
The Role of Exposomes in the Pathophysiology of Autoimmune Diseases II: Pathogens



Abstract
1. Introduction
- (1)
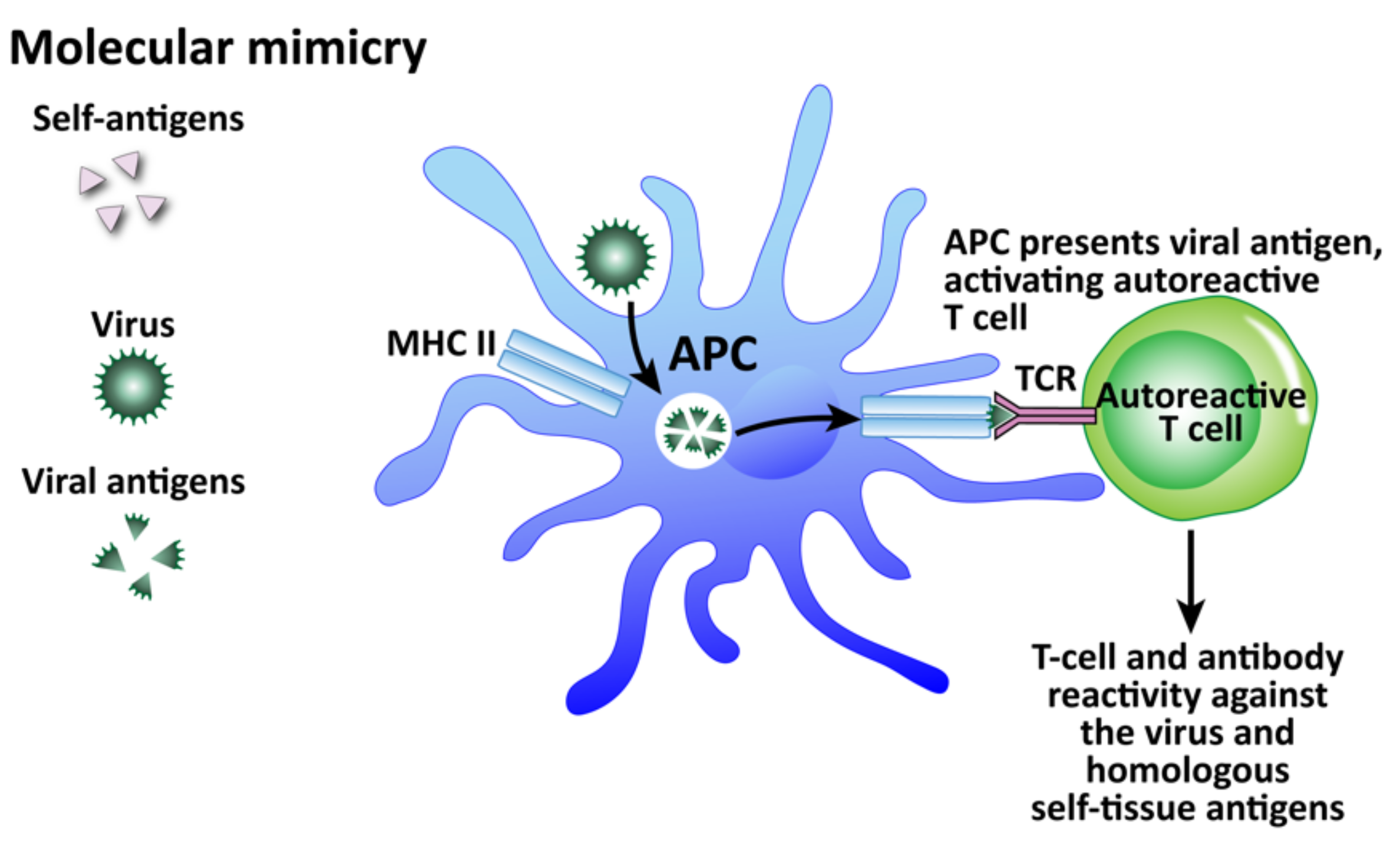
- Molecular mimicry
- (2)
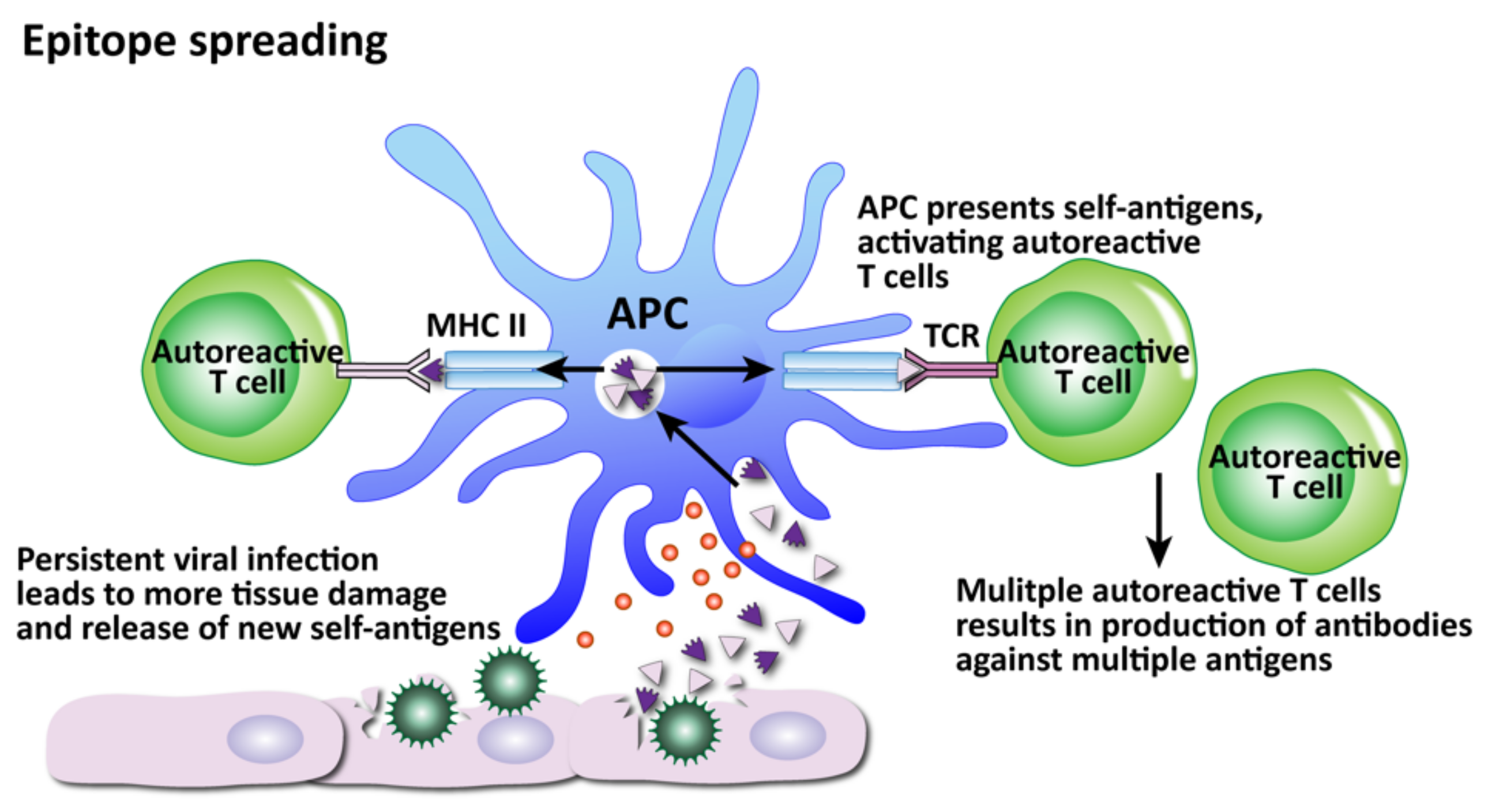
- Epitope spreading
- (3)
- Viral persistence
- (4)
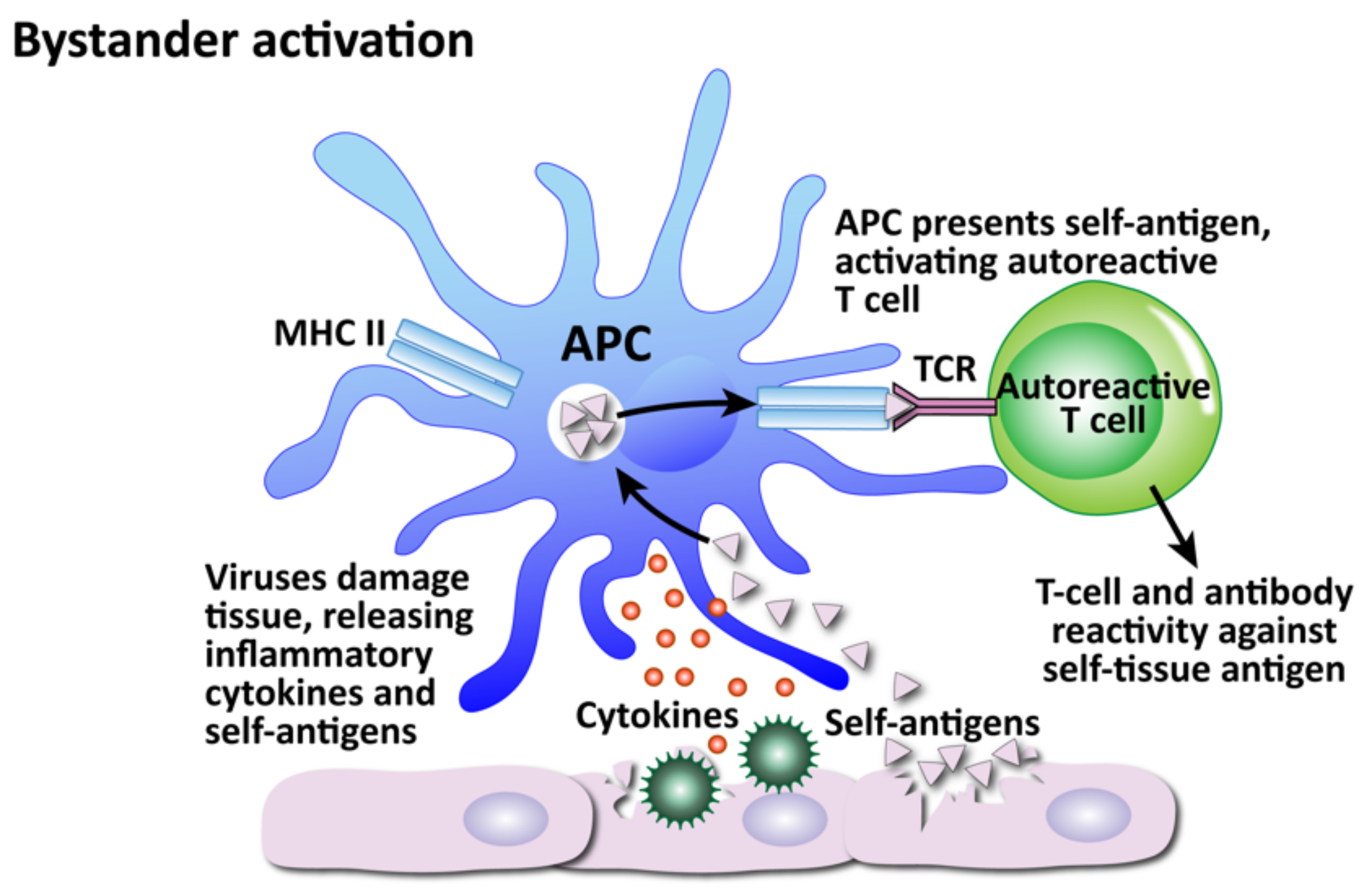
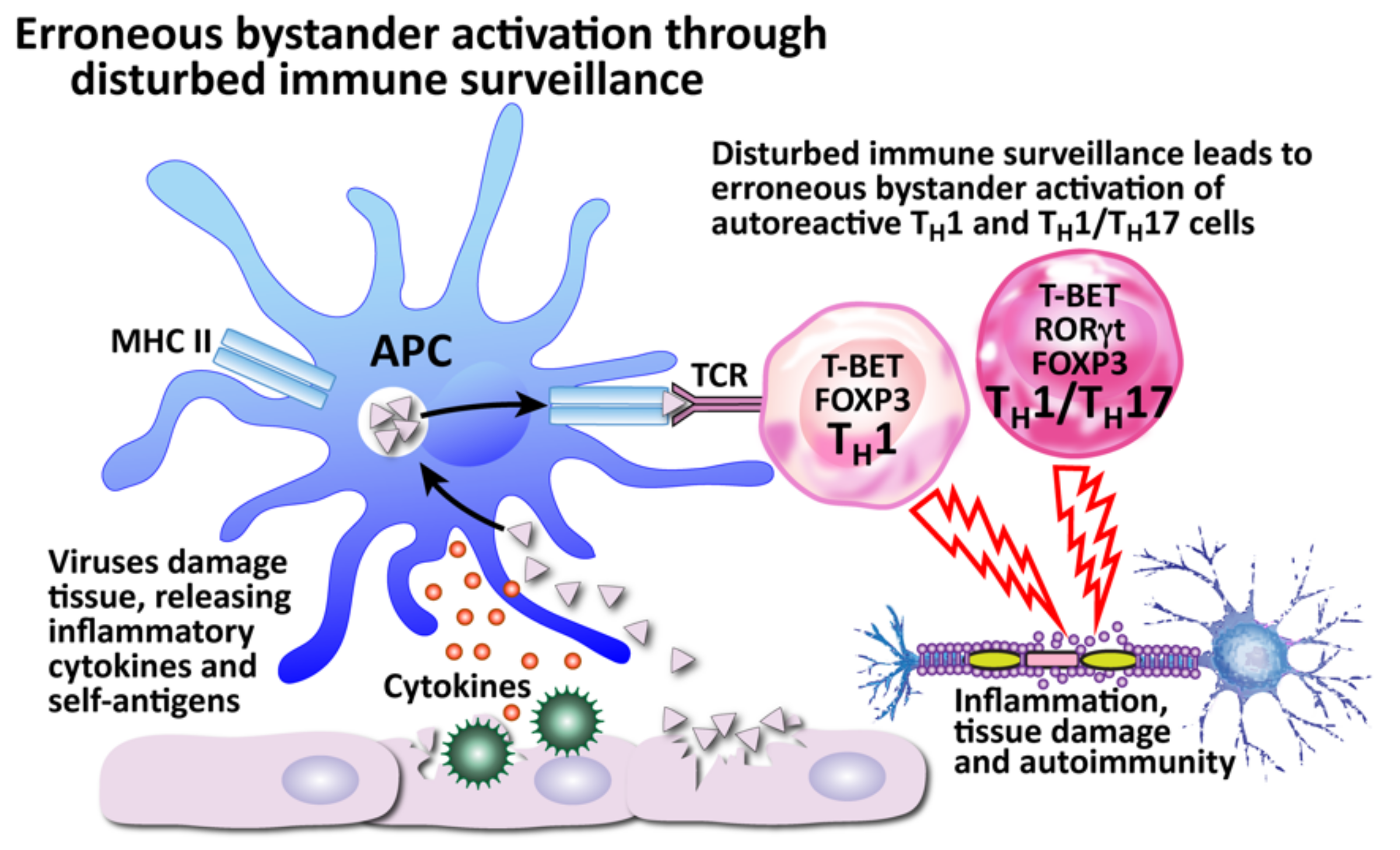
- Bystander activation
- (5)
- Polyclonal activation
- (6)
- Autoinflammatory activation of innate immunity
- (7)
- Dysregulation of immune homeostasis
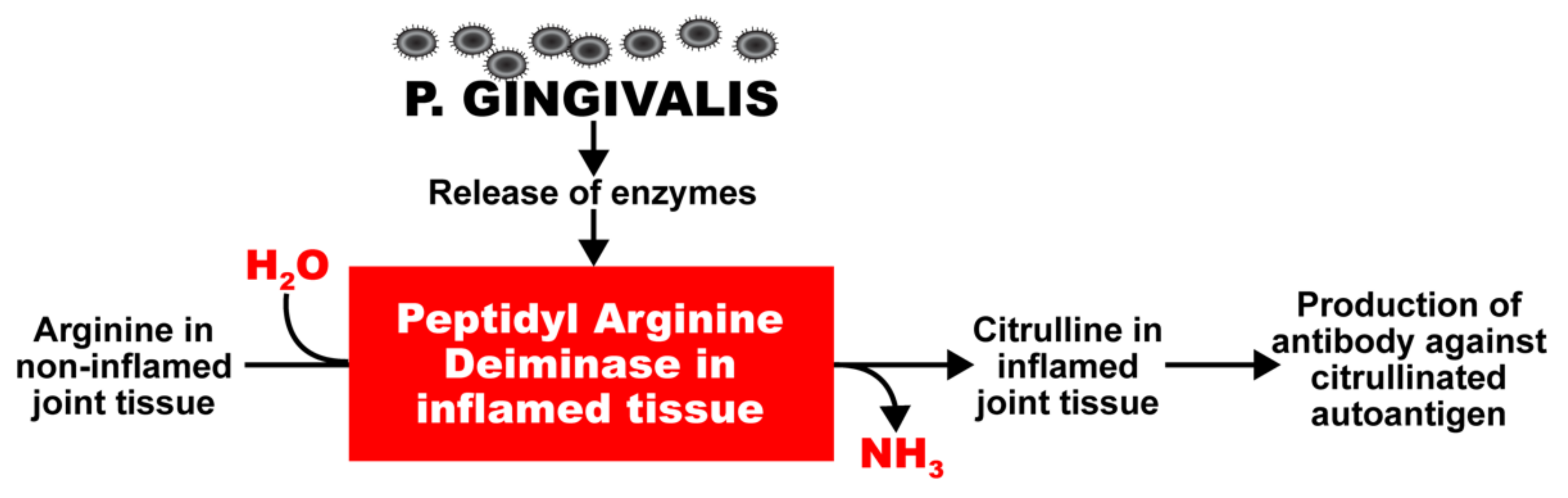
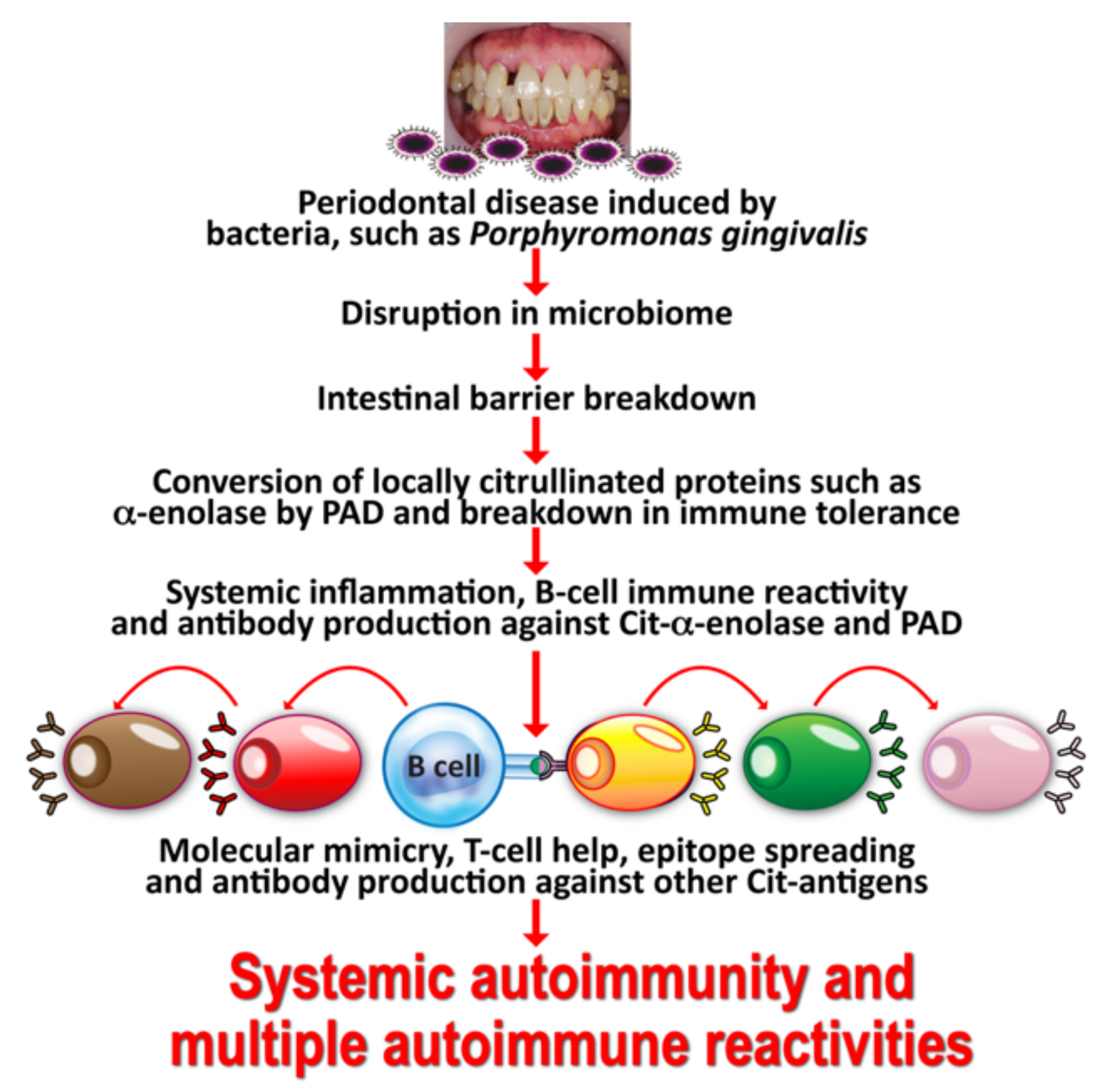
2. Oral Pathogens and Autoimmunity
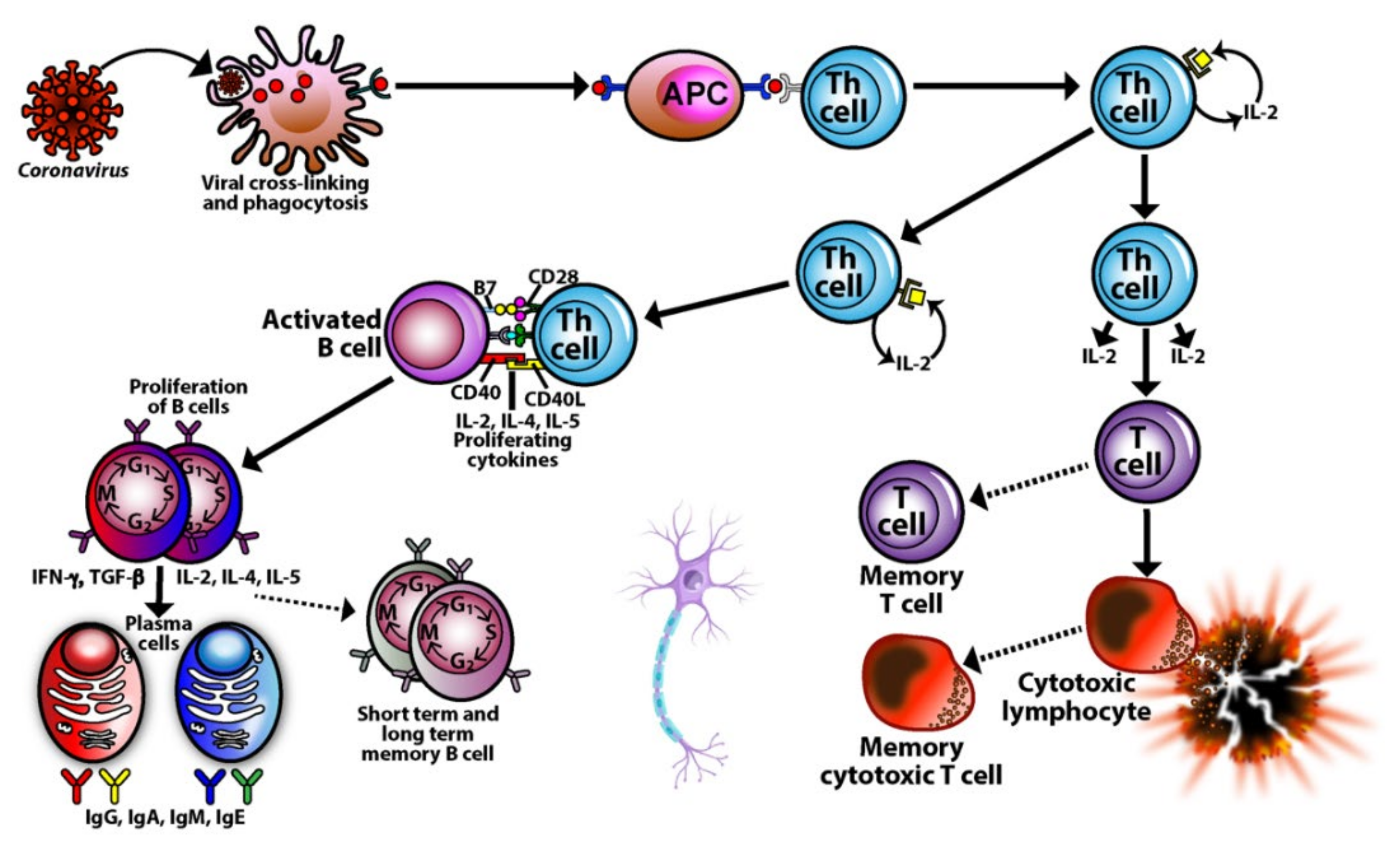
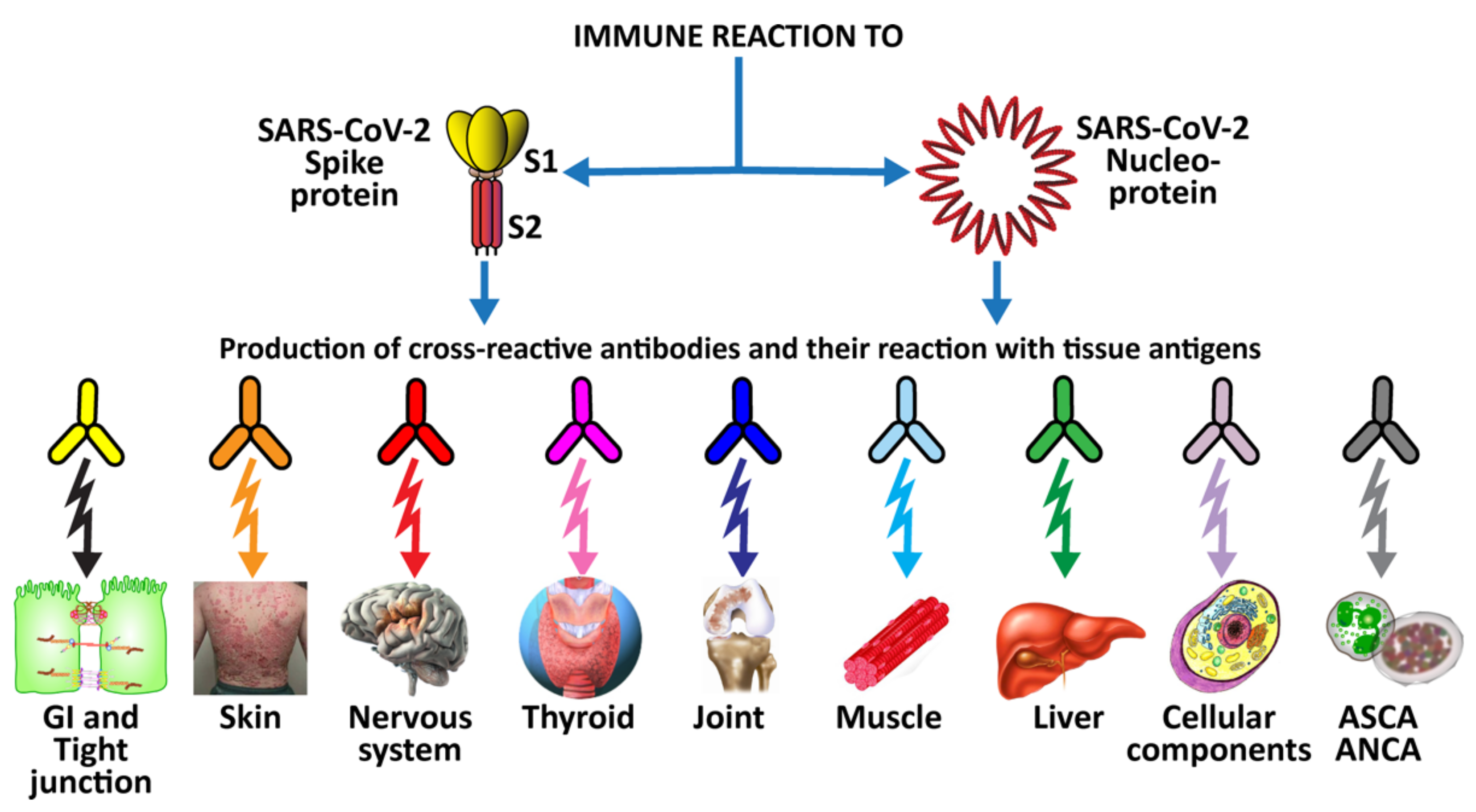
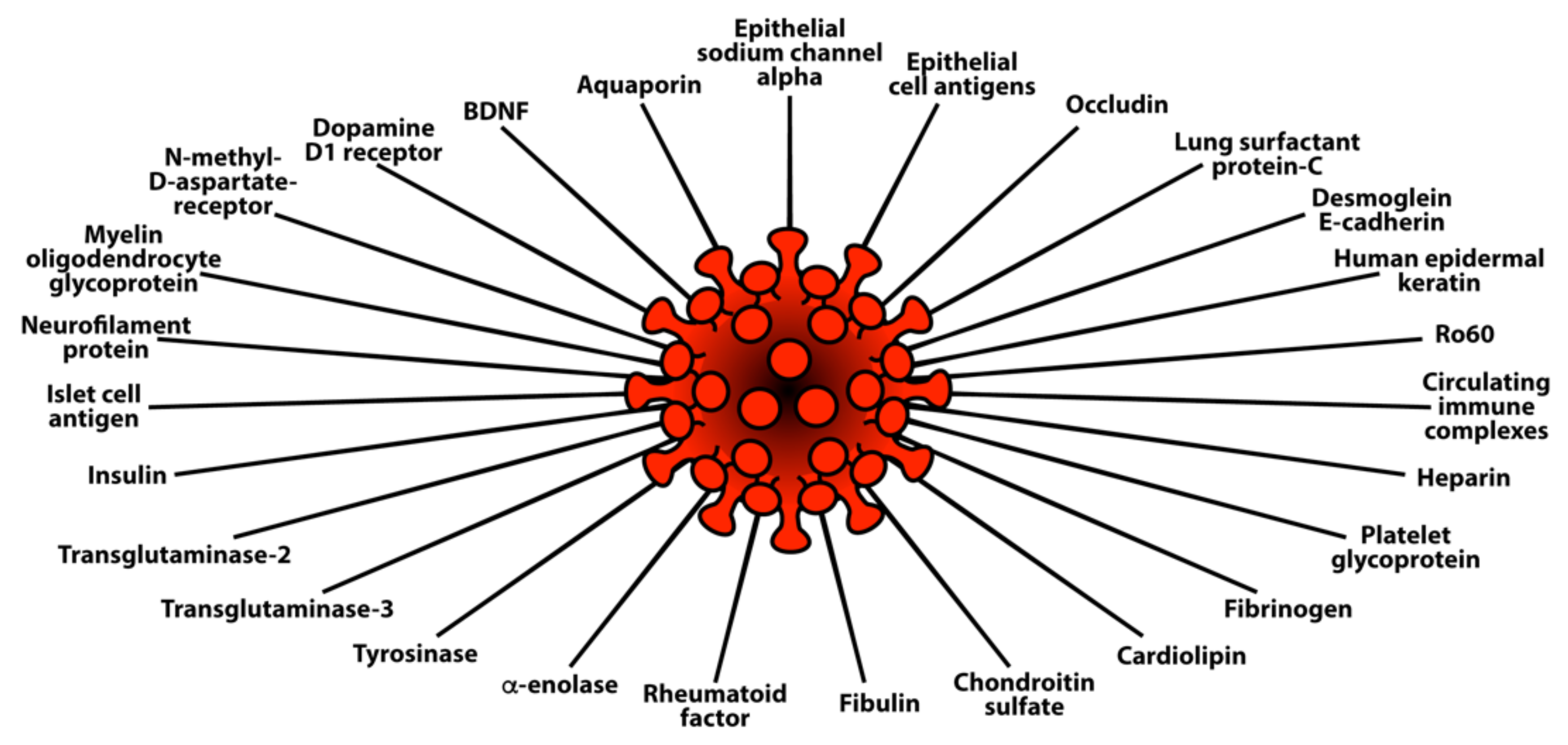
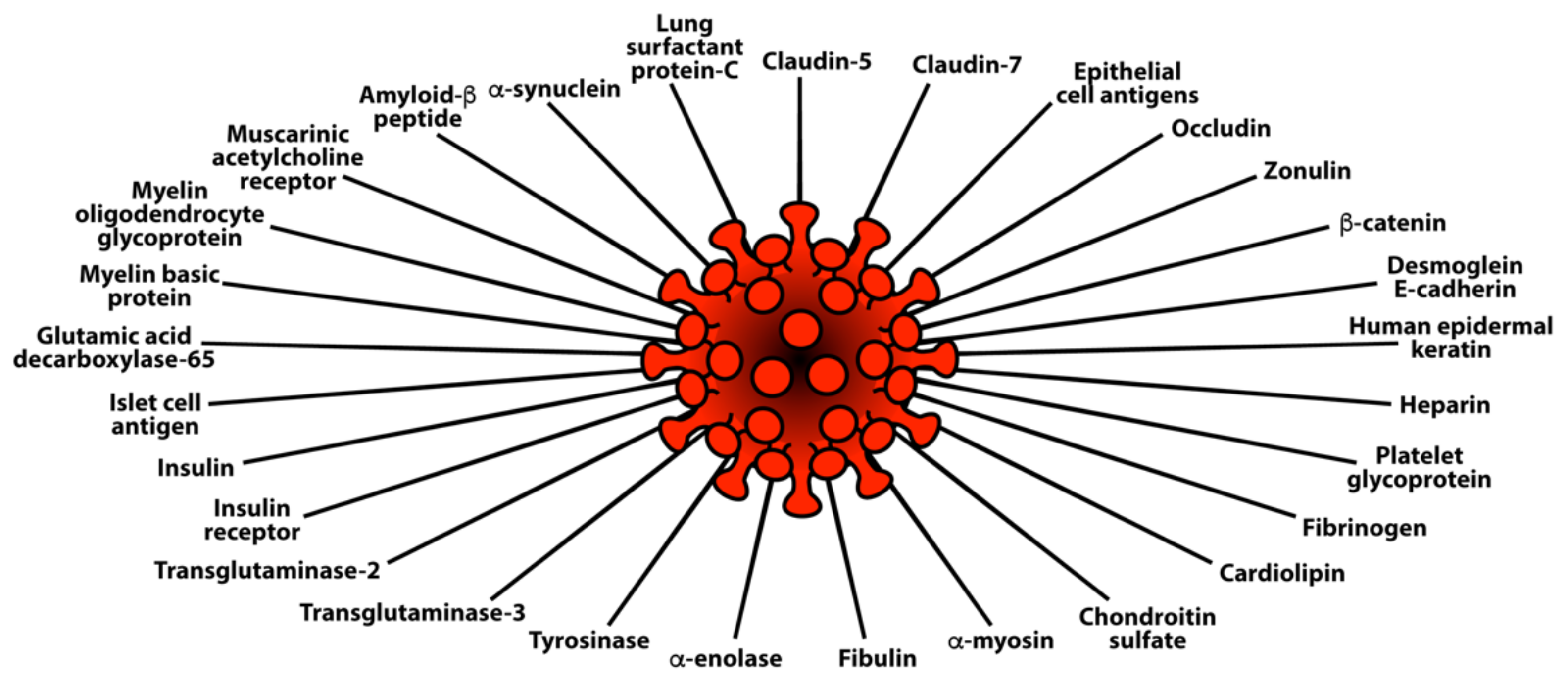
3. SARS-CoV-2: The Autoimmune Virus
- Similarities in lymphocyte map or lymphocyte subpopulation patterns between COVID-19 and autoimmune diseases
- Molecular mimicry between SARS-CoV-2 spike proteins, nucleoproteins and human autoantigens that contribute to autoimmune diseases
- Reaction of both animal and human monoclonal antibodies made against SARS-CoV-2 spike proteins and nucleoproteins with human autoantigens
- Reaction of antibodies made against human autoantigens with SARS-CoV-2 spike proteins and nucleoproteins
- Detection of autoantibodies made against human autoantigens known to cross-react with SARS-CoV-2 in the sera of patients with COVID-19
3.1. Keypoint No. 1: Similarities in Lymphocyte Map or Lymphocyte Subpopulation Patterns between COVID-19 and Autoimmune Disorders
3.2. Keypoint No. 2: Molecular Mimicry between SARS-CoV-2 Spike Proteins, Nucleoproteins, and Human Autoantigens Contributes to Autoimmune Diseases
3.3. Keypoint No. 3: Reaction of Both Animal and Human Monoclonal Antibodies Made against SARS-CoV-2 Spike Proteins and Nucleoproteins with Human Tissue Antigens
3.4. Keypoint No. 4: Reaction of Antibodies Made against Human Autoantigens with SARS-CoV-2 Spike Proteins and Nucleoproteins
3.5. Keypoint No. 5: Detection of Autoantibodies against Human Autoantigens Known to Cross-React with SARS-CoV-2 in the Sera of COVID-19 Patients
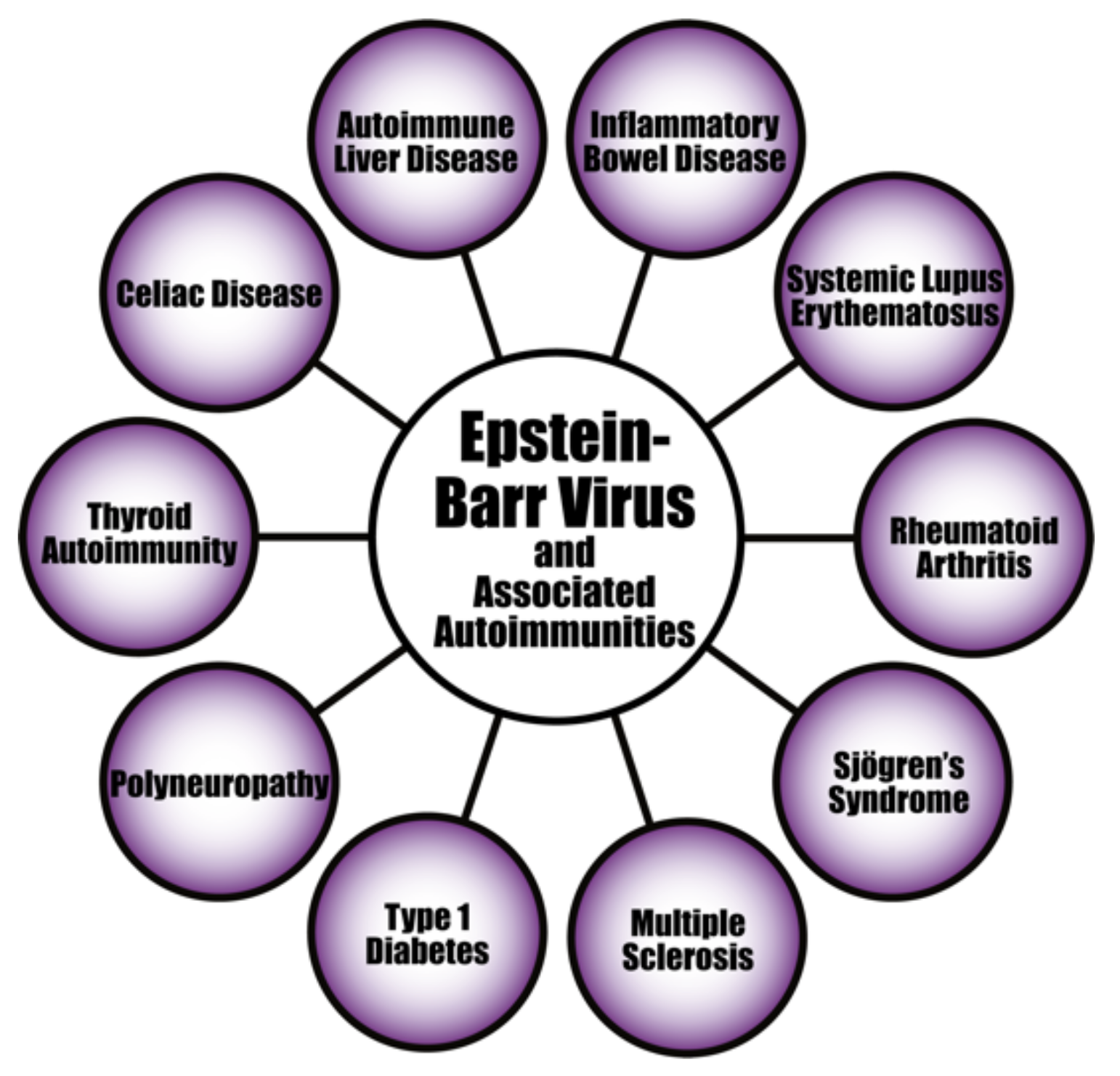
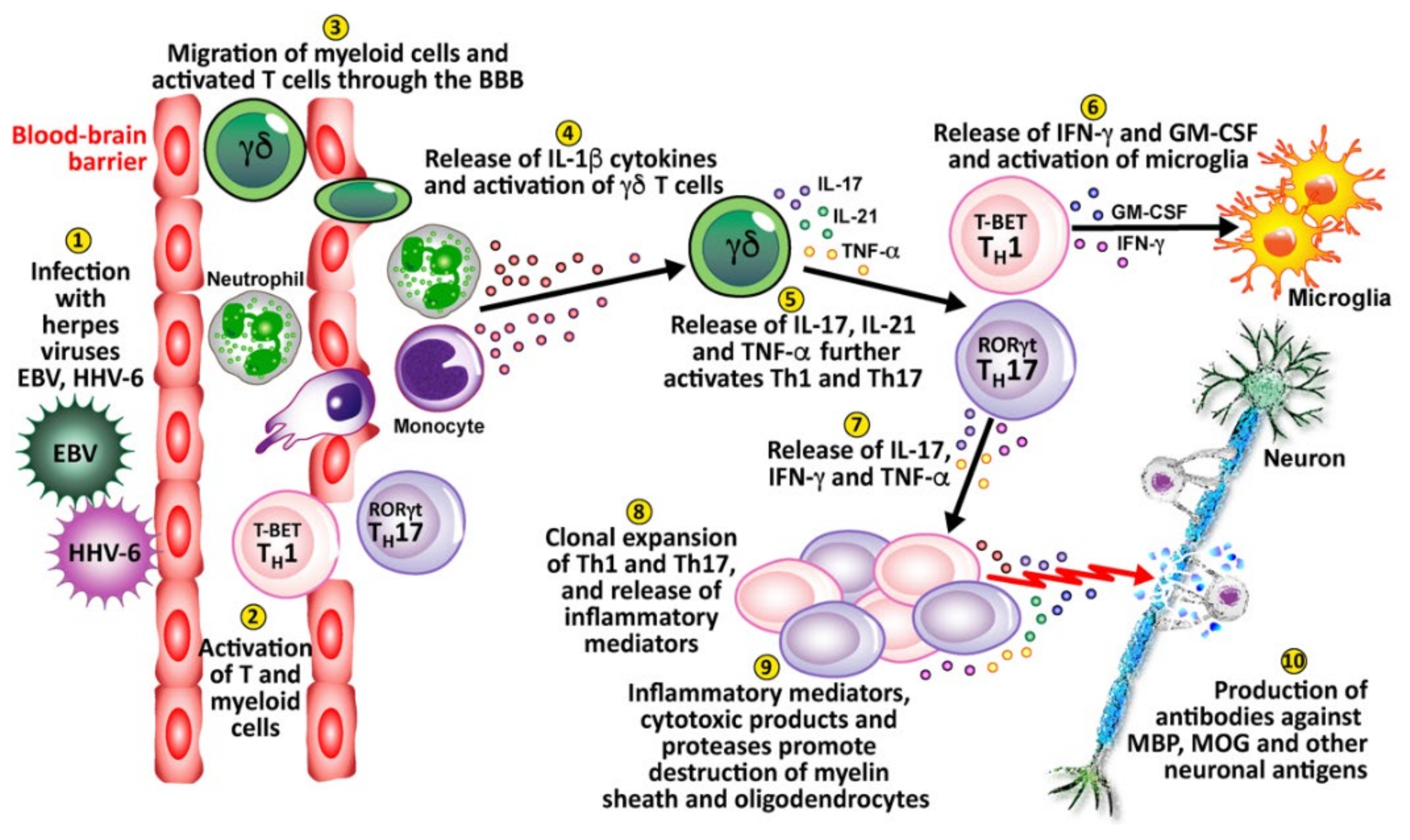
4. Herpesviruses and the Pathophysiology of Autoimmunity
Pathophysiological Mechanisms in the Induction of Autoimmunities by Herpesviruses

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Autoimmune Disease | Virus | Proposed Mechanisms | References |
|---|---|---|---|
| Autoimmune encephalitis | HSV | Molecular mimicry | [213] |
| Encephalitis (Human herpes encephalitis) | HSV | Molecular mimicry | [214] |
| Encephalitis and chronic neurological sequelae | HSV | Molecular mimicry? | [215] |
| Stromal keratitis | HSV | Bystander activation | [216] |
| Alzheimer’s | HSV | Unknown | [217] |
| Multiple sclerosis | VZV | Unknown | [218] |
| Lupus erythematosus | EBV | Molecular mimicry | [164] |
| Autoimmune hepatitis | EBV | Molecular mimicry; Persistence of EBV in B cell | [219,220] |
| Graves’ disease | EBV | EBV B-cell activation | [221] |
| Hashimoto’s disease | EBV | Unknown | [222] |
| Multiple sclerosis | EBV | Molecular mimicry; Molecular mimicry; Activation of Th1, Th17, Th1/Th17 | [176,223,224] |
| Rheumatoid arthritis | EBV | Molecular mimicry | [225] |
| Sjögren’s syndrome | EBV | B cell activation | [220] |
| Systemic sclerosis | CMV | Molecular mimicry; Induction of inflammation | [226,227] |
| Type 1 diabetes mellitus | CMV | Unknown | [172] |
| Systemic lupus erythematosus | CMV | Epitope spreading | [209] |
| Rheumatoid arthritis | CMV | Aggravation of inflammation | [228] |
| Endothelial cell autoimmunity | CMV | Molecular mimicry | [229] |
| Autoimmune thyroiditis | HHV-6A | NK cell killing of HHV-6 infected thyrocytes | [230] |
| Multiple sclerosis | HHV-6A/6B; HHV-6 | Infecting astrocytes and oligodendrocytes; Molecular mimicry; Activation of Th1, Th17, Th1/Th17 | [202,231,232] |
| Collagen vascular disease | HHV-6 | Molecular mimicry | [203] |
