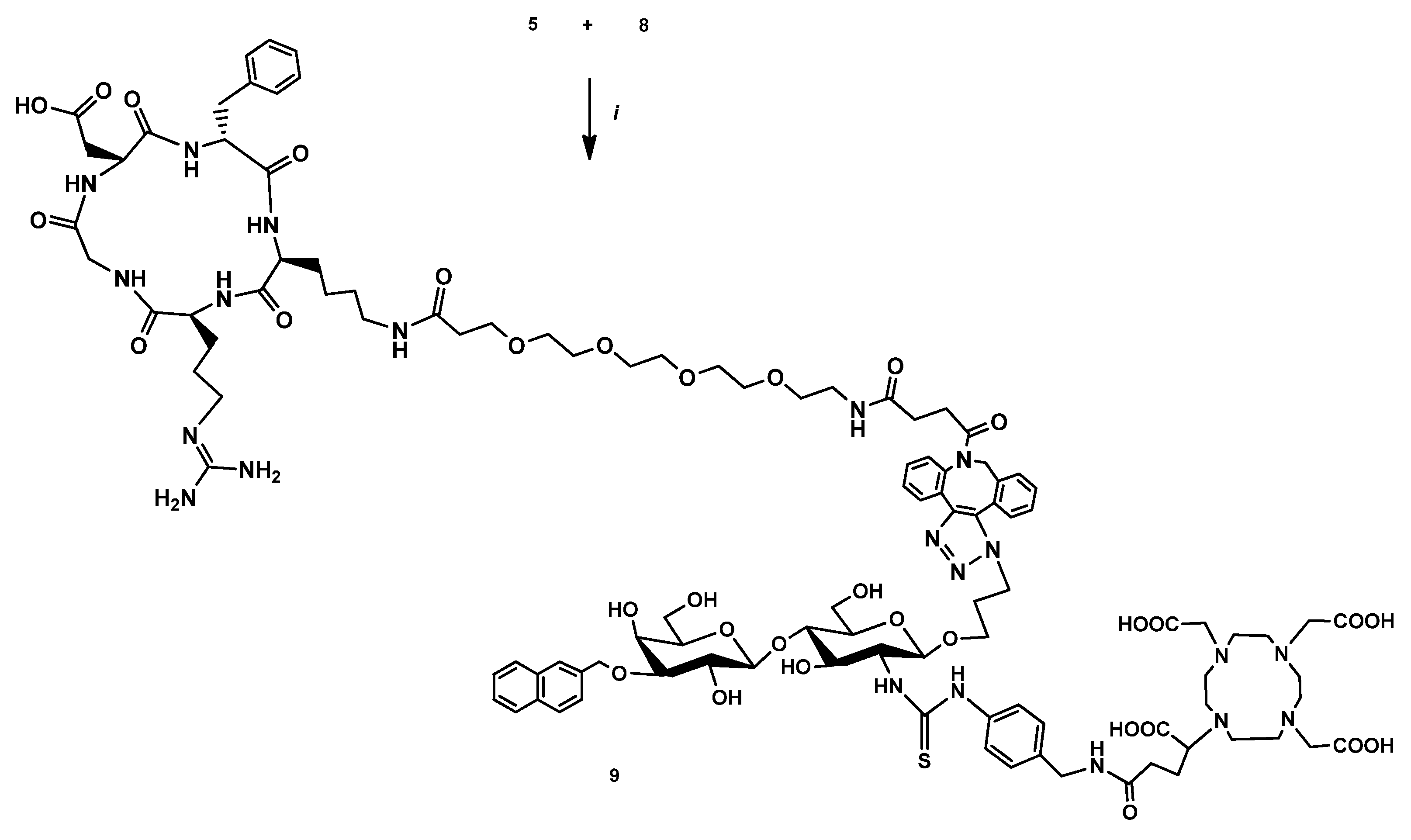
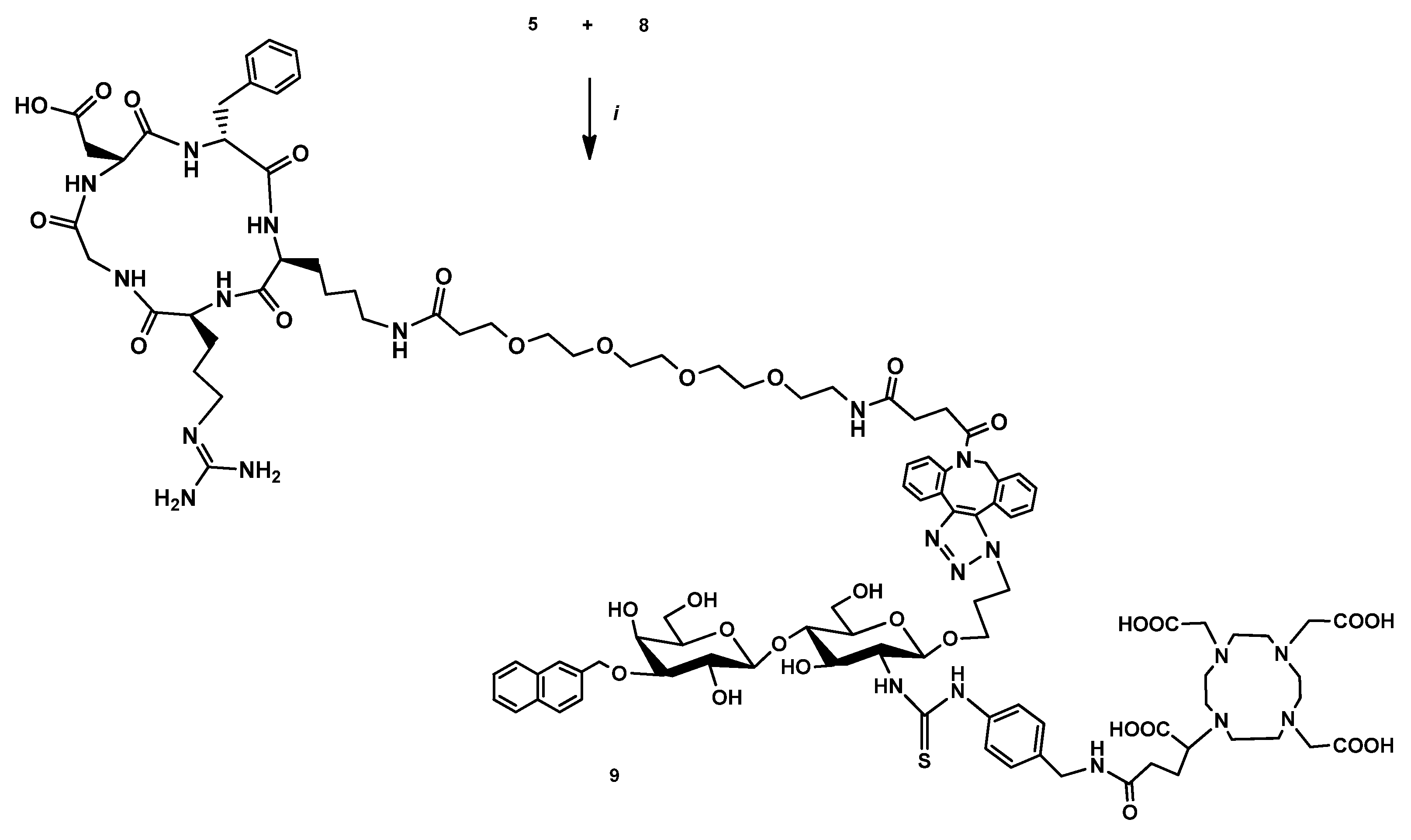
2.2.5. DOTAGA-LacN(NAP)-cRGDfK (8)
2.2.6. DOTAGA-cRGDfK (9)
2.3. Radiochemistry
2.3.1. 68Ga labeling of DOTAGA-LacN(NAP), DOTAGA-LacN(NAP)-cRGDfK, and DOTAGA-cRGDfK
2.3.2. Determination of log P Value of [68Ga]Ga-DOTAGA-LacN(NAP), [68Ga]Ga-DOTAGA-LacN(NAP)-cRGDfK, and [68Ga]Ga-DOTAGA-cRGDfK
2.3.3. Determination of In Vitro Stability of [68Ga]Ga-DOTAGA-LacN(NAP), [68Ga]Ga-DOTAGA-LacN(NAP)-cRGDfK, and [68Ga]Ga-DOTAGA-cRGDfK
2.4. Biology
2.4.1. Cell Culturing
2.4.2. Animal Housing
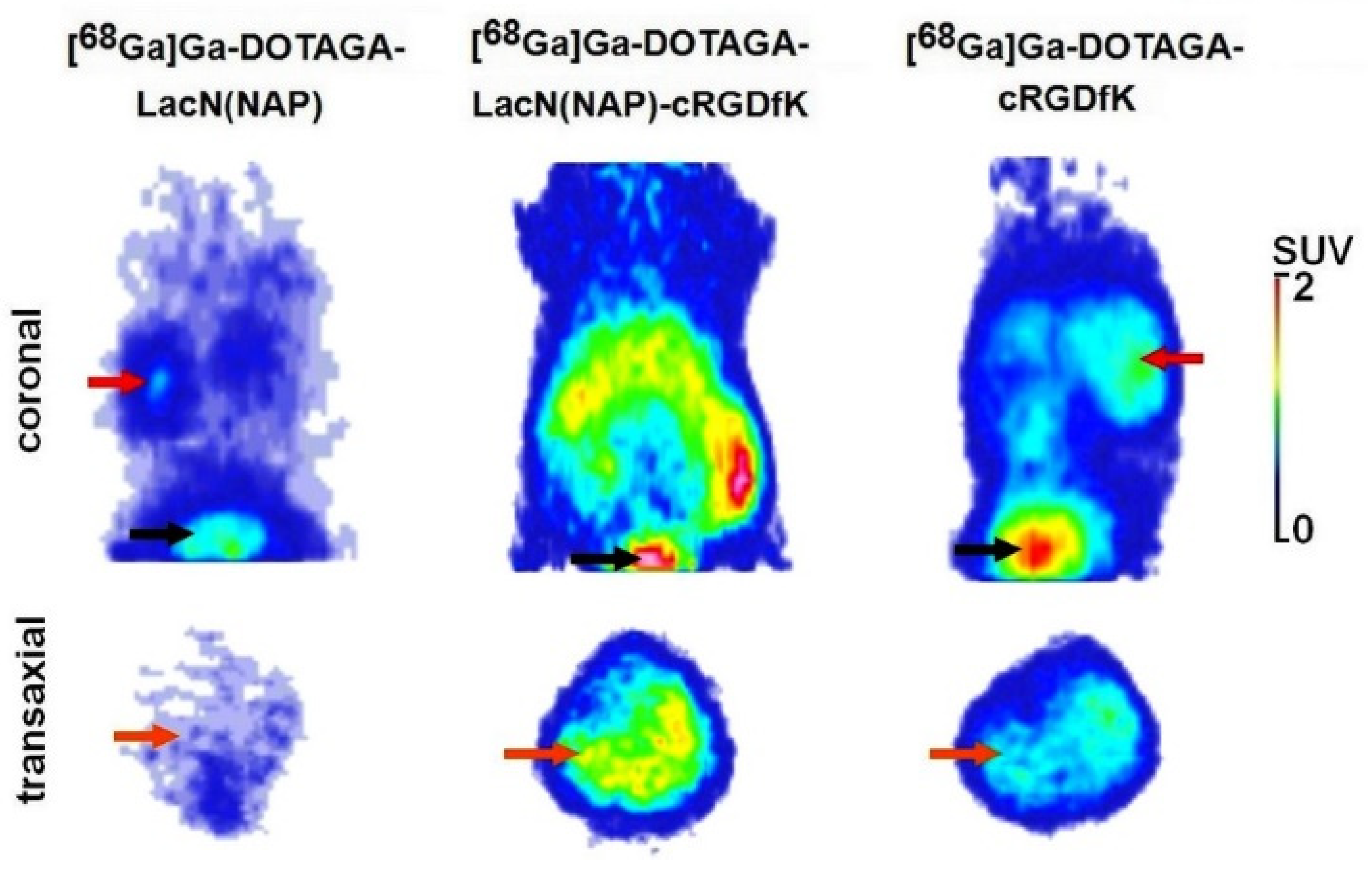
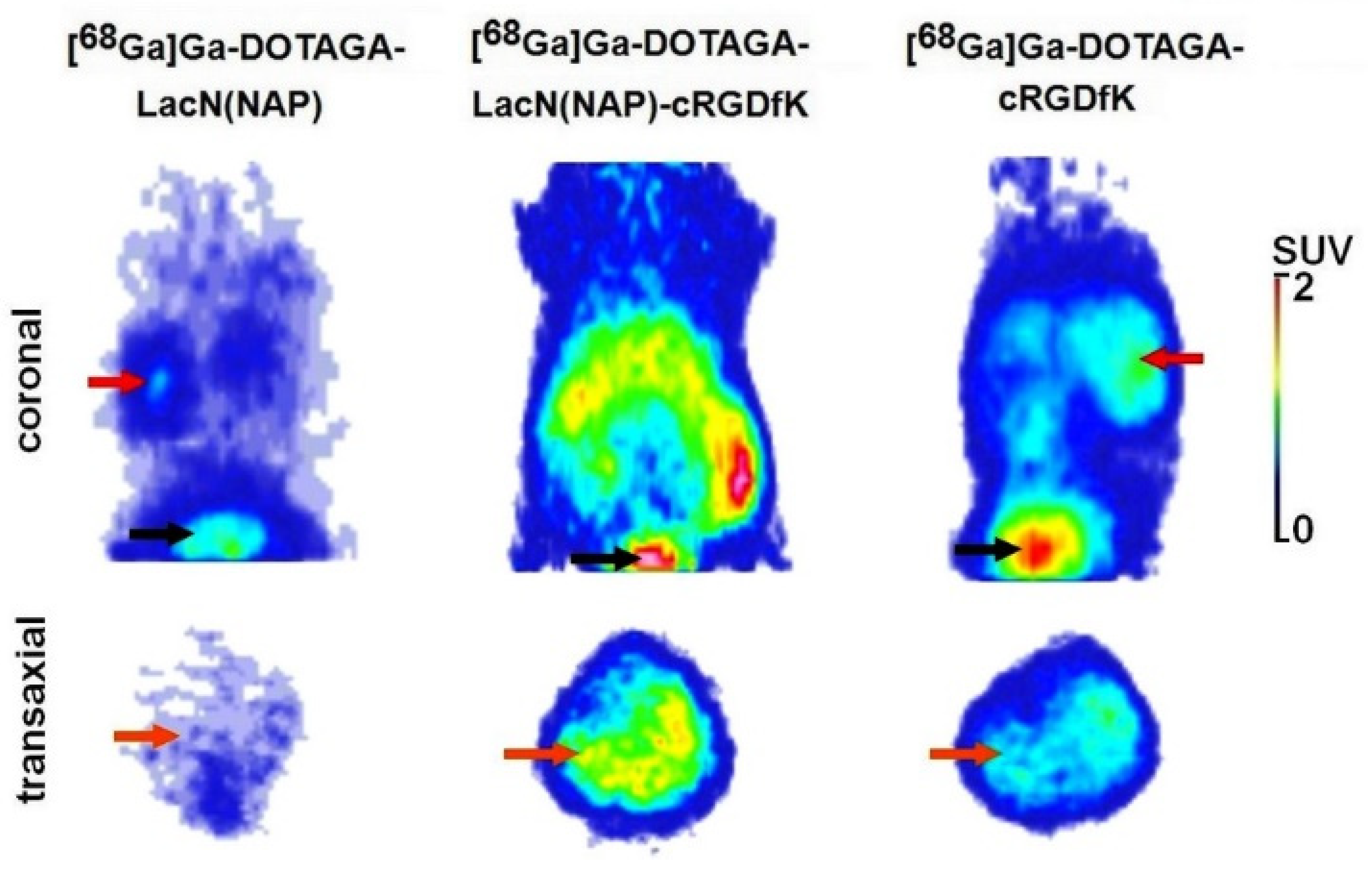
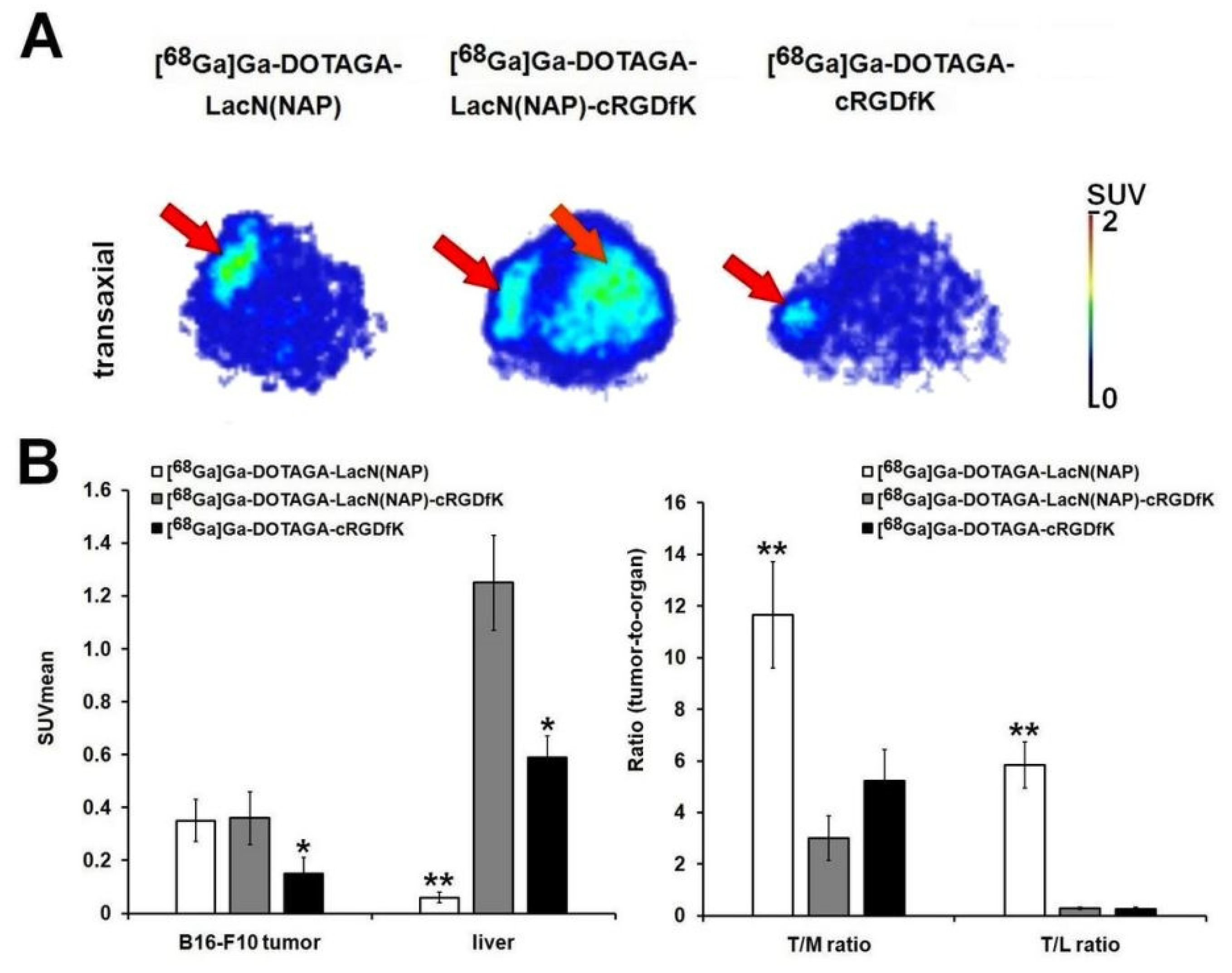
2.4.3. In Vivo PET Imaging and Image Analysis
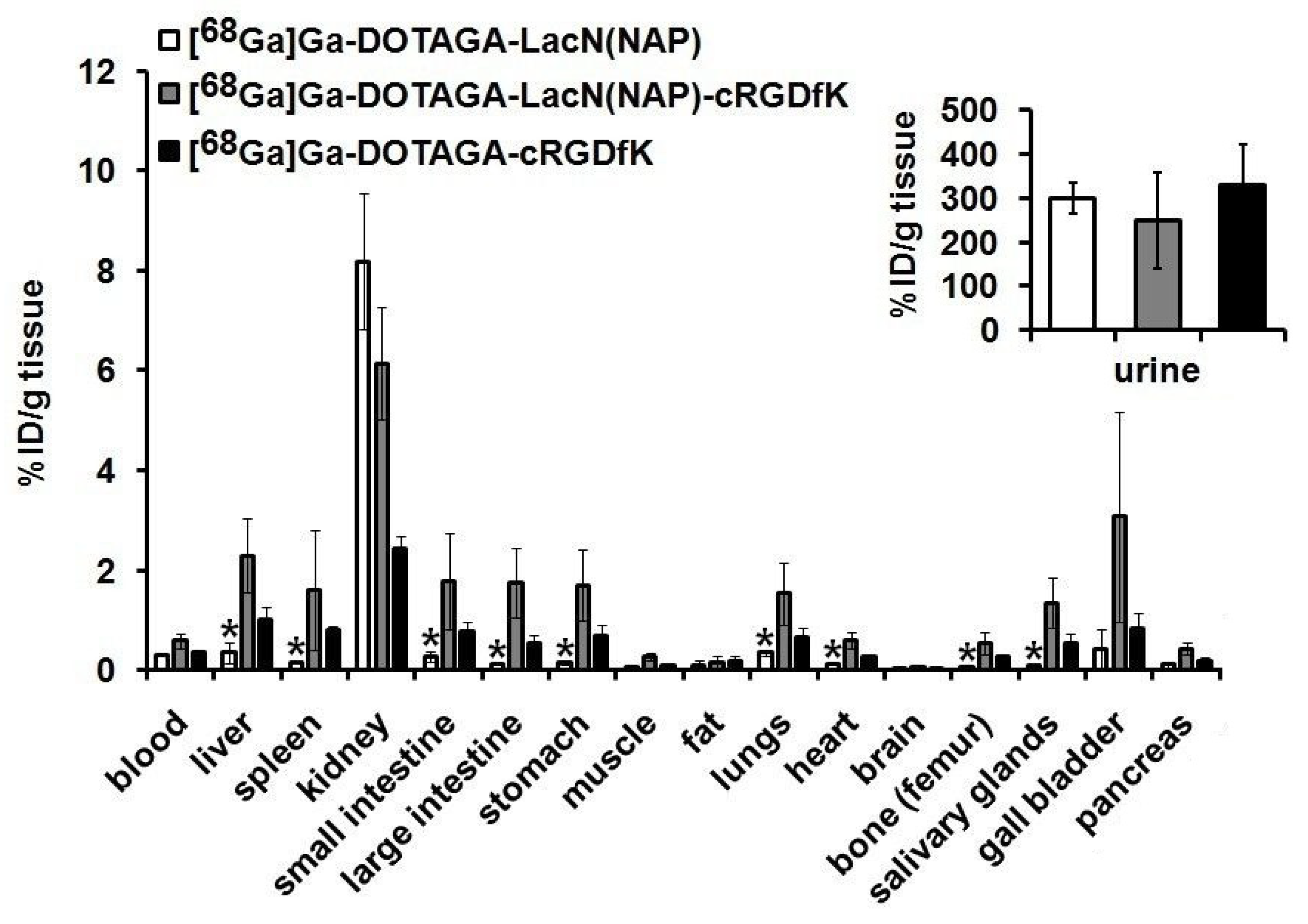
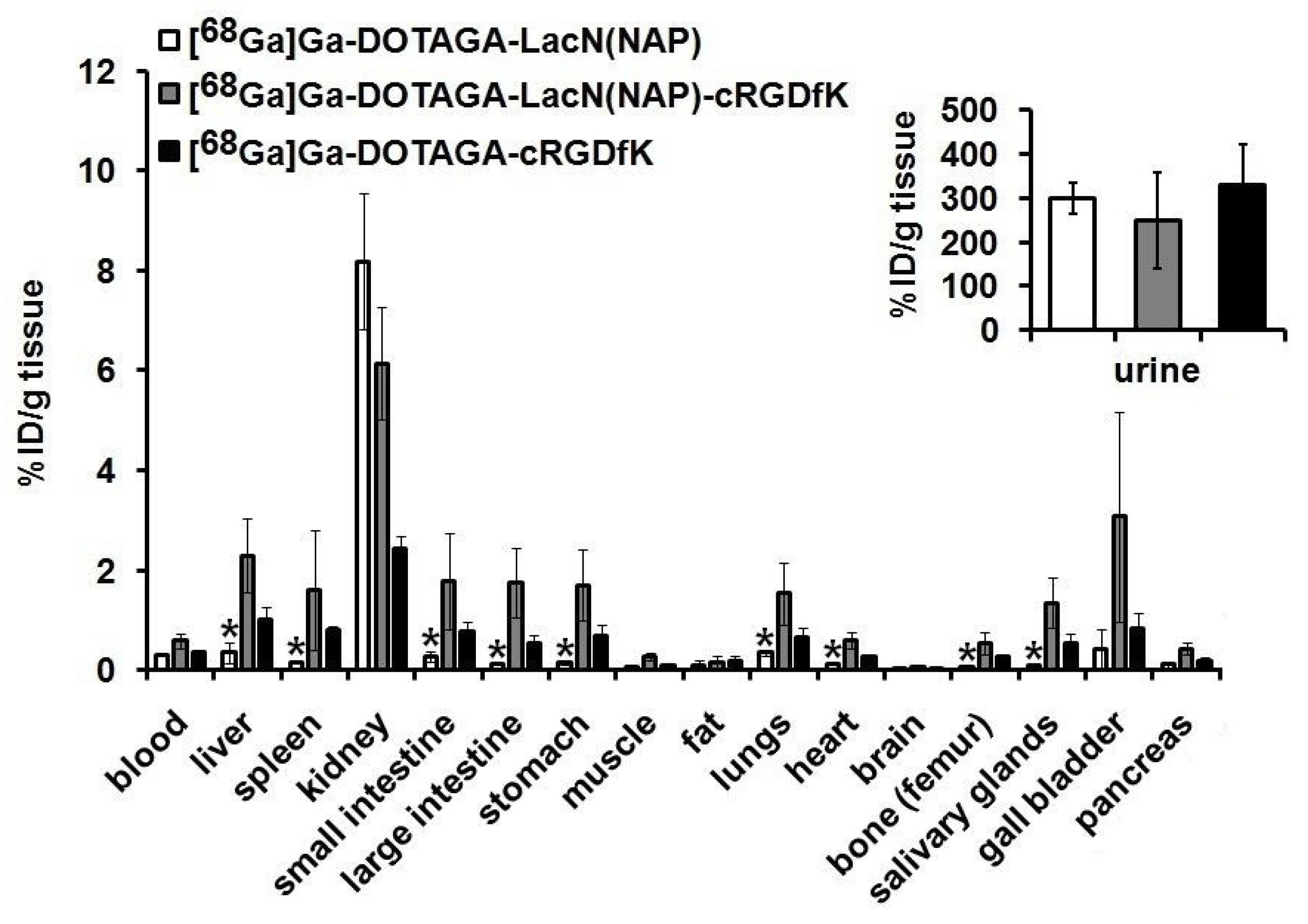
2.4.4. Ex Vivo Biodistribution Studies
2.4.5. Statistical Analysis
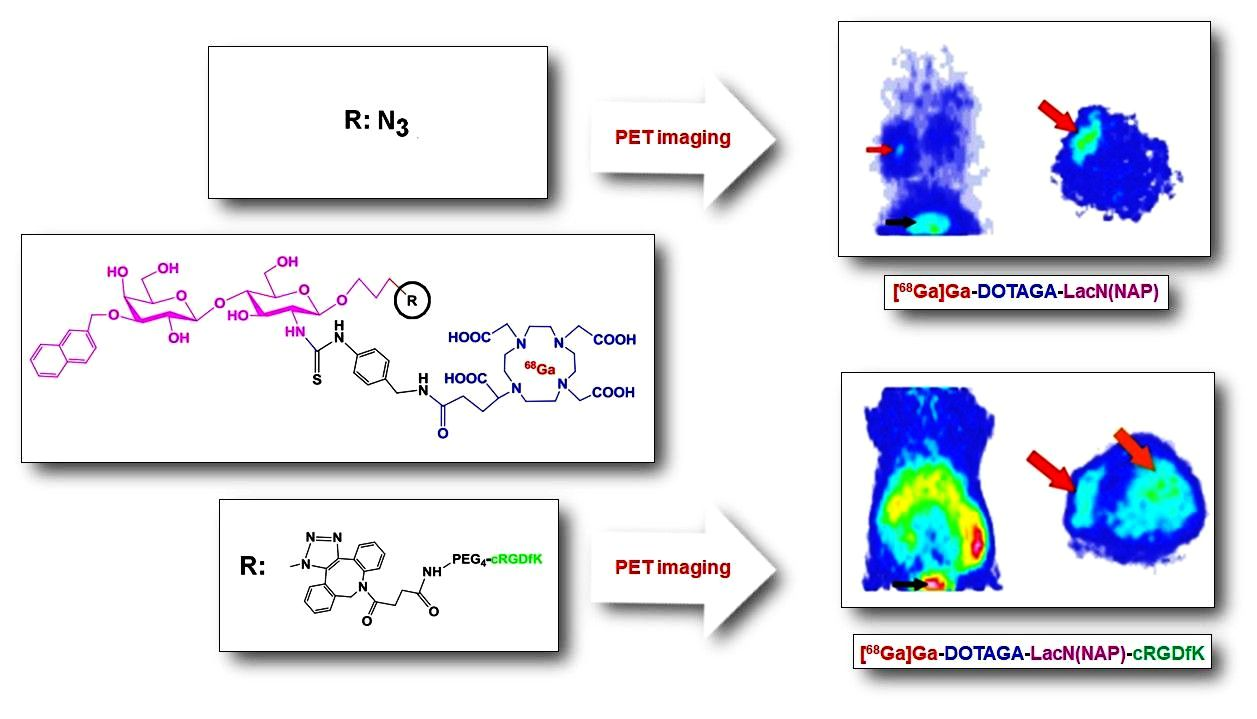
3. Results and Discussion
3.1. Chemistry
3.2. Radiochemistry
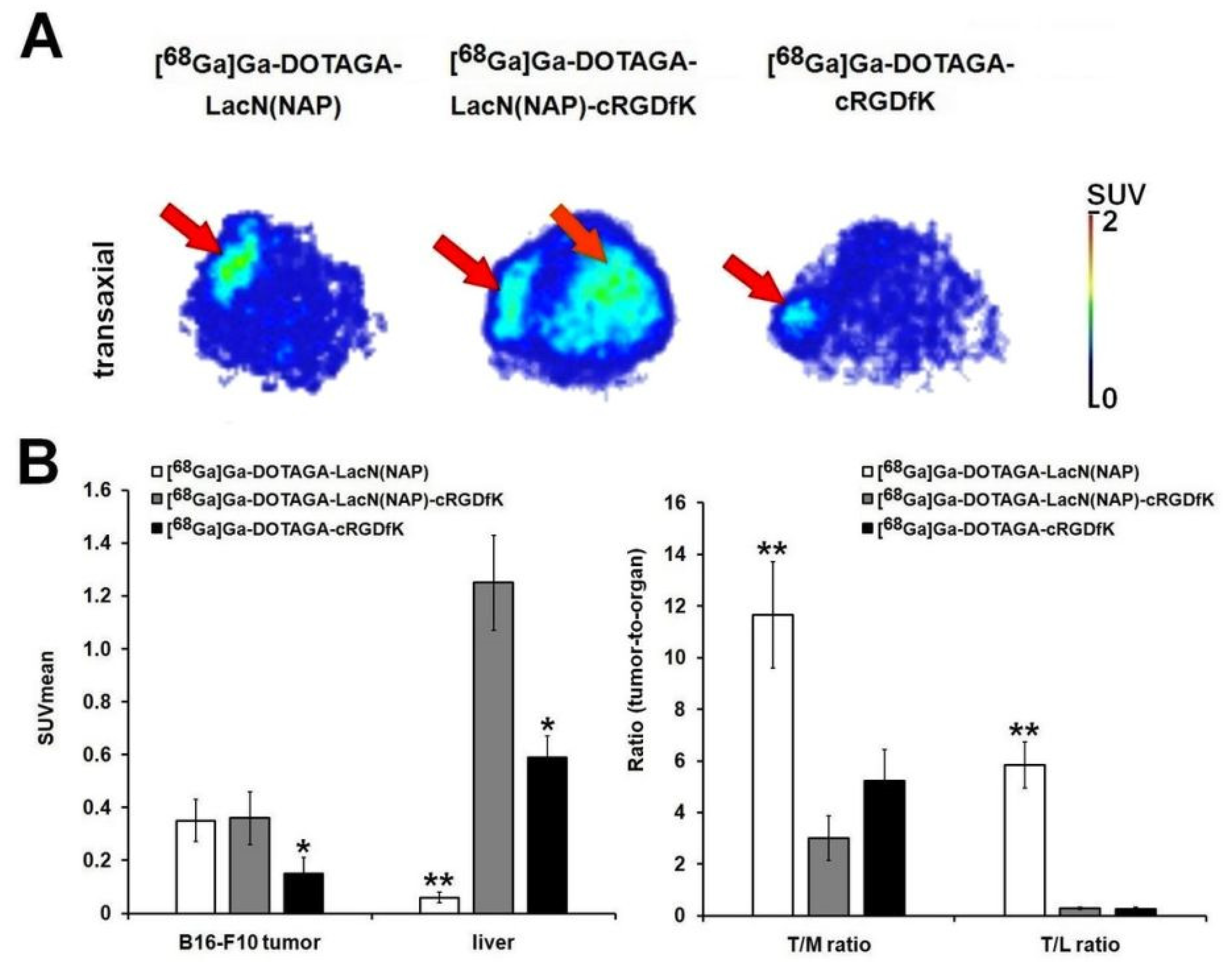
3.3. Biology
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rahman, W.T.; Wale, D.J.; Viglianti, B.L.; Townsend, D.M.; Manganaro, M.S.; Gross, M.D.; Wong, K.K.; Rubello, D. The impact of infection and inflammation in oncologic 18F-FDG PET/CT imaging. Biomed. Pharmacother. 2019, 117, 109168. [Google Scholar] [CrossRef] [PubMed]
- Ambrosini, V.; Fani, M.; Fanti, S.; Forrer, F.; Maecke, H.R. Radiopeptide Imaging and Therapy in Europe. J. Nucl. Med. 2011, 52, 42S–55S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, H.; Alsadek, D.M.M. Galectin-3 as a Potencial Target to Prevent Cancer Metastasis. Clin. Med. Insights Oncol. 2015, 9, 113. [Google Scholar] [CrossRef] [Green Version]
