

Human Melanoma Cells Differentially Express RNASEL/RNase-L and miR-146a-5p under Sex Hormonal Stimulation

 ,
,  , ,
, ,  , ,
, ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cloning of the RNase-L 3′UTR, Generation of 3′UTR Mutants and Transfections
2.3. Cell Treatment with Testosterone or 17β-Estradiol
2.3.1. RNA Extraction, Reverse Transcription and Real-Time PCR
2.3.2. Western Blot Analysis
2.4. Over-Expression of miR-146a by Mimic Transfection
2.5. Statistics
3. Results
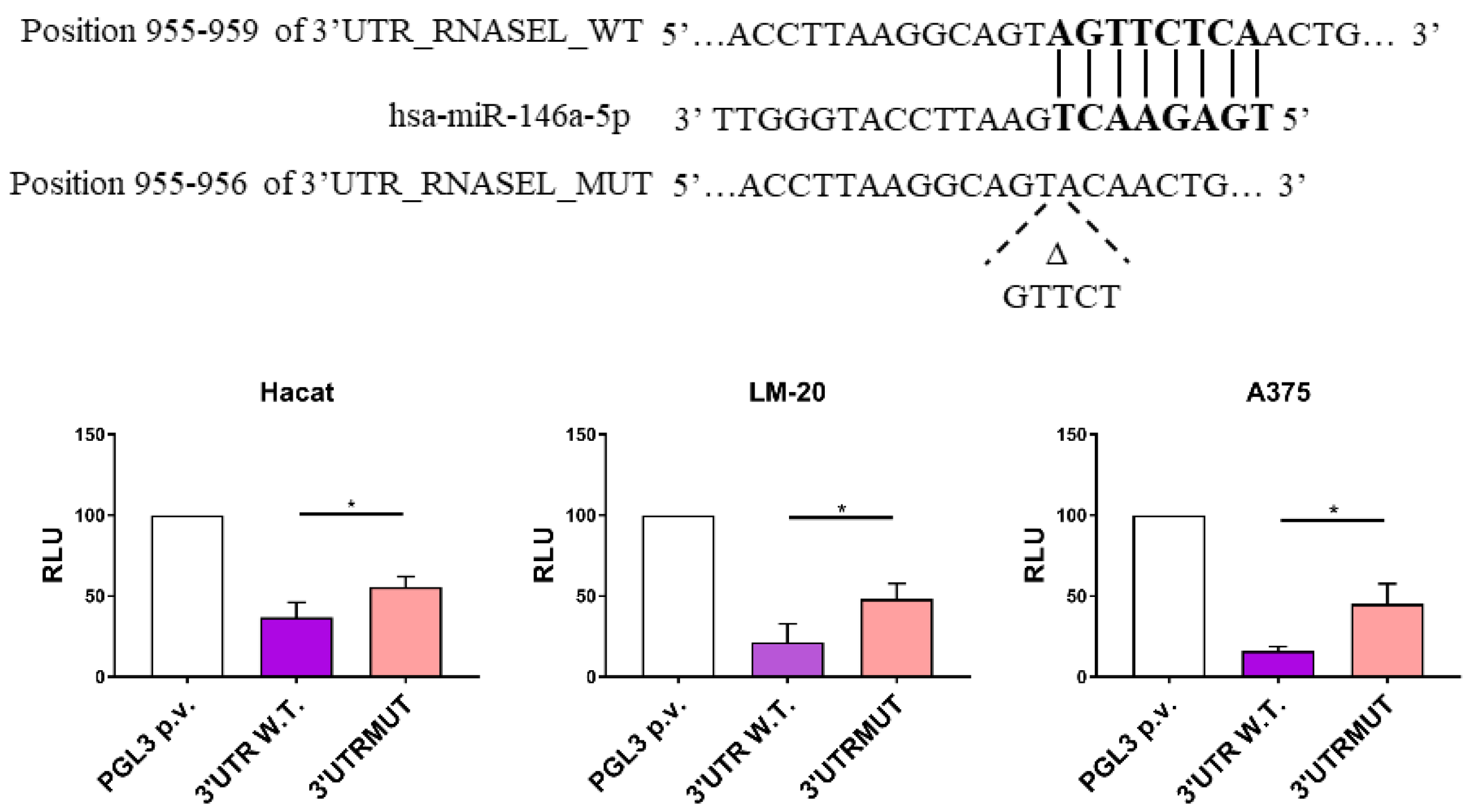
3.1. miR-146a Directly Targets RNASEL 3′UTR Region in LM-20 and A375 Melanoma Cells and in HaCaT Keratinocytes
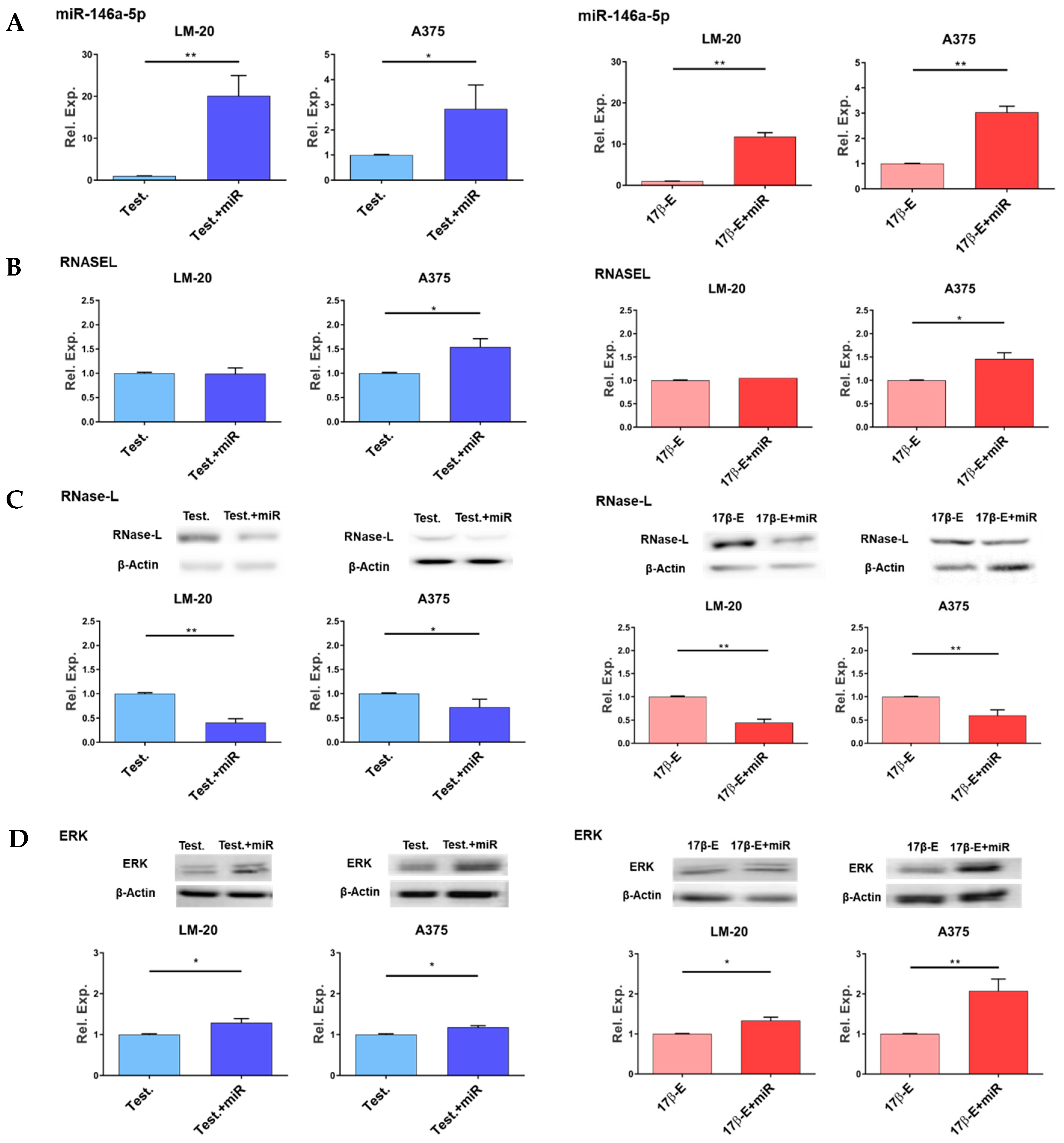
3.2. Effects of miR-146a Overexpression in the Presence of Hormones on RNASEL Gene Expression, RNase-L Protein Level and ERK1/2 Phosphorylation
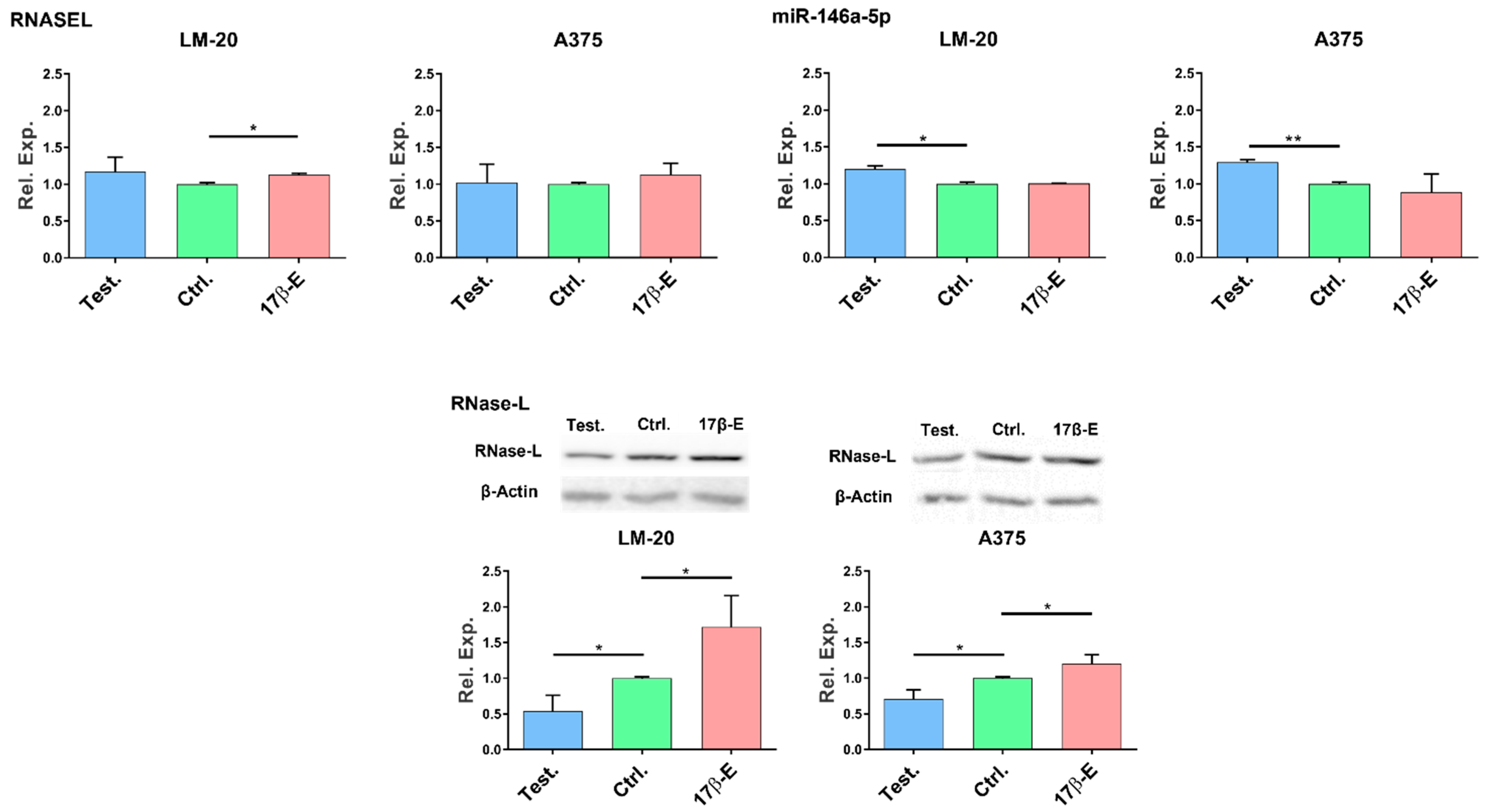
3.3. Sex Hormones Affect the Endogenous Expression of miR-146a and RNASEL Genes and RNase-L Protein in Melanoma Cells
3.3.1. Effects of Hormones on RNASEL Gene Expression
3.3.2. Effects of Hormones on miR-146a Gene Expression
3.3.3. Effects of Hormones on RNase-L Protein Expression
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arnold, M.; Singh, D.; Laversanne, M.; Vignat, J.; Vaccarella, S.; Meheus, F.; Cust, A.E.; de Vries, E.; Whiteman, D.C.; Bray, F. Global Burden of Cutaneous Melanoma in 2020 and Projections to 2040. JAMA Dermatol. 2022, 158, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Erdei, E.; Torres, S.M. A new understanding in the epidemiology of melanoma. Expert Rev. Anticancer Ther. 2010, 10, 1811–1823. [Google Scholar] [CrossRef]
- Liu, F.; Bessonova, L.; Taylor, T.H.; Ziogas, A.; Meyskens, F.L., Jr.; Anton-Culver, H. A unique gender difference in early onset melanoma implies that in addition to ultraviolet light exposure other causative factors are important. Pigm. Cell Melanoma Res. 2013, 26, 128–135. [Google Scholar] [CrossRef]
- Marzagalli, M.; Marelli, M.M.; Casati, L.; Fontana, F.; Moretti, R.M.; Limonta, P. Estrogen Receptor β in Melanoma: From Molecular Insights to Potential Clinical Utility. Front. Endocrinol. 2016, 7, 140. [Google Scholar] [CrossRef] [PubMed]
- Scoggins, C.R.; Ross, M.I.; Reintgen, D.S.; Noyes, R.D.; Goydos, J.S.; Beitsch, P.D.; Urist, M.M.; Ariyan, S.; Sussman, J.J.; Edwards, M.J.; et al. Gender-Related Differences in Outcome for Melanoma Patients. Ann. Surg. 2006, 243, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Ing, N.H. Steroid Hormones Regulate Gene Expression Posttranscriptionally by Altering the Stabilities of Messenger RNAs. Biol. Reprod. 2005, 72, 1290–1296. [Google Scholar] [CrossRef] [PubMed]
- Snyder, E.M.; Small, C.L.; Li, Y.; Griswold, M.D. Regulation of Gene Expression by Estrogen and Testosterone in the Proximal Mouse Reproductive Tract. Biol. Reprod. 2009, 81, 707–716. [Google Scholar] [CrossRef] [PubMed][Green Version]
- de Giorgi, V.; Gori, A.; Alfaioli, B.; Papi, F.; Grazzini, M.; Rossari, S.; Lotti, T.; Massi, D. Influence of Sex Hormones on Melanoma. J. Clin. Oncol. 2011, 29, e94–e95. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Portolés, C.; Payne, R.; Trautz, A.; Foskett, J.K.; Natale, C.A.; Seykora, J.T.; Ridky, T.W. ZIP9 Is a Druggable Determinant of Sex Differences in Melanoma. Cancer Res. 2021, 81, 5991–6003. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Ghosh, S.; Tavernari, D.; Katarkar, A.; Clocchiatti, A.; Mazzeo, L.; Samarkina, A.; Epiney, J.; Yu, Y.-R.; Ho, P.-C.; et al. Sustained androgen receptor signaling is a determinant of melanoma cell growth potential and tumorigenesis. J. Exp. Med. 2020, 218. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ou, Z.; Sun, Y.; Yeh, S.; Wang, X.; Long, J.; Chang, C. Androgen receptor promotes melanoma metastasis via altering the miRNA-539-3p/USP13/MITF/AXL signals. Oncogene 2016, 36, 1644–1654. [Google Scholar] [CrossRef] [PubMed]
- Natale, C.A.; Li, J.; Zhang, J.; Dahal, A.; Dentchev, T.; Stanger, B.Z.; Ridky, T.W. Activation of G protein-coupled estrogen receptor signaling inhibits melanoma and improves response to immune checkpoint blockade. eLife 2018, 7, e31770. [Google Scholar] [CrossRef] [PubMed]
- Sangalli, A.; Orlandi, E.; Poli, A.; Maurichi, A.; Santinami, M.; Nicolis, M.; Ferronato, S.; Malerba, G.; Rodolfo, M.; Lira, M.G. Sex-specific effect of RNASEL rs486907 and miR-146a rs2910164 polymorphisms’ interaction as a susceptibility factor for melanoma skin cancer. Melanoma Res. 2017, 27, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Farzan, S.F.; Karagas, M.R.; Christensen, B.C.; Li, Z.; Kuriger, J.K.; Nelson, H.H.; on behalf of the New Hampshire Skin Cancer Study. RNASEL and MIR146A SNP-SNP Interaction as a Susceptibility Factor for Non-Melanoma Skin Cancer. PLoS ONE 2014, 9, e93602. [Google Scholar] [CrossRef]
- Zhu, Z.; Gao, X.; Zhang, S. miR-146a rs2910164 polymorphism and the risk of colorectal cancer in Chinese population. J. Cancer Res. Ther. 2018, 14, S97–S99. [Google Scholar] [CrossRef]
- Zhou, X.; Zhu, J.; Zhang, H.; Zhou, G.; Huang, Y.; Liu, R. Is the microRNA-146a (rs2910164) polymorphism associated with rheumatoid arthritis? Association of microRNA-146a (rs2910164) polymorphism and rheumatoid arthritis could depend on gender. Jt. Bone Spine 2015, 82, 166–171. [Google Scholar] [CrossRef]
- Kiselev, I.; Bashinskaya, V.; Kulakova, O.; Baulina, N.; Popova, E.; Boyko, A.; Favorova, O. Variants of MicroRNA Genes: Gender-Specific Associations with Multiple Sclerosis Risk and Severity. Int. J. Mol. Sci. 2015, 16, 20067–20081. [Google Scholar] [CrossRef]
- Lee, I.; Ajay, S.S.; Yook, J.I.; Kim, H.S.; Hong, S.H.; Kim, N.H.; Dhanasekaran, S.M.; Chinnaiyan, A.M.; Athey, B.D. New class of microRNA targets containing simultaneous 5′-UTR and 3′-UTR interaction sites. Genome Res. 2009, 19, 1175–1183. [Google Scholar] [CrossRef]
- Ahmed, A.; Dai, R. Sexual dimorphism of miRNA expression: A new perspective in understanding the sex bias of autoimmune diseases. Ther. Clin. Risk Manag. 2014, 10, 151–163. [Google Scholar] [CrossRef]
- Cui, C.; Yang, W.; Shi, J.; Zhou, Y.; Yang, J.; Cui, Q.; Zhou, Y. Identification and Analysis of Human Sex-biased MicroRNAs. Genom. Proteom. Bioinform. 2018, 16, 200–211. [Google Scholar] [CrossRef]
- Syed, S.N.; Brüne, B. Exosomal and Non-Exosomal MicroRNAs: New Kids on the Block for Cancer Therapy. Int. J. Mol. Sci. 2022, 23, 4493. [Google Scholar] [CrossRef] [PubMed]
- Ali Syeda, Z.; Langden, S.S.S.; Munkhzul, C.; Lee, M.; Song, S.J. Regulatory Mechanism of MicroRNA Expression in Cancer. Int. J. Mol. Sci. 2020, 21, 1723. [Google Scholar] [CrossRef] [PubMed]
- So, A.Y.-L.; Zhao, J.L.; Baltimore, D. The Yin and Yang of microRNAs: Leukemia and immunity. Immunol. Rev. 2013, 253, 129–145. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Liu, H.; Zheng, C.; Zhao, Y.; Liao, X.; Wang, Y.; Chen, Y.; Zhao, B.; Lazartigues, E.; Yang, Y.; et al. Microvesicles Derived from Inflammation-Challenged Endothelial Cells Modulate Vascular Smooth Muscle Cell Functions. Front. Physiol. 2017, 7, 692. [Google Scholar] [CrossRef] [PubMed]
- Shahriar, A.; Shiva, G.G.-A.; Ghader, B.; Farhad, J.; Hosein, A.; Parsa, H. The dual role of mir-146a in metastasis and disease progression. Biomed. Pharmacother. 2020, 126, 110099. [Google Scholar] [CrossRef]
- Forloni, M.; Dogra, S.K.; Dong, Y.; Conte, D.; Ou, J.; Zhu, L.J.; Deng, A.; Mahalingam, M.; Green, M.R.; Wajapeyee, N. miR-146a promotes the initiation and progression of melanoma by activating Notch signaling. eLife 2014, 3, e01460. [Google Scholar] [CrossRef]
- Dong, B.; Silverman, R.H. A Bipartite Model of 2-5A-dependent RNase L. J. Biol. Chem. 1997, 272, 22236–22242. [Google Scholar] [CrossRef]
- Xiang, Y.; Wang, Z.; Murakami, J.; Plummer, S.; Klein, E.A.; Carpten, J.D.; Trent, J.M.; Isaacs, W.B.; Casey, G.; Silverman, R.H. Effects of RNase L Mutations Associated with Prostate Cancer on Apoptosis Induced by 2′,5′-Oligoadenylates. Cancer Res. 2003, 63, 6795–6801. [Google Scholar]
- Liu, W.; Liang, S.-L.; Liu, H.; Silverman, R.; Zhou, A. Tumour suppressor function of RNase L in a mouse model. Eur. J. Cancer 2007, 43, 202–209. [Google Scholar] [CrossRef]
- Liang, S.-L.; Quirk, D.; Zhou, A. RNase L: Its biological roles and regulation. IUBMB Life 2006, 58, 508–514. [Google Scholar] [CrossRef]
- Li, X.-L.; Andersen, J.B.; Ezelle, H.J.; Wilson, G.M.; Hassel, B.A. Post-transcriptional Regulation of RNase-L Expression Is Mediated by the 3′-Untranslated Region of Its mRNA. J. Biol. Chem. 2007, 282, 7950–7960. [Google Scholar] [CrossRef] [PubMed]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal Keratinization in a Spontaneously Immortalized Aneuploid Human Keratinocyte Cell Line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Colombo, I.; SanGiovanni, E.; Maggio, R.; Mattozzi, C.; Zava, S.; Corbett, Y.; Fumagalli, M.; Carlino, C.; Corsetto, P.A.; Scaccabarozzi, D.; et al. HaCaT Cells as a Reliable In Vitro Differentiation Model to Dissect the Inflammatory/Repair Response of Human Keratinocytes. Mediat. Inflamm. 2017, 2017, 7435621. [Google Scholar] [CrossRef]
- Deyrieux, A.F.; Wilson, V.G. In vitro culture conditions to study keratinocyte differentiation using the HaCaT cell line. Cytotechnology 2007, 54, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Kasela, T.; Dąbala, M.; Mistarz, M.; Wieczorek, W.; Wierzbik-Strońska, M.; Boroń, K.; Zawidlak-Węgrzyńska, B.; Grabarek, B.O. Effects of Cyclosporine A and Adalimumab on the expression profiles histaminergic system-associated genes and microRNAs regulating these genes in HaCaT cells. Cell Cycle 2022, 1–18. [Google Scholar] [CrossRef]
- Daniotti, M.; Oggionni, M.; Ranzani, T.; Vallacchi, V.; Campi, V.; Di Stasi, D.; Della Torre, G.; Perrone, F.; Luoni, C.; Suardi, S.; et al. BRAF alterations are associated with complex mutational profiles in malignant melanoma. Oncogene 2004, 23, 5968–5977. [Google Scholar] [CrossRef]
- Romanelli, M.G.; Lorenzi, P.; Sangalli, A.; Diani, E.; Mottes, M. Characterization and functional analysis of cis-acting elements of the human farnesyl diphosphate synthetase (FDPS) gene 5′ flanking region. Genomics 2009, 93, 227–234. [Google Scholar] [CrossRef]
- Kanda, N.; Watanabe, S. 17β-estradiol, Progesterone, and Dihydrotestosterone Suppress the Growth of Human Melanoma by Inhibiting Interleukin-8 Production. J. Investig. Dermatol. 2001, 117, 274–283. [Google Scholar] [CrossRef]
- Ferronato, S.; Lira, M.G.; Olivato, S.; Scuro, A.; Veraldi, G.F.; Romanelli, M.G.; Patuzzo, C.; Malerba, G.; Pignatti, P.F.; Mazzucco, S. Upregulated Expression of Toll-like Receptor 4 in Peripheral Blood of Ischaemic Stroke Patients Correlates with Cyclooxygenase 2 Expression. Eur. J. Vasc. Endovasc. Surg. 2011, 41, 358–363. [Google Scholar] [CrossRef]
- Fochi, S.; Orlandi, E.; Ceccuzzi, L.; Rodolfo, M.G.; Vergani, E.; Turco, A.; Romanelli, M.; Gomez-Lira, M. Identification of suitable mRNAs and microRNAs as reference genes for expression analyses in skin cells under sex hormone exposure. Gene 2020, 769, 145336. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Montone, R.; Romanelli, M.G.; Baruzzi, A.; Ferrarini, F.; Liboi, E.; Lievens, P.-J. Mutant FGFR3 associated with SADDAN disease causes cytoskeleton disorganization through PLCγ1/Src-mediated paxillin hyperphosphorylation. Int. J. Biochem. Cell Biol. 2017, 95, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Gershoni, M.; Pietrokovski, S. The landscape of sex-differential transcriptome and its consequent selection in human adults. BMC Biol. 2017, 15, 7. [Google Scholar] [CrossRef] [PubMed]
- Rubin, J.B.; Lagas, J.S.; Broestl, L.; Sponagel, J.; Rockwell, N.; Rhee, G.; Rosen, S.F.; Chen, S.; Klein, R.S.; Imoukhuede, P.; et al. Sex differences in cancer mechanisms. Biol. Sex Differ. 2020, 11, 17. [Google Scholar] [CrossRef]
- Lucca, I.; Klatte, T.; Fajkovic, H.; De Martino, M.; Shariat, S.F. Gender differences in incidence and outcomes of urothelial and kidney cancer. Nat. Rev. Urol. 2015, 12, 585–592. [Google Scholar] [CrossRef]
- Gabriele, L.; Buoncervello, M.; Ascione, B.; Bellenghi, M.; Matarrese, P.; Carè, A. The gender perspective in cancer research and therapy: Novel insights and on-going hypotheses. Annali Dell’istituto Superiore Di Sanita 2016, 52, 213–222. [Google Scholar] [CrossRef]
- Micheli, A.; Ciampichini, R.; Oberaigner, W.; Ciccolallo, L.; de Vries, E.; Izarzugaza, I.; Zambon, P.; Gatta, G.; De Angelis, R.; EUROCARE Working Group. The advantage of women in cancer survival: An analysis of EUROCARE-4 data. Eur. J. Cancer 2009, 45, 1017–1027. [Google Scholar] [CrossRef]
- Clocchiatti, A.; Cora, E.; Zhang, Y.; Dotto, G.P. Sexual dimorphism in cancer. Nat. Cancer 2016, 16, 330–339. [Google Scholar] [CrossRef]
- Rampen, F. Malignant melanoma: Sex differences in survival after evidence of distant metastasis. Br. J. Cancer 1980, 42, 52–57. [Google Scholar] [CrossRef]
- Kim, H.-I.; Lim, H.; Moon, A. Sex Differences in Cancer: Epidemiology, Genetics and Therapy. Biomol. Ther. 2018, 26, 335–342. [Google Scholar] [CrossRef]
- Joosse, A.; de Vries, E.; Eckel, R.; Nijsten, T.; Eggermont, A.M.; Hölzel, D.; Coebergh, J.W.W.; Engel, J. Gender Differences in Melanoma Survival: Female Patients Have a Decreased Risk of Metastasis. J. Investig. Dermatol. 2011, 131, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Ramchandran, R.; Chaluvally-Raghavan, P. miRNA-Mediated RNA Activation in Mammalian Cells. Adv. Exp. Med. Biol. 2017, 983, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Sulidankazha, C.; Han, W.; He, T.; Lin, H.; Cheng, K.; Nie, X.; Chen, Q. miR-146a Inhibited Pancreatic Cancer Cell Proliferation by Targeting SOX7. J. Healthc. Eng. 2022, 2022, 2240605. [Google Scholar] [CrossRef] [PubMed]
- Noorolyai, S.; Baghbani, E.; Shanehbandi, D.; Shahgoli, V.K.; Kojabad, A.B.; Mansoori, B.; Hajiasgharzadeh, K.; Mokhtarzadeh, A.; Baradaran, B. miR-146a-5p and miR-193a-5p Synergistically Inhibited the Proliferation of Human Colorectal Cancer Cells (HT-29 cell line) through ERK Signaling Pathway. Adv. Pharm. Bull. 2020, 11, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Dika, E.; Riefolo, M.; Porcellini, E.; Broseghini, E.; Ribero, S.; Senetta, R.; Osella-Abate, S.; Scarfì, F.; Lambertini, M.; Veronesi, G.; et al. Defining the Prognostic Role of MicroRNAs in Cutaneous Melanoma. J. Investig. Dermatol. 2020, 140, 2260–2267. [Google Scholar] [CrossRef]
- Taganov, K.D.; Boldin, M.P.; Chang, K.-J.; Baltimore, D. NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, A.; Jha, B.K.; Silverman, R.H. New insights into the role of RNase L in innate immunity. J. Interferon Cytokine Res. 2011, 37, 49–57. [Google Scholar] [CrossRef]
- Rath, S.; Donovan, J.; Whitney, G.; Chitrakar, A.; Wang, W.; Korennykh, A. Human RNase L tunes gene expression by selectively destabilizing the microRNA-regulated transcriptome. Proc. Natl. Acad. Sci. USA 2015, 112, 15916–15921. [Google Scholar] [CrossRef] [PubMed]
- Menegazzi, M.; Gotte, G. Role of the Ribonuclease ONCONASE in miRNA Biogenesis and tRNA Processing: Focus on Cancer and Viral Infections. Int. J. Mol. Sci. 2022, 23, 6556. [Google Scholar] [CrossRef] [PubMed]
- Dayal, S.; Zhou, J.; Manivannan, P.; Siddiqui, M.A.; Ahmad, O.F.; Clark, M.; Awadia, S.; Garcia-Mata, R.; Shemshedini, L.; Malathi, K. RNase L Suppresses Androgen Receptor Signaling, Cell Migration and Matrix Metalloproteinase Activity in Prostate Cancer Cells. Int. J. Mol. Sci. 2017, 18, 529. [Google Scholar] [CrossRef]
- Vellano, C.P.; White, M.G.; Andrews, M.C.; Chelvanambi, M.; Witt, R.G.; Daniele, J.R.; Titus, M.; McQuade, J.L.; Conforti, F.; Burton, E.M.; et al. Androgen receptor blockade promotes response to BRAF/MEK-targeted therapy. Nature 2022, 606, 797–803. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Genes | Primers Sequences | Products |
|---|---|---|
| Real-Time PCR (mRNA) | Primers (5′ → 3′) | Product (bp) |
| RNASEL | CAGGATCTGCAACCACAAAA | 82 |
| CCCACTTGATGCTCTTATCAAA | ||
| TBP | TGTATCCACAGTGAATCTTGG | 102 |
| ATGATTACCGCAGCAAACC | ||
| Real-Time PCR (microRNA) | Sequences (Probe) | Assay (ID) |
| hsa-miR-146a-5p | UGAGAACUGAAUUCCAUGGGUU | 478399_mir |
| hsa-miR-191-5p | CAACGGAAUCCCAAAAGCAGCUG | 478231_mir |
| Plasmid construction | Primers (5′ → 3′) | Product (bp) |
| 3′UTR RNASEL_W.T. | CCTTCTAGAAACAAGCCTCAGTGTGATGG | 1771 |
| GGATCTAGACCAGGTGCTCATTACAAATC | ||
| (−)3′UTR RNASEL_W.T. | CCTTCTAGAAACAAGCCTCAGTGTGATGG | 777 |
| GGATCTAGAACCAAAAACTTCTTCAGACTC | ||
| 3′UTR _MUTAGENESIS | ATGACCTTAAGGCAGTACAACTGGGGGGCAATTT | |
| AAATTGCCCCCCAGTTGTACTGCCTTAAGGTCAT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orlandi, E.; De Tomi, E.; Campagnari, R.; Belpinati, F.; Rodolfo, M.; Vergani, E.; Malerba, G.; Gomez-Lira, M.; Menegazzi, M.; Romanelli, M.G. Human Melanoma Cells Differentially Express RNASEL/RNase-L and miR-146a-5p under Sex Hormonal Stimulation. Curr. Issues Mol. Biol. 2022, 44, 4790-4802. https://doi.org/10.3390/cimb44100326
Orlandi E, De Tomi E, Campagnari R, Belpinati F, Rodolfo M, Vergani E, Malerba G, Gomez-Lira M, Menegazzi M, Romanelli MG. Human Melanoma Cells Differentially Express RNASEL/RNase-L and miR-146a-5p under Sex Hormonal Stimulation. Current Issues in Molecular Biology. 2022; 44(10):4790-4802. https://doi.org/10.3390/cimb44100326
Chicago/Turabian StyleOrlandi, Elisa, Elisa De Tomi, Rachele Campagnari, Francesca Belpinati, Monica Rodolfo, Elisabetta Vergani, Giovanni Malerba, Macarena Gomez-Lira, Marta Menegazzi, and Maria Grazia Romanelli. 2022. "Human Melanoma Cells Differentially Express RNASEL/RNase-L and miR-146a-5p under Sex Hormonal Stimulation" Current Issues in Molecular Biology 44, no. 10: 4790-4802. https://doi.org/10.3390/cimb44100326
APA StyleOrlandi, E., De Tomi, E., Campagnari, R., Belpinati, F., Rodolfo, M., Vergani, E., Malerba, G., Gomez-Lira, M., Menegazzi, M., & Romanelli, M. G. (2022). Human Melanoma Cells Differentially Express RNASEL/RNase-L and miR-146a-5p under Sex Hormonal Stimulation. Current Issues in Molecular Biology, 44(10), 4790-4802. https://doi.org/10.3390/cimb44100326