| Connective tissue disease | HHV-6 | Molecular mimicry; Selective reactivation | [204,233] |
| Sjögren’s syndrome | HHV-6 | Molecular mimicry; Polyclonal activation | [205] |
| Autoimmune hemolytic anemia | HHV-6 | Molecular mimicry; Polyclonal activation | [206] |
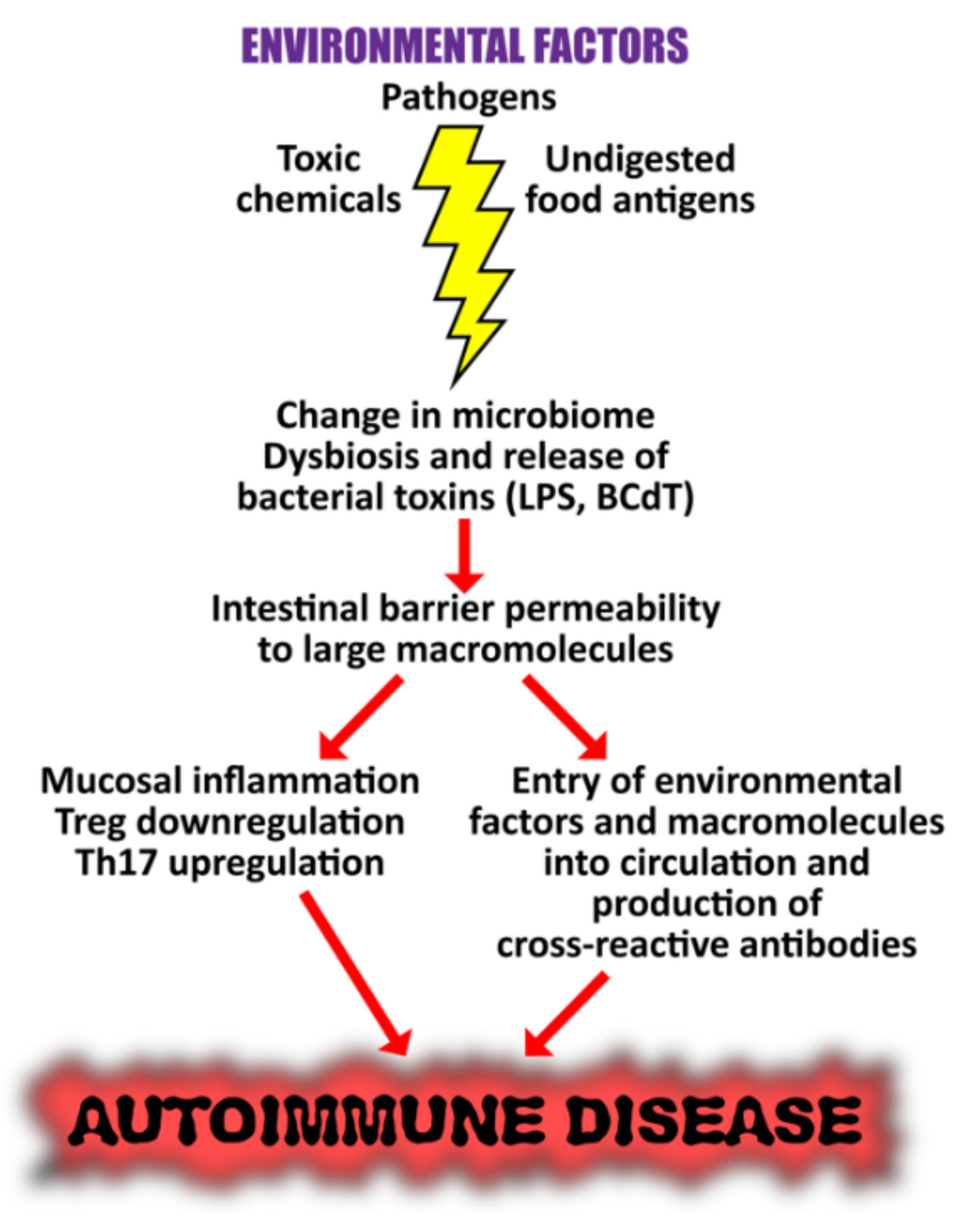
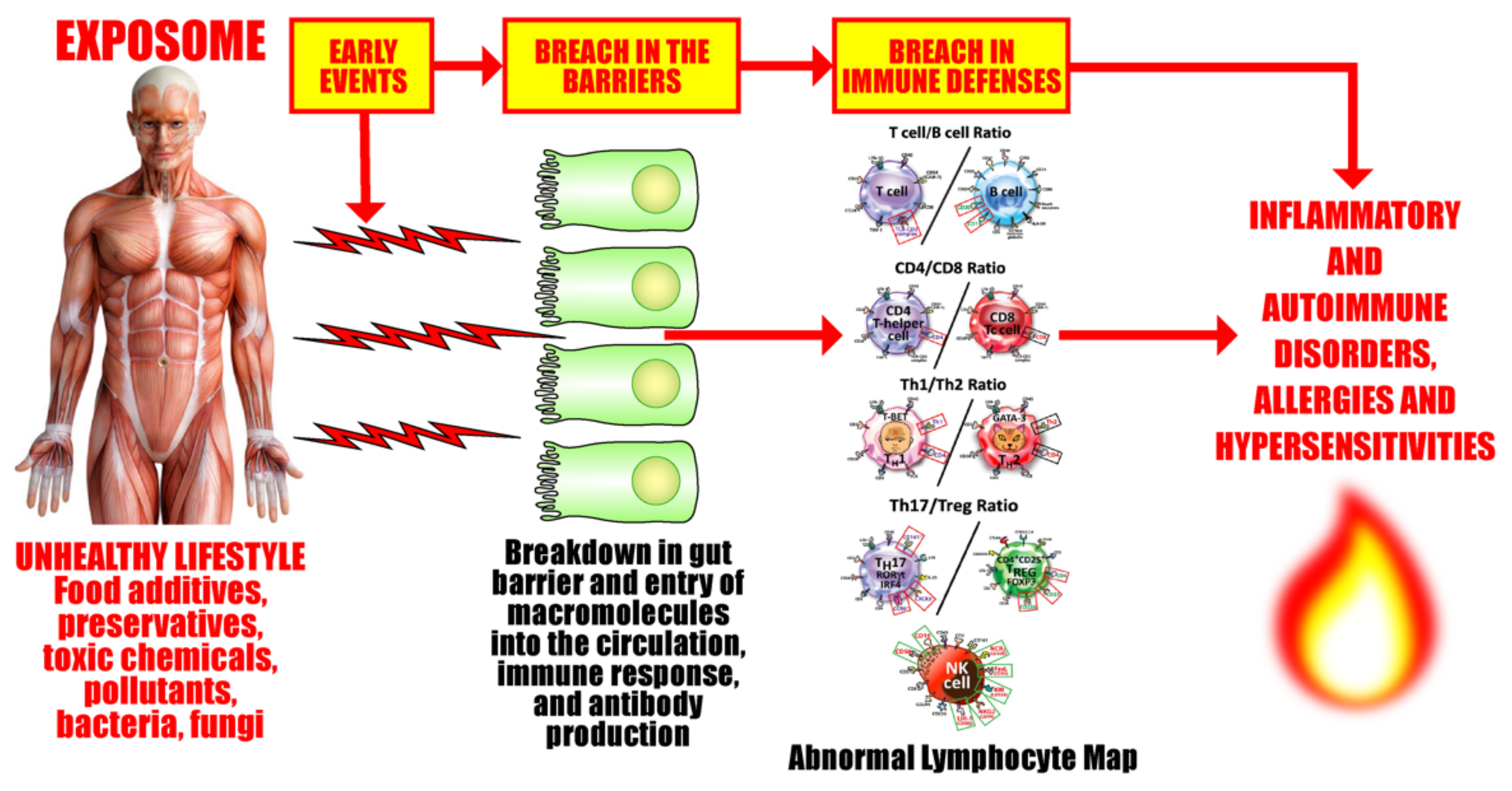
5. The Role of the Gut Microbiome in the Pathophysiology of Autoimmune Diseases
The Role of the Microbiome in the Pathophysiology of COVID-19
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vojdani, A.; Vojdani, E. The Role of Exposomes in the Pathophysiology of Autoimmune Diseases I: Toxic Chemicals and Food. Pathophysiology 2021, 28, 513–543. [Google Scholar] [CrossRef] [PubMed]
- Wild, C.P. Complementing the genome with an “exposome”: The outstanding challenge of environmental exposure measurement in molecular epidemiology. Cancer Epidemiol. Biomarkers Prev. 2005, 14, 1847–1850. [Google Scholar] [CrossRef] [PubMed]
- Vineis, P.; Barouki, R. The exposome as the science of social-to-biological transitions. Environ. Int. 2022, 165. [Google Scholar] [CrossRef] [PubMed]
- Kristo, A.; Klimis-Zacas, D.; Sikalidis, A. Protective Role of Dietary Berries in Cancer. Antioxidants 2016, 5, 37. [Google Scholar] [CrossRef] [PubMed]
- Sikalidis, A.K. Amino Acids and Immune Response: A Role for Cysteine, Glutamine, Phenylalanine, Tryptophan and Arginine in T-cell Function and Cancer? Pathol. Oncol. Res. 2015, 21, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Bogdanos, D.P.; Smyk, D.S.; Invernizzi, P.; Rigopoulou, E.I.; Blank, M.; Pouria, S.; Shoenfeld, Y. Infectome: A platform to trace infectious triggers of autoimmunity. Autoimmun. Rev. 2013, 12, 726–740. [Google Scholar] [CrossRef] [PubMed]
- Oldstone, M.B.; Nerenberg, M.; Southern, P.; Price, J.; Lewicki, H. Virus infection triggers insulin-dependent diabetes mellitus in a transgenic model: Role of anti-self (virus) immune response. Cell 1991, 65, 319–331. [Google Scholar] [CrossRef]
- Posnett, D.N.; Yarilin, D. Amplification of autoimmune disease by infection. Arthritis Res. Ther. 2005, 7, 74–84. [Google Scholar] [CrossRef][Green Version]
- Faé, K.C.; da Silva, D.D.; Oshiro, S.E.; Tanaka, A.C.; Pomerantzeff, P.M.A.; Douay, C.; Charron, D.; Toubert, A.; Cunningham, M.W.; Kalil, J.; et al. Mimicry in Recognition of Cardiac Myosin Peptides by Heart-Intralesional T Cell Clones from Rheumatic Heart Disease. J. Immunol. 2006, 176, 5662–5670. [Google Scholar] [CrossRef]
- Blank, M.; Krause, I.; Fridkin, M.; Keller, N.; Kopolovic, J.; Goldberg, I.; Tobar, A.; Shoenfeld, Y. Bacterial induction of autoantibodies to β2-glycoprotein-I accounts for the infectious etiology of antiphospholipid syndrome. J. Clin. Investig. 2002, 109, 797–804. [Google Scholar] [CrossRef]
- Kivity, S.; Agmon-Levin, N.; Blank, M.; Shoenfeld, Y. Infections and Autoimmunity—Friends or Foes? Trends Immunol. 2009, 30, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Vojdani, A. A Potential Link between Environmental Triggers and Autoimmunity. Autoimmune Dis. 2014, 2014, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Davies, T.F. Infection and Autoimmune Thyroid Disease. J. Clin. Endocrinol. Metab. 2008, 93, 674–676. [Google Scholar] [CrossRef] [PubMed]
- Blank, M.; Krause, I.; Shoenfeld, Y. Molecular Mimicry: Lessons from Experimental Models of Systemic Lupus Erythematosus and Antiphospholipid Syndrome. In Molecular Mimicry, Microbes, and Autoimmunity; Cunningham, M.W., Fujinami, R.S., Eds.; ASM Press: New York, NY, USA, 2020. [Google Scholar]
- Abu-Shakra, M.; Buskila, D.; Shoenfeld, Y. Molecular mimicry between host and pathogen: Examples from parasites and implication. Immunol. Lett. 1999, 67, 147–152. [Google Scholar] [CrossRef]
- Kanduc, D.; Shoenfeld, Y. Human Papillomavirus Epitope Mimicry and Autoimmunity: The Molecular Truth of Peptide Sharing. Pathobiology 2019, 86, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Dotan, A.; Kanduc, D.; Muller, S.; Makatsariya, A.; Shoenfeld, Y. Molecular mimicry between SARS-CoV-2 and the female reproductive system. Am. J. Reprod. Immunol. 2021, 86. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Sonthalia, N.; Kundu, S.; Ganguly, S. Autoimmune Disorders: An Overview of Molecular and Cellular Basis in Today’s Perspective. J. Clin. Cell. Immunol. 2013, 1. [Google Scholar] [CrossRef]
- Craft, J.; Fatenejad, S. Self antigens and epitope spreading in systemic autoimmunity. Arthritis Care Res. 1997, 40, 1374–1382. [Google Scholar] [CrossRef]
- James, J.A.; Harley, J.B. B-cell epitope spreading in autoimmunity. Immunol. Rev. 1998, 164, 185–200. [Google Scholar] [CrossRef]
- Sercarz, E.E. Driver Clones and Determinant Spreading. J. Autoimmun. 2000, 14, 275–277. [Google Scholar] [CrossRef]
- Monneaux, F.; Muller, S. Epitope spreading in systemic lupus erythematosus: Identification of triggering peptide sequences. Arthritis Care Res. 2002, 46, 1430–1438. [Google Scholar] [CrossRef] [PubMed]
- Ehow, K.Y.; Esong, K.P.; Echan, K.G. Porphyromonas gingivalis: An Overview of Periodontopathic Pathogen below the Gum Line. Front. Microbiol. 2016, 7, 53. [Google Scholar] [CrossRef]
- Könönen, E.; Müller, H.-P. Microbiology of aggressive periodontitis. Periodontology 2000 2014, 65, 46–78. [Google Scholar] [CrossRef] [PubMed]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019, 5, 3333. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G.; Chavakis, T. Local and systemic mechanisms linking periodontal disease and inflammatory comorbidities. Nat. Rev. Immunol. 2021, 21, 426–440. [Google Scholar] [CrossRef]
- Nara, P.L.; Sindelar, D.; Penn, M.S.; Potempa, J.; Griggin, W.S.T. Porphyromonas Gingivalis Outer Membrane Vesicles as the Major Driver of an Explanation for Neuropathogenesis, the Cholinergic Hypothesis, Iron Dyshomeostasis, and Salivary Lactoferrin in Alzheimer’s Disease. J. Alz. Dis. 2021, 82, 1417–1450. [Google Scholar] [CrossRef]
- Gao, L.; Kang, M.; Zhang, M.J.; Sailani, M.R.; Kuraji, R.; Martinez, A.; Ye, C.; Kamarajan, P.; Le, C.; Zhan, L.; et al. Polymicrobial periodontal disease triggers a wide radius of effect and unique virome. npj Biofilms Microbiomes 2020, 6, 10–13. [Google Scholar] [CrossRef]
- Farquharson, D.; Butcher, J.P.; Culshaw, S. Periodontitis, Porphyromonas, and the pathogenesis of rheumatoid arthritis. Mucosal Immunol. 2012, 5, 112–120. [Google Scholar] [CrossRef]
- Burkhardt, H.; Sehnert, B.; Bockermann, R.; Engström, Å; Kalden, J.R.; Holmdahl, R. Humoral immune response to citrullinated collagen type II determinants in early rheumatoid arthritis. Eur. J. Immunol. 2005, 35, 1643–1652. [Google Scholar] [CrossRef]
- Masson-Bessière, C.; Sebbag, M.; Girbal-Neuhauser, E.; Nogueira, L.; Vincent, C.; Senshu, T.; Serre, G. The Major Synovial Targets of the Rheumatoid Arthritis-Specific Antifilaggrin Autoantibodies Are Deiminated Forms of the α- and β-Chains of Fibrin. J. Immunol. 2001, 166, 4177–4184. [Google Scholar] [CrossRef]
- Vossenaar, E.R.; Després, N.; Lapointe, E.; Van Der Heijden, A.; Lora, M.; Senshu, T.; Van Venrooij, W.J.; Ménard, H.A. Rheumatoid arthritis specific anti-Sa antibodies target citrullinated vimentin. Arthritis Res. Ther. 2004, 6, R1142–R1150. [Google Scholar] [CrossRef]
- Kinloch, A.; Tatzer, V.; Wait, R.; Peston, D.; Lundberg, K.; Donatien, P.; Moyes, D.; Taylor, P.C.; Venables, P.J. Identification of citrullinated α-enolase as a candidate autoantigen in rheumatoid arthritis. Arthritis Res. Ther. 2005, 7, R1421–R1429. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, J.B.; Pippen, A.M.M.; Greenberg, C.S. Extravascular fibrin formation and dissolution in synovial tissue of patients with osteoarthritis and rheumatoid arthritis. Arthritis Care Res. 1991, 34, 996–1005. [Google Scholar] [CrossRef]
- Routsias, J.G.; Goules, J.D.; Goules, A.; Charalampakis, G.; Pikazis, D. Autopathogenic correlation of periodontitis and rheumatoid arthritis. Rheumatology 2011, 50, 1189–1193. [Google Scholar] [CrossRef]
- Kronzer, V.L.; Crowson, C.S.; Sparks, J.; Myasoedova, E.; Davis, J.M. Comorbidities as Risk Factors for Rheumatoid Arthritis and Their Accrual After Diagnosis. Mayo Clin. Proc. 2019, 94, 2488–2498. [Google Scholar] [CrossRef] [PubMed]
- Curran, A.M.; Naik, P.; Giles, J.T.; Darrah, E. PAD Enzymes in Rheumatoid Arthritis: Pathogenic Effectors and Autoimmune Targets. Nat. Rev. Rheumatol. 2020, 16, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.A.; Southwood, S.; Sette, A.; Jevnikar, A.M.; Bell, D.A.; Cairns, E. Cutting Edge: The Conversion of Arginine to Citrulline Allows for a High-Affinity Peptide Interaction with the Rheumatoid Arthritis-Associated HLA-DRB1*0401 MHC Class II Molecule. J. Immunol. 2003, 171, 538–541. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, K.; Wegner, N.; Yucel-Lindberg, T.; Venables, P.J. Periodontitis in RA—the citrullinated enolase connection. Nat. Rev. Rheumatol. 2010, 6, 727–730. [Google Scholar] [CrossRef]
- Lundberg, K.; Kinloch, A.; Fisher, B.A.; Wegner, N.; Wait, R.; Charles, P.; Mikuls, T.R.; Venables, P.J. Antibodies to citrullinated α-enolase peptide 1 are specific for rheumatoid arthritis and cross-react with bacterial enolase. Arthritis Care Res. 2008, 58, 3009–3019. [Google Scholar] [CrossRef]
- Kinloch, A.J.; Alzabin, S.; Brintnell, W.; Wilson, E.; Barra, L.; Wegner, N.; Bell, D.A.; Cairns, E.; Venables, P.J. Immunization with Porphyromonas gingivalis enolase induces autoimmunity to mammalian α-enolase and arthritis in DR4-IE-transgenic mice. Arthritis Care Res. 2011, 63, 3818–3823. [Google Scholar] [CrossRef]
- Ma, W.-T.; Chang, C.; Gershwin, M.E.; Lian, Z.-X. Development of autoantibodies precedes clinical manifestations of autoimmune diseases: A comprehensive review. J. Autoimmun. 2017, 83, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Cabral-Marques, O.; Halpert, G.; Schimke, L.F.; Ostrinski, Y.; Vojdani, A.; Baiocchi, G.C.; Freire, P.P.; Filgueiras, I.S.; Zyskind, I.; Lattin, M.T.; et al. Autoantibodies targeting GPCRs and RAS-related molecules associate with COVID-19 severity. Nat. Commun. 2022, 13, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Halpert, G.; Shoenfeld, Y. SARS-CoV-2, the Autoimmune Virus. Autoimmun. Rev. 2020, 19, 102695. [Google Scholar] [CrossRef] [PubMed]
- Dotan, A.; Muller, S.; Kanduc, D.; David, P.; Halpert, G.; Shoenfeld, Y. The SARS-CoV-2 as an instrumental trigger of autoimmunity. Autoimmun. Rev. 2021, 20, 102792. [Google Scholar] [CrossRef]
- Shoenfeld, Y. Corona (COVID-19) Time Musings: Our Involvement in COVID-19 Pathogenesis, Diagnosis, Treatment and Vac-cine Planning. Autoimmun. Res. 2020, 19, 102538. [Google Scholar] [CrossRef]
- Lyons-Weiler, J. Pathogenic priming likely contributes to serious and critical illness and mortality in COVID-19 via autoimmunity. J. Transl. Autoimmun. 2020, 3. [Google Scholar] [CrossRef]
- Giamarellos-Bourboulis, E.J.; Netea, M.G.; Rovina, N.; Akinosoglou, K.; Antoniadou, A.; Antonakos, N.; Damoraki, G.; Gkavogianni, T.; Adami, M.-E.; Katsaounou, P.; et al. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe 2020, 27, 992–1000.e3. [Google Scholar] [CrossRef]
- Vojdani, A.; Kharrazian, D. Potential Antigenic Cross-Reactivity Between SARS-CoV-2 and Human Tissue with a Possible Link to an Increase in Autoimmune Diseases. Clin. Immunol. 2020, 217, 108480. [Google Scholar] [CrossRef]
- Vojdani, A.; Vojdani, E.; Kharrazian, D. Reaction of Human Monoclonal Antibodies to SARS-CoV-2 Proteins With Tissue Antigens: Implications for Autoimmune Diseases. Front. Immunol. 2021, 11, 617089. [Google Scholar] [CrossRef]
- Kanduc, D.; Shoenfeld, Y. On the Molecular Determinants of the SARS-CoV-2 Attack. Clin. Immunol. 2020, 215, 108426. [Google Scholar] [CrossRef]
- Kanduc, D.; Shoenfeld, Y. Molecular mimicry between SARS-CoV-2 spike glycoprotein and mammalian proteomes: Implications for the vaccine. Immunol. Res. 2020, 68, 310–313. [Google Scholar] [CrossRef] [PubMed]
- Becker, R.C. COVID-19 update: COVID-19-associated coagulopathy. J. Thromb. Thrombolysis 2020, 50, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xiao, M.; Zhang, S.; Xia, P.; Cao, W.; Jiang, W.; Chen, H.; Ding, X.; Zhao, H.; Zhang, H.; et al. Coagulopathy and Antiphospholipid Antibodies in Patients with COVID-19. N. Engl. J. Med. 2020, 382, e38. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sawalha, A.H.; Lu, Q. COVID-19 and autoimmune diseases. Curr. Opin. Rheumatol. 2020, 33, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Ruscitti, P.; Berardicurti, O.; Barile, A.; Cipriani, P.; Shoenfeld, Y.; Iagnocco, A.; Giacomelli, R. Severe COVID-19 and related hyperferritinaemia: More than an innocent bystander? Ann. Rheum. Dis. 2020, 79, 1515–1516. [Google Scholar] [CrossRef] [PubMed]
- Ehrenfeld, M.; Tincani, A.; Andreoli, L.; Cattalini, M.; Greenbaum, A.; Kanduc, D.; Alijotas-Reig, J.; Zinserling, V.; Semenova, N.; Amital, H.; et al. COVID-19 and autoimmunity. Autoimmun. Rev. 2020, 19, 102597. [Google Scholar] [CrossRef]
- Perricone, C.; Bartoloni, E.; Bursi, R.; Cafaro, G.; Guidelli, G.M.; Shoenfeld, Y.; Gerli, R. COVID-19 as part of the hyperferritinemic syndromes: The role of iron depletion therapy. Immunol. Res. 2020, 68, 213–224. [Google Scholar] [CrossRef]
- Dahan, S.; Segal, G.; Katz, I.; Hellou, T.; Tietel, M.; Bryk, G.; Amital, H.; Shoenfeld, Y.; Dagan, A. Ferritin as a Marker of Severity in COVID-19 Patients: A Fetal Correlation. IMAJ 2020, 22, 428–434. [Google Scholar]
- Ryabkova, V.A.; Churilov, L.P.; Shoenfeld, Y. Influenza infection, SARS, MERS and COVID-19: Cytokine storm—The common denominator and the lessons to be learned. Clin. Immunol. 2020, 223, 108652. [Google Scholar] [CrossRef]
- Malkova, A.; Kudlay, D.; Kudryavtsev, I.; Starshinova, A.; Yablonskiy, P.; Shoenfeld, Y. Immunogenetic Predictors of Severe COVID-19. Vaccines 2021, 9, 211. [Google Scholar] [CrossRef]
- Mahroum, N.; Alghory, A.; Kiyak, Z.; Alwani, A.; Seida, R.; Alrais, M.; Shoenfeld, Y. Ferritin—From iron, through inflammation and autoimmunity, to COVID-19. J. Autoimmun. 2021, 126, 102778. [Google Scholar] [CrossRef] [PubMed]
- Hejrati, A.; Rafiei, A.; Soltanshahi, M.; Hosseinzadeh, S.; Dabiri, M.; Taghadosi, M.; Taghiloo, S.; Bashash, D.; Khorshidi, F.; Zafari, P. Innate immune response in systemic autoimmune diseases: A potential target of therapy. Inflammopharmacology 2020, 28, 1421–1438. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Thakur, M.; Sharma, L.K.; Chandra, K. Designing a multi-epitope peptide based vaccine against SARS-CoV-2. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Canedo-Marroquín, G.; Saavedra, F.; Andrade, C.A.; Berrios, R.V.; Rodríguez-Guilarte, L.; Opazo, M.C.; Riedel, C.A.; Kalergis, A.M. SARS-CoV-2: Immune Response Elicited by Infection and Development of Vaccines and Treatments. Front. Immunol. 2020, 11, 569760. [Google Scholar] [CrossRef]
- Conti, P.; Caraffa, A.; Galenga, C.; Ross, R.; Kritas, S.K.; Frydas, I.; Younes, A.; Ronconi, G. Coronavirus-19 (SARS-CoV-2) Induces Acute Severe Lung Inflammation via IL-1 Causing Cytokine Storm in COVID-19: A Promising Inhibitory Strategy. J. Biol. Regulat. Homeost. Agents 2020, 343, 1971–1975. [Google Scholar]
- Muskardin, T.L.W. Intravenous Anakinra for Macrophage Activation Syndrome May Hold Lessons for Treatment of Cytokine Storm in the Setting of Coronavirus Disease 2019. ACR Open Rheumatol. 2020, 2, 283–285. [Google Scholar] [CrossRef]
- Setiati, S.; Harimurti, K.; Safitri, E.D.; Ranakusuma, R.W.; Saldi, S.R.F.; Azwar, M.K.; Marsigit, J.; Pitoyo, Y.; Widyaningsih, W. Risk factors and laboratory test results associated with severe illness and mortality in COVID-19 patients: A systematic review. Acta Med Indones 2020, 52, 227–245. [Google Scholar]
- Ziadi, A.; Hachimi, A.; Admou, B.; Hazime, R.; Brahim, I.; Douirek, F.; Zarrouki, Y.; El Adib, A.R.; Younous, S.; Samkaoui, A.M. Lymphopenia in critically ill COVID-19 patients: A predictor factor of severity and mortality. Int. J. Lab. Hematol. 2020, 43. [Google Scholar] [CrossRef]
- Ciceri, F.; Castagna, A.; Rovere-Querini, P.; De Cobelli, F.; Ruggeri, A.; Galli, L.; Conte, C.; De Lorenzo, R.; Poli, A.; Ambrosio, A.; et al. Early predictors of clinical outcomes of COVID-19 outbreak in Milan, Italy. Clin. Immunol. 2020, 217, 108509. [Google Scholar] [CrossRef]
- Vassallo, M.; Manni, S.; Pini, P.; Blanchouin, E.; Ticchioni, M.; Seitz-Polski, B.; Puchois, A.; Sindt, A.; Lotte, L.; Fauque, P.; et al. Patients with COVID-19 exhibit different immunological profiles according to their clinical presentation. Int. J. Infect. Dis. 2020, 101, 174–179. [Google Scholar] [CrossRef]
- Azar, M.M.; Shin, J.J.; Kang, I.; Landry, M. Diagnosis of SARS-CoV-2 infection in the setting of the cytokine release syndrome. Expert Rev. Mol. Diagn. 2020, 20, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, Q.; Yin, Y.; Zhang, Y.; Cao, Y.; Lin, X.; Huang, L.; Hoffmann, D.; Lu, M.; Qiu, Y. Excessive Neutrophils and Neutrophil Extracellular Traps in COVID-19. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Satış, H.; Özger, H.S.; Yıldız, P.A.; Hızel, K.; Gulbahar, Ö; Erbaş, G.; Aygencel, G.; Tunccan, O.G.; Öztürk, M.A.; Dizbay, M.; et al. Prognostic value of interleukin-18 and its association with other inflammatory markers and disease severity in COVID-19. Cytokine 2020, 137, 155302. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Dong, Y.; Wang, L.; Xie, H.; Li, B.; Chang, C.; Wang, F.-S. Characteristics and prognostic factors of disease severity in patients with COVID-19: The Beijing experience. J. Autoimmun. 2020, 112, 102473. [Google Scholar] [CrossRef]
- Woodruff, M.C.; Ramonell, R.P.; Nguyen, D.C.; Cashman, K.S.; Saini, A.S.; Haddad, N.S.; Ley, A.M.; Kyu, S.; Howell, J.C.; Ozturk, T.; et al. Extrafollicular B cell responses correlate with neutralizing antibodies and morbidity in COVID-19. Nat. Immunol. 2020, 21, 1506–1516. [Google Scholar] [CrossRef]
- Oliviero, B.; Varchetta, S.; Mele, D.; Mantovani, S.; Cerino, A.; Perotti, C.G.; Ludovisi, S.; Mondelli, M.U. Expansion of atypical memory B cells is a prominent feature of COVID-19. Cell. Mol. Immunol. 2020, 17, 1101–1103. [Google Scholar] [CrossRef]
- Varchetta, S.; Mele, D.; Oliviero, B.; Mantovani, S.; Ludovisi, S.; Cerino, A.; Bruno, R.; Castelli, A.; Mosconi, M.; Vecchia, M.; et al. Unique immunological profile in patients with COVID-19. Cell. Mol. Immunol. 2020, 18, 604–612. [Google Scholar] [CrossRef]
- Neidleman, J.; Luo, X.; George, A.F.; McGregor, M.; Yang, J.; Yun, C.; Murray, V.; Gill, G.; Greene, W.C.; Vasquez, J.; et al. Distinctive features of SARS-CoV-2-specific T cells predict recovery from severe COVID-19. Cell Rep. 2021, 36, 109414. [Google Scholar] [CrossRef]
- Mathew, D.; Giles, J.R.; Baxter, A.E.; Oldridge, D.A.; Greenplate, A.R.; Wu, J.E.; Alanio, C.; Kuri-Cervantes, L.; Pampena, M.B.; D’Andrea, K.; et al. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science 2020, 369. [Google Scholar] [CrossRef]
- Liu, Q.; Fang, X.; Tokuno, S.; Chung, U.; Chen, X.; Dai, X.; Liu, X.; Xu, F.; Wang, B.; Peng, P. A web visualization tool using T cell subsets as the predictor to evaluate COVID-19 patient’s severity. PLoS ONE 2020, 15, e0239695. [Google Scholar] [CrossRef]
- Maucourant, C.; Filipovic, I.; Ponzetta, A.; Aleman, S.; Cornillet, M.; Hertwig, L.; Strunz, B.; Lentini, A.; Reinius, B.; Brownlie, D.; et al. Natural killer cell immunotypes related to COVID-19 disease severity. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, M.A.; Sunny, S.; Balabayova, K.; Kaur, A.; Gupta, A.; Abdallah, M.; Quale, J. Tocilizumab therapy for COVID-19: A comparison of subcutaneous and intravenous therapies. Int. J. Infect. Dis. 2020, 101, 59–64. [Google Scholar] [CrossRef]
- Liu, Y.L.; Chang, C.; Lu, Q. Management strategies for patients with autoimmune diseases during the COVID-19 pandemic: A perspective from China. Eur. J. Rheumatol. 2020, 7, S94–S96. [Google Scholar] [CrossRef] [PubMed]
- Rojas, M.; Rodríguez, Y.; Monsalve, D.M.; Acosta-Ampudia, Y.; Camacho, B.; Gallo, J.E.; Rojas-Villarraga, A.; Ramírez-Santana, C.; Díaz-Coronado, J.C.; Manrique, R.; et al. Convalescent plasma in COVID-19: Possible mechanisms of action. Autoimmun. Rev. 2020, 19, 102554. [Google Scholar] [CrossRef] [PubMed]
- Solun, B.; Shoenfeld, Y. Inhibition of metalloproteinases in therapy for severe lung injury due to COVID-19. Med. Drug Discov. 2020, 7, 100052. [Google Scholar] [CrossRef] [PubMed]
- Paula, D.; Shoenfeld, Y. Bacillus Calmette-Guerin Protective Factor for COVID-19? IMAJ 2020, 22, 448–449. [Google Scholar]
- Starshinova, A.; Malkova, A.; Zinchenko, U.; Kudlay, D.; Glushkova, A.; Dovgalyk, I.; Yablonskiy, P.; Shoenfeld, Y. Efficacy of Different Types of Therapy for COVID-19: A Comprehensive Review. Life 2021, 11, 753. [Google Scholar] [CrossRef]
- Mahroum, N.; Watad, A.; Bridgewood, C.; Mansour, M.; Nasr, A.; Hussein, A.; Khamisy-Farah, R.; Farah, R.; Gendelman, O.; Lidar, M.; et al. Systematic Review and Meta-Analysis of Tocilizumab Therapy versus Standard of Care in over 15,000 COVID-19 Pneumonia Patients during the First Eight Months of the Pandemic. Int. J. Environ. Res. Public Health 2021, 18, 9149. [Google Scholar] [CrossRef]
- Danieli, M.G.; Piga, M.A.; Paladini, A.; Longhi, E.; Mezzanotte, C.; Moroncini, G.; Shoenfeld, Y. Intravenous immunoglobulin as an important adjunct in the prevention and therapy of coronavirus 2019 disease. Scand. J. Immunol. 2021, 94, e13101. [Google Scholar] [CrossRef]
- Bluestone, J.A.; Seyfert Margolis, V. Foreword: The Bedside is the Bench. In Measuring Immunity: Basic Biology and Clinical Assessment; Lotze, M.T., Thomson, A.W., Eds.; Elsevier Academic Press: Cambridge, MA, USA, 2004. [Google Scholar]
- Vora, S.M.; Lieberman, J.; Wu, H. Inflammasome activation at the crux of severe COVID-19. Nat. Rev. Immunol. 2021, 21, 694–703. [Google Scholar] [CrossRef]
- Hussein, M.R.; Fathi, N.A.; El-Din, A.M.E.; Hassan, H.I.; Abdullah, F.; Al-Hakeem, E.; Backer, E.A. Alterations of the CD4+, CD8+ T Cell Subsets, Interleukins-1β, IL-10, IL-17, Tumor Necrosis Factor-α and Soluble Intercellular Adhesion Molecule-1 in Rheumatoid Arthritis and Osteoarthritis: Preliminary Observations. Pathol. Oncol. Res. 2008, 14, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Vojdani, A.; Ghoneum, M.; Brautbar, N. Immune Alteration Associated with Exposure to Toxic Chemicals. Toxicol. Ind. Health 1992, 8, 239–254. [Google Scholar] [CrossRef]
- Vojdani, A.; Campbell, A.; Brautbar, N. Immune Functional Impairment in Patients with Clinical Abnormalities and Silicone Breast Implants. Toxicol. Ind. Health 1992, 8, 415–429. [Google Scholar] [CrossRef]
- Brautbar, N.; Vojdani, A. Silicone implants and systemic immunological disease: Review of the literature and preliminary re-sults. Toxicol. Ind. Health 1992, 8, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Boldt, A.; Borte, S.; Fricke, S.; Kentouche, K.; Emmrich, F.; Borte, M.; Kahlenberg, F.; Sack, U. Eight-color immunophenotyping of T-, B-, and NK-cell subpopulations for characterization of chronic immunodeficiencies. Cytom. Part B Clin. Cytom. 2014, 86, 191–206. [Google Scholar] [CrossRef]
- Kaczorowski, K.J.; Shekhar, K.; Nkulikiyimfura, D.; Dekker, C.L.; Maecker, H.; Davis, M.M.; Chakraborty, A.K.; Brodin, P. Continuous immunotypes describe human immune variation and predict diverse responses. Proc. Natl. Acad. Sci. USA 2017, 114, E6097–E6106. [Google Scholar] [CrossRef]
- Voskuhl, R.R.; Martin, R.; Bergman, C.; Dalal, M.; Ruddle, N.H.; McFarland, H.F. T Helper 1 (TH1) Functional Phenotype of Human Myelin Basic Protein-Specific T Lymphocytes. Autoimmunity 1993, 15, 137–143. [Google Scholar] [CrossRef]
- Skapenko, A.; Leipe, J.; Lipsky, P.E.; Schulze-Koops, H. The role of the T cell in autoimmune inflammation. Arthritis Res. Ther. 2005, 7, S4–S14. [Google Scholar] [CrossRef]
- Georas, S.N.; Guo, J.; De Fanis, U.; Casolaro, V. T-helper cell type-2 regulation in allergic disease. Eur. Respir. J. 2005, 26, 1119–1137. [Google Scholar] [CrossRef]
- Bosnjak, B.; Stelzmueller, B.; Erb, K.J.; Epstein, M.M. Treatment of allergic asthma: Modulation of Th2 cells and their responses. Respir. Res. 2011, 12, 114. [Google Scholar] [CrossRef]
- Lee, G.R. The balance of Th17 versus treg cells in autoimmunity. Int. J. Mol. Sci. 2018, 19, 730. [Google Scholar] [CrossRef] [PubMed]
- Oukka, M. Th17 cells in immunity and autoimmunity. Ann. Rheum. Dis. 2008, 67, iii26–iii29. [Google Scholar] [CrossRef] [PubMed]
- Mohr, A.; Atif, M.; Balderas, R.; Gorochov, G.; Miyara, M. The role of FOXP3+ regulatory T cells in human autoimmune and inflammatory diseases. Clin. Exp. Immunol. 2019, 197, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Rocamora-Reverte, L.; Melzer, F.L.; Würzner, R.; Weinberger, B. The Complex Role of Regulatory T Cells in Immunity and Aging. Front. Immunol. 2021, 11. [Google Scholar] [CrossRef]
- Brown, C.Y.; Sadlon, T.; Hope, C.M.; Wong, Y.Y.; Wong, S.; Liu, N.; Withers, H.; Brown, K.; Bandara, V.; Gundsambuu, B.; et al. Molecular Insights Into Regulatory T-Cell Adaptation to Self, Environment, and Host Tissues: Plasticity or Loss of Function in Autoimmune Disease. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Liu, W.; Liu, S.; Verma, M.; Zafar, I.; Good, J.T.; Rollins, D.; Groshong, S.; Gorska, M.M.; Martin, R.J.; Alam, R. Mechanism of TH2/TH17-predominant and neutrophilic TH2/TH17-low subtypes of asthma. J. Allergy Clin. Immunol. 2016, 139, 1548–1558.e4. [Google Scholar] [CrossRef]
- Choy, D.F.; Hart, K.M.; Borthwick, L.A.; Shikotra, A.; Nagarkar, D.R.; Siddiqui, S.; Jia, G.; Ohri, C.M.; Doran, E.; Vannella, K.M.; et al. TH2 and TH17 inflammatory pathways are reciprocally regulated in asthma. Sci. Transl. Med. 2015, 7, 301ra129. [Google Scholar] [CrossRef]
- Wu, L.; Van Kaer, L. Natural killer T cells in health and disease. Front. Biosci. 2011, 3, 236–251. [Google Scholar] [CrossRef]
- Wu, L.; Kaer, L. Natural Killer T Cells and Autoimmune Disease. Curr. Mol. Med. 2009, 9, 4–14. [Google Scholar] [CrossRef]
- Krijgsman, D.; Hokland, M.; Kuppen, P.J.K. The role of natural killer T cells in cancer—A phenotypical and functional approach. Front. Immunol. 2018, 9, 367. [Google Scholar] [CrossRef]
- Tang, Y.; Li, X.; Wang, M.; Zou, Q.; Zhao, S.; Sun, B.; Xu, L.; Jiang, Y. Increased Numbers of NK Cells, NKT-Like Cells, and NK Inhibitory Receptors in Peripheral Blood of Patients with Chronic Obstructive Pulmonary Disease. Clin. Dev. Immunol. 2013, 2013, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Krijgsman, D.; de Vries, N.L.; Skovbo, A.; Andersen, M.N.; Swets, M.; Bastiaannet, E.; Vahrmeijer, A.L.; van de Velde, C.J.H.; Heemskerk, M.H.M.; Hokland, M.; et al. Characterization of circulating T-, NK-, and NKT cell subsets in patients with colorectal cancer: The peripheral blood immune cell profile. Cancer Immunol. Immunother. 2019, 68, 1011–1024. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, B.; Hassouneh, F.; Delgado, E.; Casado, J.G.; Tarazona, R. Natural killer cells in recurrent miscarriage: An overview. J. Reprod. Immunol. 2020, 142, 103209. [Google Scholar] [CrossRef] [PubMed]
- Dolff, S.; Bijl, M.; Huitema, M.G.; Limburg, P.C.; Kallenberg, C.G.; Abdulahad, W.H. Disturbed Th1, Th2, Th17 and Treg balance in patients with systemic lupus erythematosus. Clin. Immunol. 2011, 141, 197–204. [Google Scholar] [CrossRef]
- Álvarez-Rodríguez, L.; Martínez-Taboada, V.; Calvo-Alén, J.; Beares, I.; Villa, I.; López-Hoyos, M. Altered Th17/Treg Ratio in Peripheral Blood of Systemic Lupus Erythematosus but Not Primary Antiphospholipid Syndrome. Front. Immunol. 2019, 10, 391. [Google Scholar] [CrossRef]
- Yuliasih, Y.; Rahmawati, L.D.; Putri, R.M. Th17/Treg ratio and disease activity in systemic lupus erythematosus. Caspian J. Intern. Med. 2019, 10, 65–72. [Google Scholar] [CrossRef]
- Covas, M.I.; Esquerda, A.; García-Rico, A.; Mahy, N. Peripheral blood T-lymphocyte subsets in autoimmune thyroid disease. J. Investig. Allergy Clin. Immunol. 1992, 2, 131–135. [Google Scholar]
- Vitales-Noyola, M.; Ramos-Levi, A.M.; Martínez-Hernández, R.; Serrano-Somavilla, A.; Sampedro-Nuñez, M.; González-Amaro, R.; Marazuela, M. Pathogenic Th17 and Th22 cells are increased in patients with autoimmune thyroid disorders. