
The Emerging Role of Heat Shock Factor 1 (HSF1) and Heat Shock Proteins (HSPs) in Ferroptosis

Abstract
1. Introduction
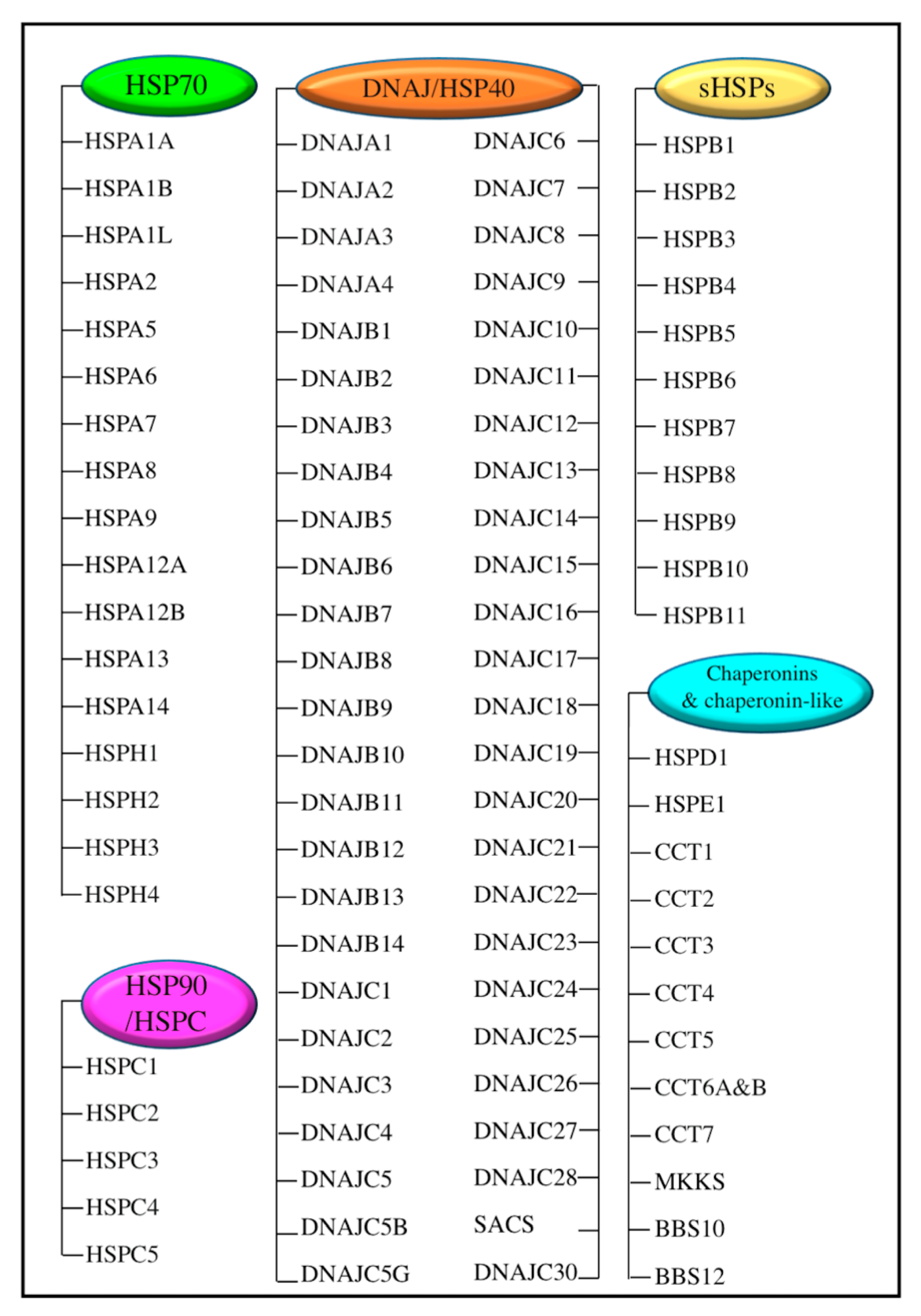
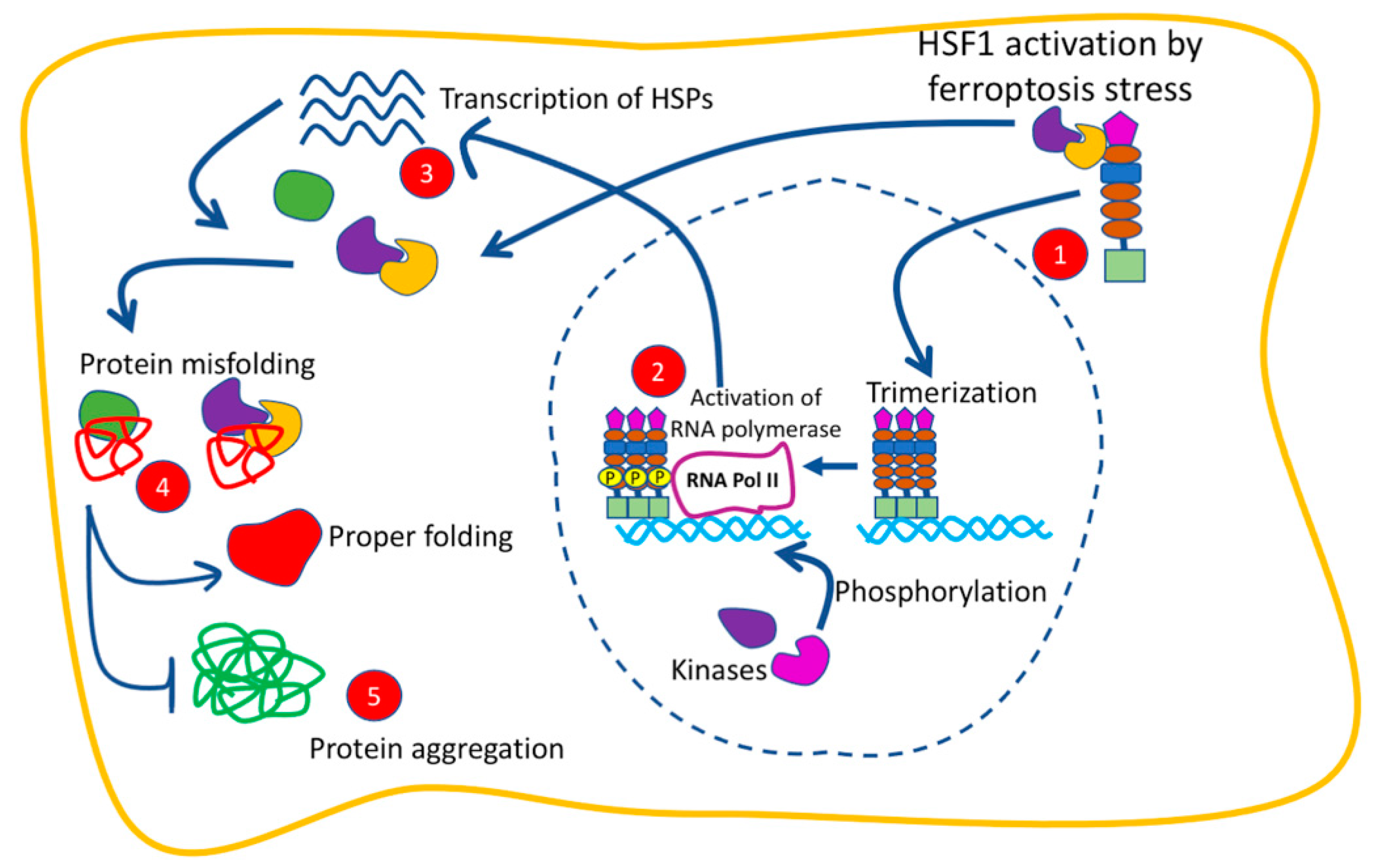
2. Heat Shock Proteins (HSPs) and Their Transcriptional Regulator Heat Shock Factor 1 (HSF1)
3. Ferroptosis
3.1. Overview
3.2. Basic Features of Ferroptosis
3.3. Regulation Mechanism Ferroptosis
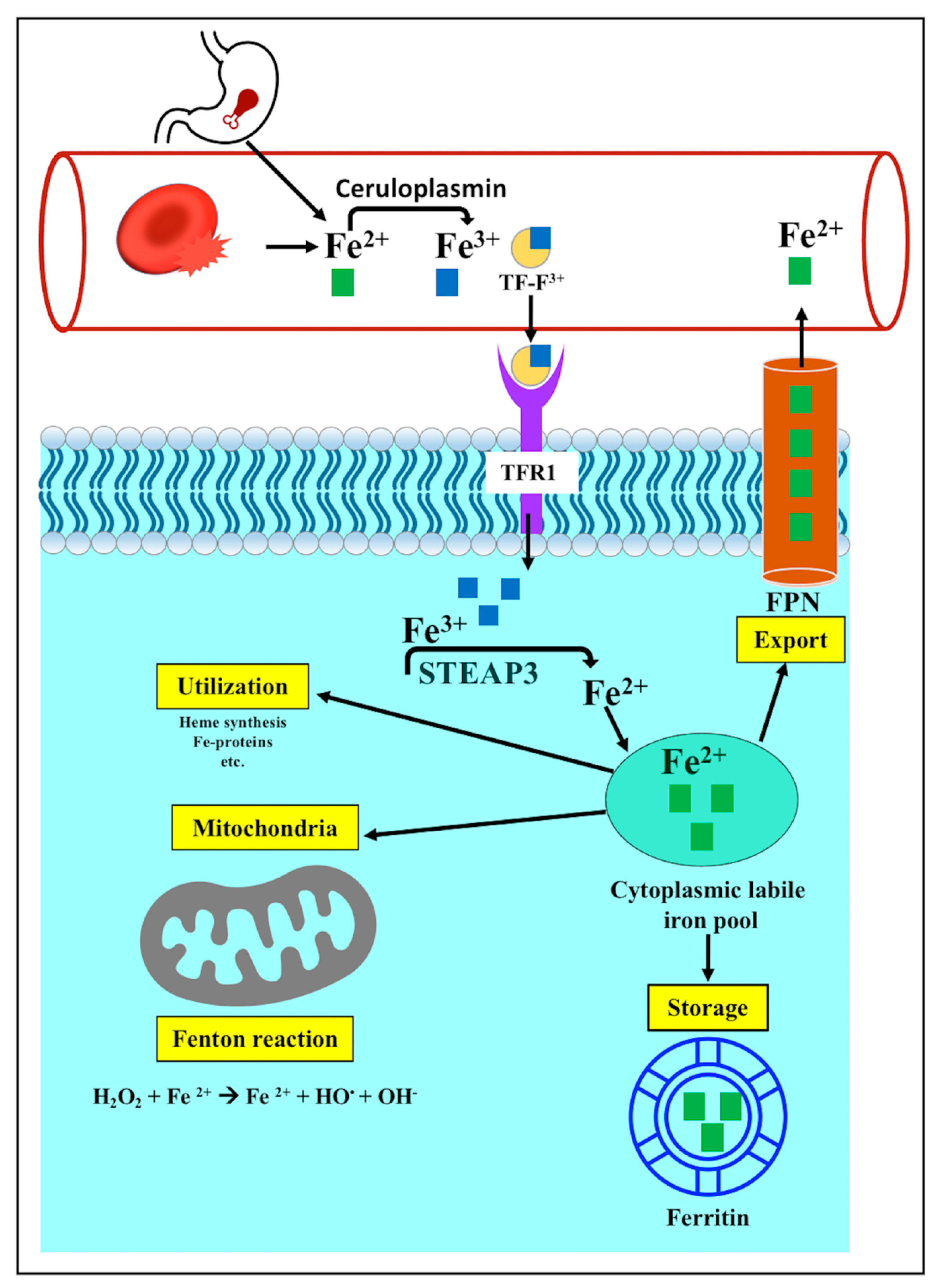
3.3.1. Iron Metabolism
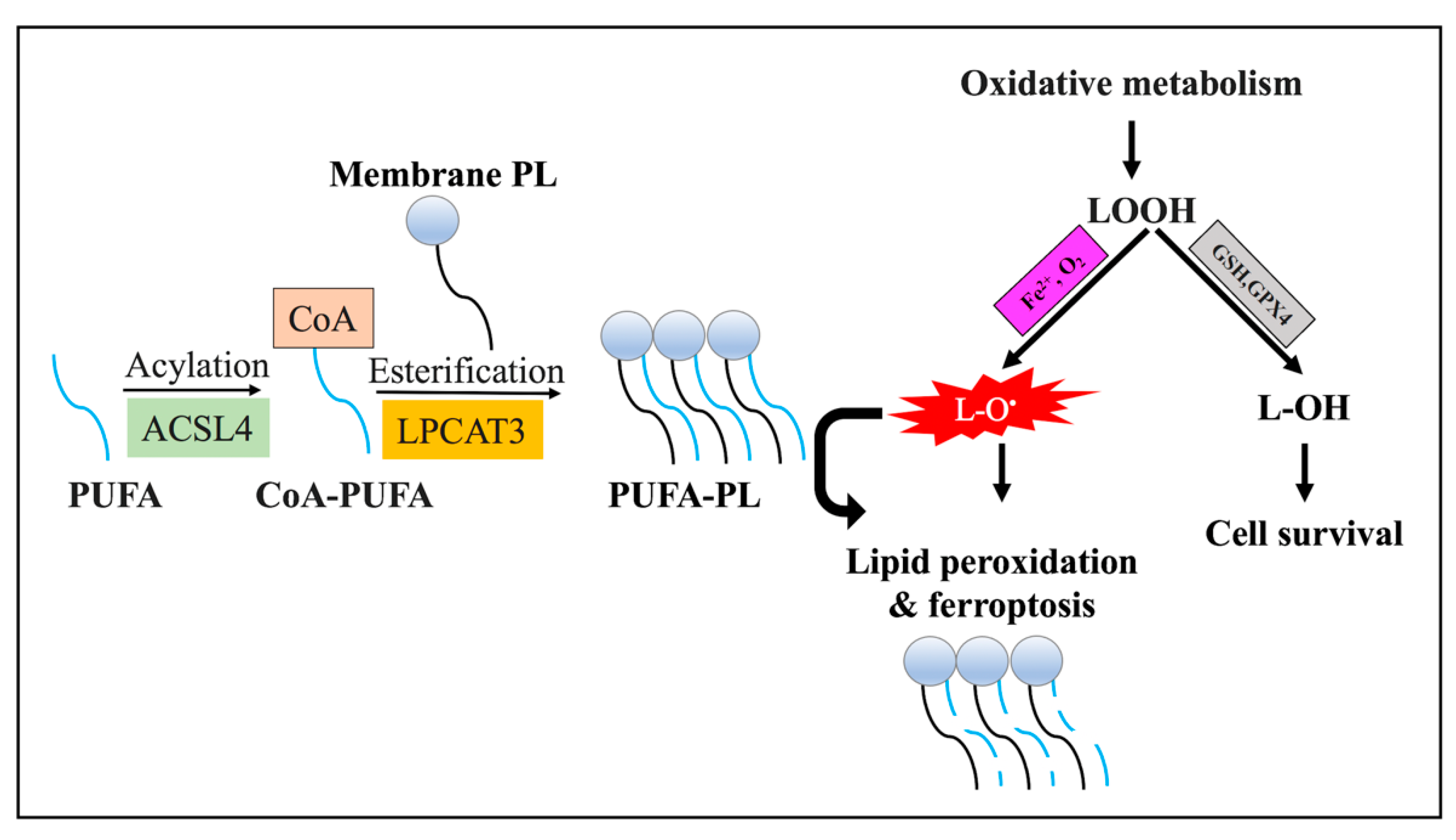
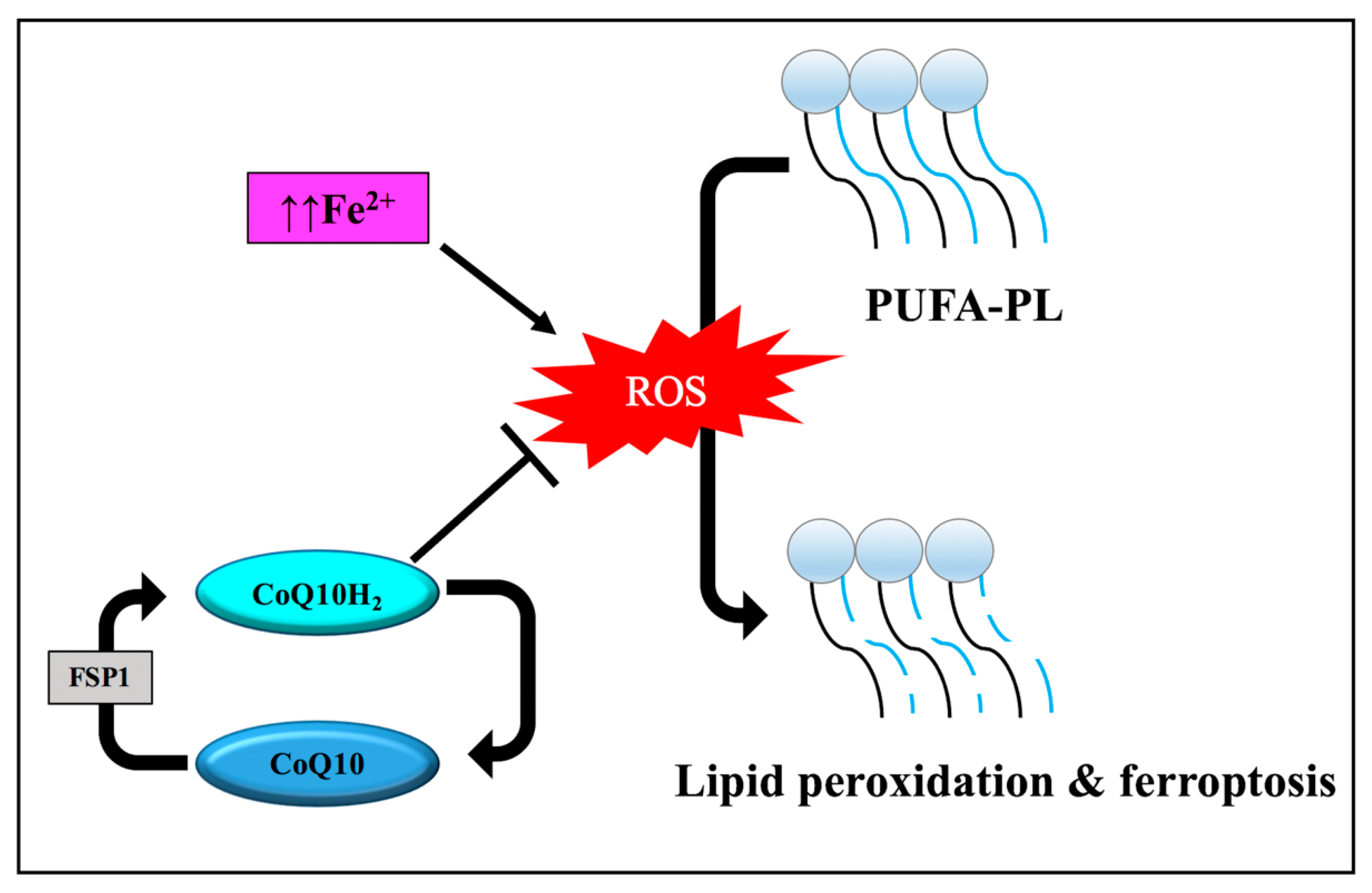
3.3.2. Lipid Metabolism
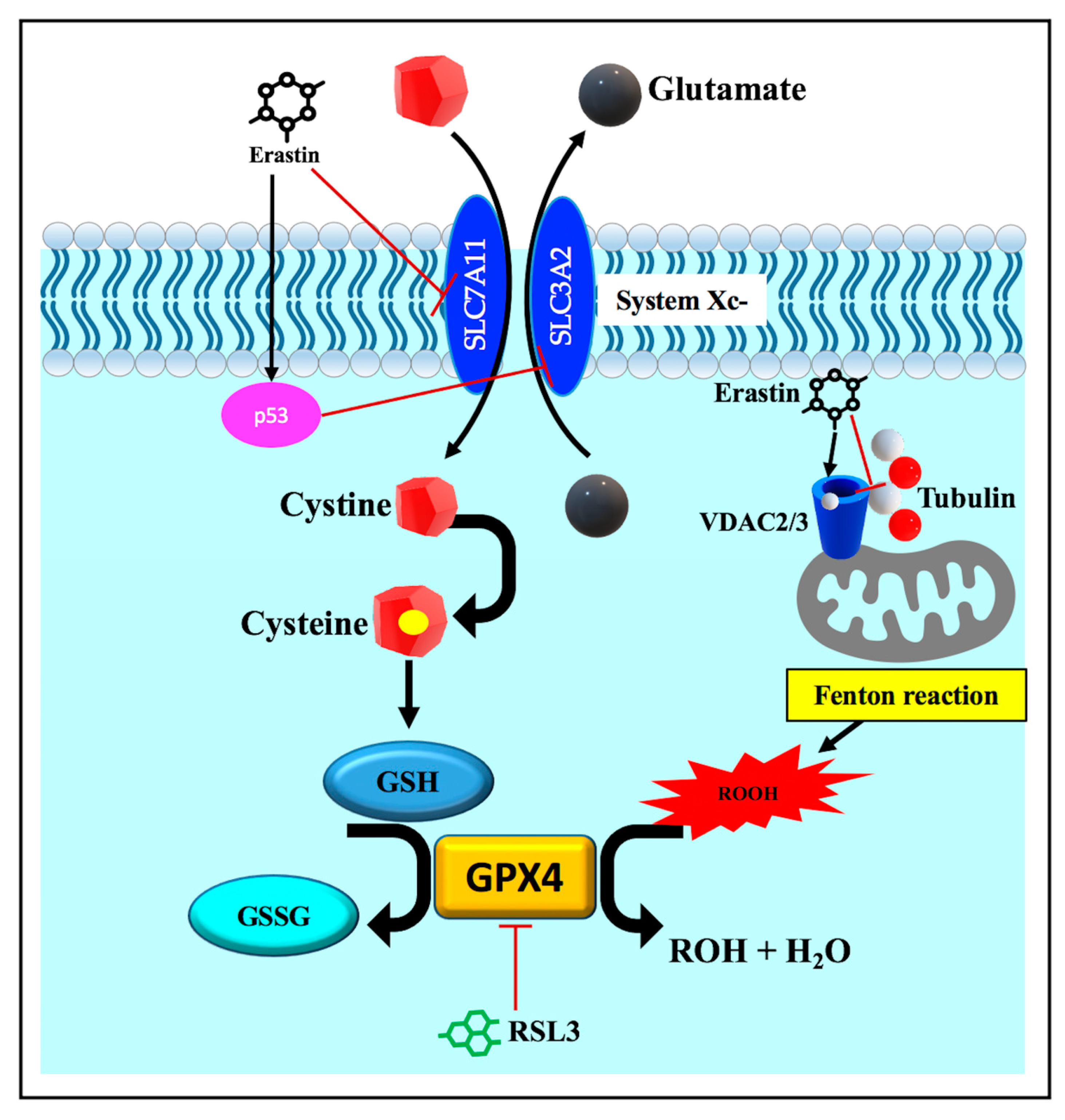
3.3.3. Disruption of Endogenous Redox Homeostasis System, the Xc-GSH-GPX4 Axis
3.3.4. Other Regulators of Ferroptosis
4. The Role of Heat Shock Factor 1 (HSF1) and Heat Shock Proteins (HSPs) in Ferroptosis
4.1. The Function of HSF1 in Ferroptosis
4.2. The Function of HSPs in Ferroptosis
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Joutsen, J.; Sistonen, L. Tailoring of Proteostasis Networks with Heat Shock Factors. Cold Spring Harb. Perspect. Biol. 2019, 11, a034066. [Google Scholar] [CrossRef] [PubMed]
- Carra, S.; Alberti, S.; Arrigo, P.A.; Benesch, J.L.; Benjamin, I.J.; Boelens, W.; Bartelt-Kirbach, B.; Brundel, B.J.J.M.; Buchner, J.; Bukau, B.; et al. The Growing World of Small Heat Shock Proteins: From Structure to Functions. Cell Stress Chaperones 2017, 22, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Jee, H. Size Dependent Classification of Heat Shock Proteins: A Mini-Review. J. Exerc. Rehabil. 2016, 12, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Shan, Q.; Ma, F.; Wei, J.; Li, H.; Ma, H.; Sun, P. Physiological Functions of Heat Shock Proteins. Curr. Protein Pept. Sci. 2020, 21, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular Mechanisms and Health Implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef]
- Lanneau, D.; Wettstein, G.; Bonniaud, P.; Garrido, C. Heat Shock Proteins: Cell Protection through Protein Triage. Sci. World J. 2010, 10, 1543–1552. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Hageman, J.; Vos, M.J.; Kubota, H.; Tanguay, R.M.; Bruford, E.A.; Cheetham, M.E.; Chen, B.; Hightower, L.E. Guidelines for the Nomenclature of the Human Heat Shock Proteins. Cell Stress Chaperones 2009, 14, 105–111. [Google Scholar] [CrossRef]
- Sumi, M.P.; Ghosh, A. Hsp90 in Human Diseases: Molecular Mechanisms to Therapeutic Approaches. Cells 2022, 11, 976. [Google Scholar] [CrossRef]
- Shiber, A.; Ravid, T. Chaperoning Proteins for Destruction: Diverse Roles of Hsp70 Chaperones and Their Co-Chaperones in Targeting Misfolded Proteins to the Proteasome. Biomolecules 2014, 4, 704–724. [Google Scholar] [CrossRef]
- Kriehuber, T.; Rattei, T.; Weinmaier, T.; Bepperling, A.; Haslbeck, M.; Buchner, J. Independent Evolution of the Core Domain and Its Flanking Sequences in Small Heat Shock Proteins. FASEB J. 2010, 24, 3633–3642. [Google Scholar] [CrossRef] [PubMed]
- Strauch, A.; Haslbeck, M. The Function of Small Heat-Shock Proteins and Their Implication in Proteostasis. Essays Biochem. 2016, 60, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, Y.; Zhang, R.; Wang, F.; Wang, T.; Jiao, Y. The Role of Erastin in Ferroptosis and Its Prospects in Cancer Therapy. Onco. Targets. Ther. 2020, 13, 5429–5441. [Google Scholar] [CrossRef]
- Hu, X.; Li, Z.; Lin, R.; Shan, J.; Yu, Q.; Wang, R.; Liao, L.; Yan, W.; Wang, Z.; Shang, L.; et al. Guidelines for Regulated Cell Death Assays: A Systematic Summary, a Categorical Comparison, a Prospective. Front. Cell Dev. Biol. 2021, 9, 634690. [Google Scholar] [CrossRef] [PubMed]
- Hao, S.; Liang, B.; Huang, Q.; Dong, S.; Wu, Z.; He, W.; Shi, M. Metabolic Networks in Ferroptosis (Review). Oncol. Lett. 2018, 15, 5405–5411. [Google Scholar] [CrossRef] [PubMed]
- Fenton, H.J.H. Oxidation of Tartaric Acid in Presence of Iron. J. Chem. Soc., Trans. 1894, 65, 899–910. [Google Scholar] [CrossRef]
- Nie, Q.; Hu, Y.; Yu, X.; Li, X.; Fang, X. Induction and Application of Ferroptosis in Cancer Therapy. Cancer Cell Int. 2022, 22, 12. [Google Scholar] [CrossRef]
- Shintoku, R.; Takigawa, Y.; Yamada, K.; Kubota, C.; Yoshimoto, Y.; Takeuchi, T.; Koshiishi, I.; Torii, S. Lipoxygenase-mediated Generation of Lipid Peroxides Enhances Ferroptosis Induced by Erastin and RSL3. Cancer Sci. 2017, 108, 2187–2194. [Google Scholar] [CrossRef]
- Zou, Y.; Li, H.; Graham, E.T.; Deik, A.A.; Eaton, J.K.; Wang, W.; Sandoval-Gomez, G.; Clish, C.B.; Doench, J.G.; Schreiber, S.L. Cytochrome P450 Oxidoreductase Contributes to Phospholipid Peroxidation in Ferroptosis. Nat. Chem. Biol. 2020, 16, 302–309. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. Identification of ACSL4 as a Biomarker and Contributor of Ferroptosis. Biochem. Biophys. Res. Commun. 2016, 478, 1338–1343. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 Dictates Ferroptosis Sensitivity by Shaping Cellular Lipid Composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Chu, B.; Kon, N.; Chen, D.; Li, T.; Liu, T.; Jiang, L.; Song, S.; Tavana, O.; Gu, W. ALOX12 Is Required for P53-Mediated Tumour Suppression through a Distinct Ferroptosis Pathway. Nat. Cell Biol. 2019, 21, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ Oxidoreductase FSP1 Acts Parallel to GPX4 to Inhibit Ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, M.; Freigang, S.; Schneider, C.; Conrad, M.; Bornkamm, G.W.; Kopf, M. T Cell Lipid Peroxidation Induces Ferroptosis and Prevents Immunity to Infection. J. Exp. Med. 2015, 212, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Yiannikourides, A.; Latunde-Dada, G. A Short Review of Iron Metabolism and Pathophysiology of Iron Disorders. Medicines 2019, 6, 85. [Google Scholar] [CrossRef]
- Xu, T.; Ding, W.; Ji, X.; Ao, X.; Liu, Y.; Yu, W.; Wang, J. Molecular Mechanisms of Ferroptosis and Its Role in Cancer Therapy. J. Cell. Mol. Med. 2019, 23, 4900–4912. [Google Scholar] [CrossRef]
- Pantopoulos, K.; Porwal, S.K.; Tartakoff, A.; Devireddy, L. Mechanisms of Mammalian Iron Homeostasis. Biochemistry 2012, 51, 5705–5724. [Google Scholar] [CrossRef]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the P62-Keap1-NRF2 Pathway Protects against Ferroptosis in Hepatocellular Carcinoma Cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.-Y.; Park, E.; Lee, S.-J.; Chung, S.W. Heme Oxygenase-1 Accelerates Erastin-Induced Ferroptotic Cell Death. Oncotarget 2015, 6, 24393–24403. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Henson, E.S.; Chen, Y.; Gibson, S.B. Ferroptosis Is Induced Following Siramesine and Lapatinib Treatment of Breast Cancer Cells. Cell Death Dis. 2016, 7, e2307. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-H.; Wu, J.; Ding, C.-K.C.; Lin, C.-C.; Pan, S.; Bossa, N.; Xu, Y.; Yang, W.-H.; Mathey-Prevot, B.; Chi, J.-T. Kinome Screen of Ferroptosis Reveals a Novel Role of ATM in Regulating Iron Metabolism. Cell Death Differ. 2020, 27, 1008–1022. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Xu, F.; Lu, H. LncRNA PVT1 Regulates Ferroptosis through MiR-214-Mediated TFR1 and P53. Life Sci. 2020, 260, 118305. [Google Scholar] [CrossRef]
- Wu, H.; Wang, F.; Ta, N.; Zhang, T.; Gao, W. The Multifaceted Regulation of Mitochondria in Ferroptosis. Life 2021, 11, 222. [Google Scholar] [CrossRef]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Autophagy Promotes Ferroptosis by Degradation of Ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef]
- Park, E.; Chung, S.W. ROS-Mediated Autophagy Increases Intracellular Iron Levels and Ferroptosis by Ferritin and Transferrin Receptor Regulation. Cell Death Dis. 2019, 10, 822. [Google Scholar] [CrossRef]
- Dixon, S.J.; Stockwell, B.R. The Role of Iron and Reactive Oxygen Species in Cell Death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized Arachidonic and Adrenic PEs Navigate Cells to Ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Li, Y.; Feng, D.; Wang, Z.; Zhao, Y.; Sun, R.; Tian, D.; Liu, D.; Zhang, F.; Ning, S.; Yao, J.; et al. Ischemia-Induced ACSL4 Activation Contributes to Ferroptosis-Mediated Tissue Injury in Intestinal Ischemia/Reperfusion. Cell Death Differ. 2019, 26, 2284–2299. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Winter, G.E.; Musavi, L.S.; Lee, E.D.; Snijder, B.; Rebsamen, M.; Superti-Furga, G.; Stockwell, B.R. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem. Biol. 2015, 10, 1604–1609. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.A.; Magtanong, L.; Dixon, S.J.; Watts, J.L. Dietary Lipids Induce Ferroptosis in Caenorhabditis Elegans and Human Cancer Cells. Dev. Cell 2020, 54, 447–454.e4. [Google Scholar] [CrossRef]
- Magtanong, L.; Ko, P.-J.; To, M.; Cao, J.Y.; Forcina, G.C.; Tarangelo, A.; Ward, C.C.; Cho, K.; Patti, G.J.; Nomura, D.K.; et al. Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem. Biol. 2019, 26, 420–432.e9. [Google Scholar] [CrossRef]
- Ubellacker, J.M.; Tasdogan, A.; Ramesh, V.; Shen, B.; Mitchell, E.C.; Martin-Sandoval, M.S.; Gu, Z.; McCormick, M.L.; Durham, A.B.; Spitz, D.R.; et al. Lymph Protects Metastasizing Melanoma Cells from Ferroptosis. Nature 2020, 585, 113–118. [Google Scholar] [CrossRef]
- Magtanong, L.; Ko, P.J.; Dixon, S.J. Emerging Roles for Lipids in Non-Apoptotic Cell Death. Cell Death Differ. 2016, 23, 1099–1109. [Google Scholar] [CrossRef]
- Chen, X.; Comish, P.B.; Tang, D.; Kang, R. Characteristics and Biomarkers of Ferroptosis. Front. Cell Dev. Biol. 2021, 9, 637162. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Liu, T.; Wang, H.; Xue, L.; Wang, J. Fatty Acids Metabolism: The Bridge between Ferroptosis and Ionizing Radiation. Front. Cell Dev. Biol. 2021, 9, 675617. [Google Scholar] [CrossRef]
- Kuang, F.; Liu, J.; Xie, Y.; Tang, D.; Kang, R. MGST1 Is a Redox-Sensitive Repressor of Ferroptosis in Pancreatic Cancer Cells. Cell Chem. Biol. 2021, 28, 765–775.e5. [Google Scholar] [CrossRef]
- Yan, B.; Ai, Y.; Sun, Q.; Ma, Y.; Cao, Y.; Wang, J.; Zhang, Z.; Wang, X. Membrane Damage during Ferroptosis Is Caused by Oxidation of Phospholipids Catalyzed by the Oxidoreductases POR and CYB5R1. Mol. Cell 2021, 81, 355–369.e10. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Kim, W.K.; Bae, K.-H.; Lee, S.C.; Lee, E.-W. Lipid Metabolism and Ferroptosis. Biology 2021, 10, 184. [Google Scholar] [CrossRef] [PubMed]
- Gaschler, M.M.; Stockwell, B.R. Lipid Peroxidation in Cell Death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Yu, Y.; Yan, Y.; Niu, F.; Wang, Y.; Chen, X.; Su, G.; Liu, Y.; Zhao, X.; Qian, L.; Liu, P.; et al. Ferroptosis: A Cell Death Connecting Oxidative Stress, Inflammation and Cardiovascular Diseases. Cell Death Discov. 2021, 7, 193. [Google Scholar] [CrossRef] [PubMed]
- Pedrera, L.; Espiritu, R.A.; Ros, U.; Weber, J.; Schmitt, A.; Stroh, J.; Hailfinger, S.; von Karstedt, S.; García-Sáez, A.J. Ferroptotic Pores Induce Ca2+ Fluxes and ESCRT-III Activation to Modulate Cell Death Kinetics. Cell Death Differ. 2021, 28, 1644–1657. [Google Scholar] [CrossRef]
- Hassannia, B.; Vandenabeele, P.; Vanden Berghe, T. Targeting Ferroptosis to Iron out Cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef]
- Li, F.-J.; Long, H.-Z.; Zhou, Z.-W.; Luo, H.-Y.; Xu, S.-G.; Gao, L.-C. System Xc−/GSH/GPX4 Axis: An Important Antioxidant System for the Ferroptosis in Drug-Resistant Solid Tumor Therapy. Front. Pharmacol. 2022, 13, 910292. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.-J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a P53-Mediated Activity during Tumour Suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological Inhibition of Cystine–Glutamate Exchange Induces Endoplasmic Reticulum Stress and Ferroptosis. Elife 2014, 3, e02523. [Google Scholar] [CrossRef]
- Woo, J.H.; Shimoni, Y.; Yang, W.S.; Subramaniam, P.; Iyer, A.; Nicoletti, P.; Rodríguez Martínez, M.; López, G.; Mattioli, M.; Realubit, R.; et al. Elucidating Compound Mechanism of Action by Network Perturbation Analysis. Cell 2015, 162, 441–451. [Google Scholar] [CrossRef]
- Seiler, A.; Schneider, M.; Förster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Rådmark, O.; Wurst, W.; et al. Glutathione Peroxidase 4 Senses and Translates Oxidative Stress into 12/15-Lipoxygenase Dependent- and AIF-Mediated Cell Death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Imai, H.; Matsuoka, M.; Kumagai, T.; Sakamoto, T.; Koumura, T. Lipid Peroxidation-Dependent Cell Death Regulated by GPx4 and Ferroptosis. In Current Topics in Microbiology and Immunology; Springer: Cham, Swizterland, 2016; pp. 143–170. [Google Scholar]
- Ursini, F.; Maiorino, M. Lipid Peroxidation and Ferroptosis: The Role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Hadian, K. Ferroptosis Suppressor Protein 1 (FSP1) and Coenzyme Q 10 Cooperatively Suppress Ferroptosis. Biochemistry 2020, 59, 637–638. [Google Scholar] [CrossRef]
- DeHart, D.N.; Fang, D.; Heslop, K.; Li, L.; Lemasters, J.J.; Maldonado, E.N. Opening of Voltage Dependent Anion Channels Promotes Reactive Oxygen Species Generation, Mitochondrial Dysfunction and Cell Death in Cancer Cells. Biochem. Pharmacol. 2018, 148, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, E.N.; Sheldon, K.L.; DeHart, D.N.; Patnaik, J.; Manevich, Y.; Townsend, D.M.; Bezrukov, S.M.; Rostovtseva, T.K.; Lemasters, J.J. Voltage-Dependent Anion Channels Modulate Mitochondrial Metabolism in Cancer Cells. J. Biol. Chem. 2013, 288, 11920–11929. [Google Scholar] [CrossRef]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS–RAF–MEK-Dependent Oxidative Cell Death Involving Voltage-Dependent Anion Channels. Nature 2007, 447, 865–869. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Synthetic Lethal Screening Identifies Compounds Activating Iron-Dependent, Nonapoptotic Cell Death in Oncogenic-RAS-Harboring Cancer Cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef]
- Nagpal, A.; Redvers, R.P.; Ling, X.; Ayton, S.; Fuentes, M.; Tavancheh, E.; Diala, I.; Lalani, A.; Loi, S.; David, S.; et al. Neoadjuvant Neratinib Promotes Ferroptosis and Inhibits Brain Metastasis in a Novel Syngeneic Model of Spontaneous HER2+ve Breast Cancer Metastasis. Breast Cancer Res. 2019, 21, 94. [Google Scholar] [CrossRef]
- Hassannia, B.; Wiernicki, B.; Ingold, I.; Qu, F.; Van Herck, S.; Tyurina, Y.Y.; Bayır, H.; Abhari, B.A.; Angeli, J.P.F.; Choi, S.M.; et al. Nano-Targeted Induction of Dual Ferroptotic Mechanisms Eradicates High-Risk Neuroblastoma. J. Clin. Investig. 2018, 128, 3341–3355. [Google Scholar] [CrossRef]
- Lou, J.-S.; Zhao, L.-P.; Huang, Z.-H.; Chen, X.-Y.; Xu, J.-T.; TAI, W.C.-S.; Tsim, K.W.K.; Chen, Y.-T.; Xie, T. Ginkgetin Derived from Ginkgo Biloba Leaves Enhances the Therapeutic Effect of Cisplatin via Ferroptosis-Mediated Disruption of the Nrf2/HO-1 Axis in EGFR Wild-Type Non-Small-Cell Lung Cancer. Phytomedicine 2021, 80, 153370. [Google Scholar] [CrossRef]
- Mao, C.; Wang, X.; Liu, Y.; Wang, M.; Yan, B.; Jiang, Y.; Shi, Y.; Shen, Y.; Liu, X.; Lai, W.; et al. A G3BP1-Interacting LncRNA Promotes Ferroptosis and Apoptosis in Cancer via Nuclear Equestration of P53. Cancer Res. 2018, 78, 3484–3496. [Google Scholar] [CrossRef] [PubMed]
- Probst, L.; Dächert, J.; Schenk, B.; Fulda, S. Lipoxygenase Inhibitors Protect Acute Lymphoblastic Leukemia Cells from Ferroptotic Cell Death. Biochem. Pharmacol. 2017, 140, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Ou, Z.; Xie, M.; Kang, R.; Fan, Y.; Niu, X.; Wang, H.; Cao, L.; Tang, D. HSPB1 as a Novel Regulator of Ferroptotic Cancer Cell Death. Oncogene 2015, 34, 5617–5625. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Zhang, Q.; Sun, X.; Zeh, H.J.; Lotze, M.T.; Kang, R.; Tang, D. HSPA5 Regulates Ferroptotic Cell Death in Cancer Cells. Cancer Res. 2017, 77, 2064–2077. [Google Scholar] [CrossRef]
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent. Sci. 2018, 4, 387–396. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, W.; Li, Y.; Xiao, Y.; Cheng, J.; Jia, J. The 5-Lipoxygenase Inhibitor Zileuton Confers Neuroprotection against Glutamate Oxidative Damage by Inhibiting Ferroptosis. Biol. Pharm. Bull. 2015, 38, 1234–1239. [Google Scholar] [CrossRef]
- Wu, Z.; Geng, Y.; Lu, X.; Shi, Y.; Wu, G.; Zhang, M.; Shan, B.; Pan, H.; Yuan, J. Chaperone-Mediated Autophagy Is Involved in the Execution of Ferroptosis. Proc. Natl. Acad. Sci. USA 2019, 116, 2996–3005. [Google Scholar] [CrossRef]
- Toma-Jonik, A.; Vydra, N.; Janus, P.; Widłak, W. Interplay between HSF1 and P53 Signaling Pathways in Cancer Initiation and Progression: Non-Oncogene and Oncogene Addiction. Cell. Oncol. 2019, 42, 579–589. [Google Scholar] [CrossRef]
- Jiang, B.; Zhao, Y.; Shi, M.; Song, L.; Wang, Q.; Qin, Q.; Song, X.; Wu, S.; Fang, Z.; Liu, X. DNAJB6 Promotes Ferroptosis in Esophageal Squamous Cell Carcinoma. Dig Dis Sci. 2020, 65, 999–2008. [Google Scholar] [CrossRef]
- Cao, H.; Zuo, C.; Huang, Y.; Zhu, L.; Zhao, J.; Yang, Y.; Jiang, Y.; Wang, F. Hippocampal Proteomic Analysis Reveals Activation of Necroptosis and Ferroptosis in a Mouse Model of Chronic Unpredictable Mild Stress-Induced Depression. Behav. Brain Res. 2021, 407, 113261. [Google Scholar] [CrossRef]
- Liu, M.; Fan, Y.; Li, D.; Han, B.; Meng, Y.; Chen, F.; Liu, T.; Song, Z.; Han, Y.; Huang, L.; et al. Ferroptosis Inducer Erastin Sensitizes NSCLC Cells to Celastrol through Activation of the ROS–Mitochondrial Fission–Mitophagy Axis. Mol. Oncol. 2021, 15, 2084–2105. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Ma, H.; Li, J.; Meng, C.; Zou, J.; Wang, H.; Liu, K.; Liu, M.; Xiao, X.; Zhang, H.; et al. HSF1 Functions as a Key Defender against Palmitic Acid-Induced Ferroptosis in Cardiomyocytes. J. Mol. Cell. Cardiol. 2021, 150, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Mi, Y.; Zhang, X.; Ma, Q.; Song, Y.; Zhang, L.; Wang, D.; Xing, J.; Hou, B.; Li, H.; et al. Dihydroartemisinin-Induced Unfolded Protein Response Feedback Attenuates Ferroptosis via PERK/ATF4/HSPA5 Pathway in Glioma Cells. J. Exp. Clin. Cancer Res. 2019, 38, 402. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wang, D.; Yu, Y.; Zhao, T.; Min, N.; Wu, Y.; Kang, L.; Zhao, Y.; Du, L.; Zhang, M.; et al. Legumain Promotes Tubular Ferroptosis by Facilitating Chaperone-Mediated Autophagy of GPX4 in AKI. Cell Death Dis. 2021, 12, 65. [Google Scholar] [CrossRef]
- Chen, H.; Zheng, C.; Zhang, Y.; Chang, Y.-Z.; Qian, Z.-M.; Shen, X. Heat Shock Protein 27 Downregulates the Transferrin Receptor 1-Mediated Iron Uptake. Int. J. Biochem. Cell Biol. 2006, 38, 1402–1416. [Google Scholar] [CrossRef]
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X.; et al. Ferroptosis as a Target for Protection against Cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680. [Google Scholar] [CrossRef]
- Lin, H.; Chen, X.; Zhang, C.; Yang, T.; Deng, Z.; Song, Y.; Huang, L.; Li, F.; Li, Q.; Lin, S.; et al. EF24 Induces Ferroptosis in Osteosarcoma Cells through HMOX1. Biomed. Pharmacother. 2021, 136, 111202. [Google Scholar] [CrossRef]
- Tang, Z.; Ju, Y.; Dai, X.; Ni, N.; Liu, Y.; Zhang, D.; Gao, H.; Sun, H.; Zhang, J.; Gu, P. HO-1-Mediated Ferroptosis as a Target for Protection against Retinal Pigment Epithelium Degeneration. Redox Biol. 2021, 43, 101971. [Google Scholar] [CrossRef]
- Meng, Z.; Liang, H.; Zhao, J.; Gao, J.; Liu, C.; Ma, X.; Liu, J.; Liang, B.; Jiao, X.; Cao, J.; et al. HMOX1 Upregulation Promotes Ferroptosis in Diabetic Atherosclerosis. Life Sci. 2021, 284, 119935. [Google Scholar] [CrossRef]
- Adedoyin, O.; Boddu, R.; Traylor, A.; Lever, J.M.; Bolisetty, S.; George, J.F.; Agarwal, A. Heme Oxygenase-1 Mitigates Ferroptosis in Renal Proximal Tubule Cells. Am. J. Physiol. Physiol. 2018, 314, F702–F714. [Google Scholar] [CrossRef]
- Nitti, M.; Piras, S.; Brondolo, L.; Marinari, U.; Pronzato, M.; Furfaro, A. Heme Oxygenase 1 in the Nervous System: Does It Favor Neuronal Cell Survival or Induce Neurodegeneration? Int. J. Mol. Sci. 2018, 19, 2260. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gong, W.; Wu, S.; Perrett, S. Hsp70 in Redox Homeostasis. Cells 2022, 11, 829. [Google Scholar] [CrossRef]
- Picard, D. Heat-Shock Protein 90, a Chaperone for Folding and Regulation. Cell. Mol. Life Sci. 2002, 59, 1640–1648. [Google Scholar] [CrossRef] [PubMed]
- Wettstein, G.; Bellaye, P.S.; Micheau, O.; Bonniaud, P. Small Heat Shock Proteins and the Cytoskeleton: An Essential Interplay for Cell Integrity? Int. J. Biochem. Cell Biol. 2012, 44, 1680–1686. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Gong, Y.; Wan, J.; Tang, J.; Zhang, Q. Trends in HSPB5 Research: A 36-Year Bibliometric Analysis. Cell Stress Chaperones 2021, 26, 799–810. [Google Scholar] [CrossRef] [PubMed]
- Alam, J.; Cook, J.L. How Many Transcription Factors Does It Take to Turn On the Heme Oxygenase-1 Gene? Am. J. Respir. Cell Mol. Biol. 2007, 36, 166–174. [Google Scholar] [CrossRef]
- Syapin, P.J. Regulation of Haeme Oxygenase-1 for Treatment of Neuroinflammation and Brain Disorders. Br. J. Pharmacol. 2008, 155, 623–640. [Google Scholar] [CrossRef]
- Poss, K.D.; Tonegawa, S. Heme Oxygenase 1 Is Required for Mammalian Iron Reutilization. Proc. Natl. Acad. Sci. USA 1997, 94, 10919–10924. [Google Scholar] [CrossRef]
- Bolisetty, S.; Zarjou, A.; Agarwal, A. Heme Oxygenase 1 as a Therapeutic Target in Acute Kidney Injury. Am. J. Kidney Dis. 2017, 69, 531–545. [Google Scholar] [CrossRef]
- Chiang, S.-K.; Chen, S.-E.; Chang, L.-C. A Dual Role of Heme Oxygenase-1 in Cancer Cells. Int. J. Mol. Sci. 2018, 20, 39. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year of Identification | Ferroptosis Modulator | Function | Reference | Example on Pharmacological Modulator |
|---|---|---|---|---|
| 2007 | VDAC2/3 | Mitochondrial transmembrane channels | [67] | Erastin & Piperazine [5,21] |
| Mutated RAS | Oncogene | [67] | Erastin [5] | |
| 2008 | TF | Cellular iron uptake | [68] | Siramesine [33] Lapatinib [33] Neratinib [69] |
| 2012 (Identification of ferroptosis) | SLC7A11 | Drives cystine glutamate exchange through System Xc- | [5] | Erastin & Piperazine [37] Sulfasalazine [5] Sorafenib & Glutamate [59] |
| 2014 | GPX4 | Glutathione peroxidase involved in reduction in lipid peroxides | [21] | RSL3 [42] Altretamine [60] Withaferin A [70] DPI7 & Cisplatin [71] |
| 2015 | P53 | Transcription factor | [58] | LncRNA P53RRA [72] |
| ALOX12 | Lipoxygenase | [24] | Baicalein [73] | |
| HSPB1 | Molecular chaperone | [74] | Erastin [74] | |
| FTH1 | Intracellular iron storage protein | [31] | - | |
| LPCAT | Lipogenesis | [42] | RSL3 & DPI7 [42] | |
| 2016 | ACSL4 | Lipogenesis | [22] | RSL3 & DPI7 [42] |
| 2017 | HSPA5 | Molecular chaperone | [75] | - |
| FPN | Iron transporter | [33] | - | |
| 2018 | ALOXs | Lipoxygenases | [76] | Zileuton [77] |
| 2019 | HSP90 | Molecular chaperone | [78] | 2-amino-5-chloro-N,3-dimethylbenzamide (CDDO) [78] |
| HSF1 | Heat shock transcription factor | [79] | Erastin [74] | |
| 2020 | DNAJB6 | Molecular chaperone | [80] | - |
| FSP1 & CoQ10 | Antioxidant system | [64] | Altretamine & FIN56 [60] | |
| 2021 | HSPB5 | Molecular chaperone | [81] | - |
| Protein | Effect | Mechanism of Action | Reference |
|---|---|---|---|
| HSF1 | Anti-ferroptotic | Erastin- and celastro-mediated ferroptosis is associated with increased ROS production, mitochondrial fission, autophagy, mitophagy, and transcriptional activation of HSF1. HSF1 reduces cellular sensitivity to ferroptosis induced by erastin and celastrol. | [82] |
| HSF1 interacts with p53 transcription factor, supporting its transcriptional regulatory function in the cell cycle. | [79] | ||
| Palmitic acid-induced ferroptosis is accompanied by reduced expression of HSF1 and GPX4. HSF1 protects against ferroptosis via maintenance of iron homeostasis and activation of GPX4 in palmitic acid-induced ferroptosis. | [83] | ||
| HSP70 | Anti-ferroptotic | HSP70 protects GPX4 from degradation and potentiates its expression. This happens alongside the increased expression of HSPA5 (also known as BIP or GRP78). | [75,84] |
| HSP40 | Pro-ferroptotic | HSP40 induction in esophageal squamous cell carcinoma is associated with decline in GSH levels and downregulation of GPX4. | [80] |
| HSP90 | Pro-ferroptotic | HSP90, alongside HSPA8 (also known as HSC70), stimulates CMA-dependent lysosomal degradation of GPX4. | [78,85] |
| HSPB1 (also known as HSP27 and HSP25) | Anti-ferroptotic | HSPB1 mediates stabilization of the cytoskeleton and reduces cellular iron uptake through TFR1. | [86] |
| Hsp27 phosphorylation by PKC inhibits ferroptosis by affecting iron metabolism. It decreases iron-dependent production of lipid ROS. | [74] | ||
| HSPB5 (also known as CRYAB) | Probably anti-ferroptotic | Chronic depression is linked to ferroptotic neuronal death and reduced expression of HSPB5. The mechanism of HSPB5 involvement in ferroptotic neuronal death is still unknown. | [81] |
| HO-1 (also known as Hsp 32) | Pro-ferroptotic | Doxorubicin-mediated ferroptosis in cardiomyocytes induces HO-1, which increases free iron production causing further lipid peroxidation of mitochondrial membrane. | [87] |
| EF24-mediated ferroptosis in osteogenic sarcoma cells induces HMOX1 and increases malondialdehyde and ROS generation. | [88] | ||
| Sodium-iodate-induced ferroptosis in retinal pigmented epithelium upregulates HO-1 with consequent free iron buildup and lipid peroxidation. | [89] | ||
| Diabetic atherosclerosis is accompanied by HMOX1 induction that causes free iron accumulation, and further ROS production, and lipid peroxidation. | [90] | ||
| Anti-ferroptotic | HO-1 inhibits erastin- or RSL3-induced ferroptosis in renal epithelial cells. The potential mechanism for HO-1 anti-ferroptotic effect in this model is that HO-1 upregulation provides antioxidant and cytoprotective effects in response to stressful stimuli. | [91] | |
| In neuronal cells, activation of HO-1 shows anti-ferroptotic function due to its antioxidant and anti-inflammatory function. | [92] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aolymat, I.; Hatmal, M.M.; Olaimat, A.N. The Emerging Role of Heat Shock Factor 1 (HSF1) and Heat Shock Proteins (HSPs) in Ferroptosis. Pathophysiology 2023, 30, 63-82. https://doi.org/10.3390/pathophysiology30010007
Aolymat I, Hatmal MM, Olaimat AN. The Emerging Role of Heat Shock Factor 1 (HSF1) and Heat Shock Proteins (HSPs) in Ferroptosis. Pathophysiology. 2023; 30(1):63-82. https://doi.org/10.3390/pathophysiology30010007
Chicago/Turabian StyleAolymat, Iman, Ma’mon M. Hatmal, and Amin N. Olaimat. 2023. "The Emerging Role of Heat Shock Factor 1 (HSF1) and Heat Shock Proteins (HSPs) in Ferroptosis" Pathophysiology 30, no. 1: 63-82. https://doi.org/10.3390/pathophysiology30010007
APA StyleAolymat, I., Hatmal, M. M., & Olaimat, A. N. (2023). The Emerging Role of Heat Shock Factor 1 (HSF1) and Heat Shock Proteins (HSPs) in Ferroptosis. Pathophysiology, 30(1), 63-82. https://doi.org/10.3390/pathophysiology30010007