- Dong, R.; Zhang, M.; Hu, Q.; Zheng, S.; Soh, A.; Zheng, Y.; Yuan, H. Galectin-3 as a novel biomarker for disease diagnosis and a target for therapy (Review). Int. J. Mol. Med. 2018, 41, 599–614. [Google Scholar] [CrossRef] [Green Version]
- Wdowiak, K.; Francuz, T.; Gallego-Colon, E.; Ruiz-Agamez, N.; Kubeczko, M.; Grochoła, I.; Wojnar, J. Galectin Targeted Therapy in Oncology: Current Knowledge and Perspectives. Int. J. Mol. Sci. 2018, 19, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ram, T.J.; Lekshmi, A.; Somanathan, T.; Sujathan, K. Galectin-3: A factotum in carcinogenesis bestowing an archery for prevention. Tumor Biol. 2021, 43, 77–96. [Google Scholar]
- Takenaka, Y.; Fukumori, T.; Raz, A. Galectin-3 and metastasis. Glycoconj. J. 2002, 19, 543–549. [Google Scholar] [CrossRef]
- Seetharaman, J.; Kanigsberg, A.; Slaaby, R.; Leffler, H.; Barondes, S.H.; Rini, J.M. X-ray Crystal Structure of the Human Galectin-3 Carbohydrate Recognition Domain at 2.1-Å Resolution. J. Biol. Chem. 1998, 273, 13047–13052. [Google Scholar] [CrossRef] [Green Version]
- Funasaka, T.; Raz, A.; Nangia-Makker, P. Galectin-3 in angiogenesis and metastasis. Glycobiology 2014, 24, 886–891. [Google Scholar] [CrossRef] [Green Version]
- Farhad, M.; Rolig, A.S.; Redmond, W.L. The role of Galectin-3 in modulating tumor growth and immunosuppression within the tumor microenviroment. Oncoimmunology 2018, 7, e1434467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.; Tang, J.-W.; Owusu, L.; Sun, M.-Z.; Wu, J.; Zhang, J. Galectin-3 in cancer. Clin. Chim. Acta 2014, 431, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.-S.; Weng, D.-S.; Wang, Q.-J.; Pan, K.; Zhang, Y.-J.; Li, Y.-Q.; Li, J.-J.; Zhao, J.-J.; He, J.; Lv, L.; et al. Galectin-3 is associated with a poor prognosis in primary hepatocellular carcinoma. J. Transl. Med. 2014, 12, 273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endo, K.; Kohnoe, S.; Tsujita, E.; Watanabe, A.; Nakashima, H.; Baba, H.; Maehara, Y. Galectin-3 expression is a potent prognostic marker in colorectal cancer. Anticancer Res. 2005, 25, 3117–3121. [Google Scholar] [PubMed]
- Sun, W.; Li, L.; Yang, Q.; Shan, W.; Zhang, Z.; Huang, Y. G3-C12 Peptide Reverses Galectin-3 from Foe to Friend for Active Targeting Cancer Treatment. Mol. Pharm. 2015, 12, 4124–4136. [Google Scholar] [CrossRef] [PubMed]
- Heine, V.; Hovorková, M.; Valchová, M.; Filipová, M.; Bumba, L.; Janousková, O.; Hubálek, M.; Cvacka, J.; Petrásková, L.; Pelantová, H.; et al. Immunoprotective neo-glycoproteins: Chemoenzymatic synthesis of multivalent glycomimetics for inhibition of cancer-related galectin-3. Eur. J. Med. Chem. 2021, 220, 113500. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Li, L.; Li, L.-J.; Yang, Q.-Q.; Zhang, Z.-R.; Huang, Y. Two birds, one stone: Dual targeting of the cancer cell surface and subcellular mitochondria by the galectin-3-binding peptide G3-C12. Acta Pharmacol. Sin. 2017, 38, 806–822. [Google Scholar] [CrossRef] [Green Version]
- Bertuzzi, S.; Quintana, J.I.; Ardá, A.; Gimeno, A.; Jiménez-Barbero, J. Targeting Galectins with Glycomimetics. Front. Chem. 2020, 8, 593. [Google Scholar] [CrossRef]
- Denavit, V.; Lainé, D.; Tremblay, T.; St-Gelais, J.; Giguère, D. Synthetic Inhibitors of Galectins: Structures and Syntheses. Trends Glycosci. Glycotechnol. 2018, 30, SE21–SE40. [Google Scholar] [CrossRef] [Green Version]
- Zou, J.; Glinsky, V.V.; Landon, L.A.; Matthews, L.; Deutscher, S.L. Peptides specific to the galectin-3 carbohydrate recognition domain inhibit metastasis-associated cancer cell adhesion. Carcinogenesis 2005, 26, 309–318. [Google Scholar] [CrossRef] [Green Version]
- Chan, Y.C.; Lin, H.Y.; Tu, Z.; Kuo, Y.H.; Hsu, S.T.D.; Lin, C.H. Dissecting the structure–activity relationship of galectin–ligand interactions. Int. J. Mol. Sci. 2018, 19, 392. [Google Scholar] [CrossRef] [Green Version]
- Klyosov, A.A.; Zomer, E.; Platt, D. DAVANT® (GM-CT-01) and Colon Cancer: Preclinical and Clinical (Phase I and II) Studies. In Glycobiology and Drug Design, 1st ed.; Klyosov, A.A., Ed.; American Chemical Society: Washington, DC, USA, 2012; Volume 1102, pp. 89–130. [Google Scholar]
- Campo, V.L.; Marchiori, M.F.; Rodrigues, L.C.; Dias-Baruffi, M. Synthetic glycoconjugates inhibitors of tumor-related galectin-3: An update. Glycoconj. J. 2016, 33, 853–876. [Google Scholar] [CrossRef] [PubMed]
- Vuong, L.; Kouverianou, E.; Rooney, C.M.; McHugh, B.J.; Howie, S.E.; Gregory, C.D.; Forbes, S.J.; Henderson, N.C.; Zetterberg, F.R.; Nilsson, U.J.; et al. An Orally Active Galectin-3 Antagonist Inhibits Lung Adenocarcinoma Growth and Augments Response to PD-L1 BlockadeNovel Galectin-3 Antagonist Inhibits Lung Cancer Progression. Cancer Res. 2019, 79, 1480–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyuricza, B.; Szabó, J.P.; Arató, V.; Dénes, N.; Szűcs, Á.; Berta, K.; Kis, A.; Szücs, D.; Forgács, V.; Szikra, D.; et al. Synthesis of 68Ga-labeled cNGR-Based Glycopeptides and In Vivo Evaluation by PET Imaging. Pharmaceutics 2021, 13, 2103. [Google Scholar] [CrossRef]
- van Hattum, H.; Branderhorst, H.M.; Moret, E.E.; Nilsson, U.J.; Leffler, H.; Pieters, R.J. Tuning the Preference of Thiodigalactozide- and Lactosamine-Based Ligands to Galectin-3 over Galectin-1. J. Med. Chem. 2013, 56, 1350–1354. [Google Scholar] [CrossRef] [PubMed]
- Sörme, P.; Qian, Y.; Nyholm, P.-G.; Leffler, H.; Nilsson, U.J. Low Micromolar Inhibitors of Galectin-3 Based on 3′-Dervivatization of N-acetyllactosamine. ChemBioChem 2002, 3, 183–189. [Google Scholar] [CrossRef]
- Sörme, P.; Arnoux, P.; Kahl-Knuttsson, B.; Leffler, H.; Rini, J.M.; Nilsson, U.J. Structural and Thermodynamic Studies on Cation-Π Interactions in Lectin-Ligand Complexes: High-Affinity Galectin-3 Inhibitors through Fine-Tuning of an Arginine-Arene Interaction. J. Am. Chem. Soc. 2005, 127, 1737–1743. [Google Scholar] [CrossRef]
- Kumar, S.R.; Deutscher, S.L. 111In-labeled galectin-3-targeting peptide as a SPECT agent for imaging breast tumors. J. Nucl. Med. 2008, 49, 796–803. [Google Scholar] [CrossRef] [Green Version]
- Deutscher, S.L.; Figueroa, S.D.; Kumar, S.R. Tumor targeting and SPECT imaging properties of an 111In-labeled galectin-3 binding peptide in prostate carcinoma. Nucl. Med. Biol. 2009, 36, 137–146. [Google Scholar] [CrossRef]
- D’Alessandria, C.; Braesch-Andersen, S.; Bejo, K.; Reder, S.; Blechert, B.; Schwaiger, M.; Bartolazzi, A. Noninvasive In Vivo Imaging and Biologic Characterization of Thyroid Tumors by ImmunoPET Targeting of Galectin-3. Cancer Res. 2016, 76, 3583–3592. [Google Scholar] [CrossRef] [Green Version]
- Bratteby, K.; Torkelsson, E.; L’Estrade, E.T.; Peterson, K.; Shalgunov, V.; Xiong, M.; Leffler, H.; Zetterberg, F.R.; Olsson, T.G.; Gillings, N.; et al. In Vivo Veritas: 18F-Radiolabeled Glycomimetics Allow Insights into the Pharmacological Fate of Galectin-3 Inhibitors. J. Med. Chem. 2019, 63, 747–755. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, F.; Chen, X. Integrin αvβ3-targeted cancer therapy. Drug Dev. Res. 2008, 69, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Danhier, F.; Le Breton, A.; Préat, V. RGD-Based Strategies To Target Alpha(v) Beta(3) Integrin in Cancer Therapy and Diagnosis. Mol. Pharm. 2012, 9, 2961–2973. [Google Scholar] [CrossRef] [PubMed]
- Gyuricza, B.; Szabó, J.P.; Arató, V.; Szücs, D.; Vágner, A.; Szikra, D.; Fekete, A. Synthesis of Novel, Dual-Targeting 68Ga-NODAGA-LacN-E[c(RGDfK)]2 Glycopeptide as a PET Imaging Agent for Cancer Diagnosis. Pharmaceutics 2021, 13, 796. [Google Scholar] [CrossRef] [PubMed]
- Sörme, P.; Kahl-Knutsson, B.; Wellmar, U.; Magnusson, B.G.; Leffler, H.; Nilsson, U.J. Design and synthesis of galectin inhibitors. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2003; Volume 363, pp. 157–169. [Google Scholar]
- Li, L.; Chen, X.; Yu, J.; Yuan, S. Preliminary Clinical Application of RGD-Containing Peptides as PET Radiotracers for Imaging Tumors. Front. Oncol. 2022, 12, 837952. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.C.; Kuo, P.L. The Role of Galectin-3 in the Kidneys. Int. J. Mol. Sci. 2016, 17, 565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, D.K.; Dowling, C.A.; Jeng, K.C.; Chen, J.T.; Yang, R.Y.; Liu, F.T. Galectin-3 expression is induced in cirrhotic liver and hepatocellular carcinoma. Int. J. Cancer 1999, 81, 519–526. [Google Scholar] [CrossRef]
- Rathinam, R.; Alahari, S.K. Important role of integrins in the cancer biology. Cancer Metastasis Rev. 2010, 29, 223–237. [Google Scholar] [CrossRef]
- Dayan, A.; Fleminger, G.; Ashur-Fabian, O. RGD-modified dihydrolipoamide dehydrogenase conjugated to titanium dioxide nanoparticles—Switchable integrin-targeted photodynamic treatment of melanoma cells. RSC Adv. 2018, 8, 9112–9119. [Google Scholar] [CrossRef] [Green Version]
- Comodo, A.N.; Bachi, A.L.L.; Soares, M.F.; Franco, M.; Teixeira, V.D.P.C. Galectin-3 expression favors metastasis in murine melanoma. Adv. Biosci. Biotechnol. 2013, 4, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-G.; Kim, S.-J.; Baek, J.-H.; Lee, H.-W.; Jeong, S.-Y.; Chun, K.-H. Galectin-3 increases the motility of mouse melanoma cells by regulating matrix metalloproteinase-1 expression. Exp. Mol. Med. 2012, 44, 387–393. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor | [68Ga]Ga-DOTAGA-LacN(NAP) | [68Ga]Ga-DOTAGA-LacN(NAP)-cRGDfK | [68Ga]Ga-DOTAGA-cRGDfK |
|---|---|---|---|
| B16-F10 (n = 5) | 0.81 ± 0.27 | 0.80 ± 0.23 | 0.35 ± 0.12 |
| B16-F10/Muscle ratio | 13.97 ± 2.58 ** | 3.43 ± 1.09 | 4.54 ± 1.24 |
| B16-F10/Liver ratio | 2.66 ± 0.04 ** | 0.45 ± 0.11 | 0.35 ± 0.14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gyuricza, B.; Szűcs, Á.; Szabó, J.P.; Arató, V.; Képes, Z.; Szücs, D.; Szikra, D.; Trencsényi, G.; Fekete, A. The Synthesis and Preclinical Investigation of Lactosamine-Based Radiopharmaceuticals for the Detection of Galectin-3-Expressing Melanoma Cells. Pharmaceutics 2022, 14, 2504. https://doi.org/10.3390/pharmaceutics14112504
Gyuricza B, Szűcs Á, Szabó JP, Arató V, Képes Z, Szücs D, Szikra D, Trencsényi G, Fekete A. The Synthesis and Preclinical Investigation of Lactosamine-Based Radiopharmaceuticals for the Detection of Galectin-3-Expressing Melanoma Cells. Pharmaceutics. 2022; 14(11):2504. https://doi.org/10.3390/pharmaceutics14112504
Chicago/Turabian StyleGyuricza, Barbara, Ágnes Szűcs, Judit P. Szabó, Viktória Arató, Zita Képes, Dániel Szücs, Dezső Szikra, György Trencsényi, and Anikó Fekete. 2022. "The Synthesis and Preclinical Investigation of Lactosamine-Based Radiopharmaceuticals for the Detection of Galectin-3-Expressing Melanoma Cells" Pharmaceutics 14, no. 11: 2504. https://doi.org/10.3390/pharmaceutics14112504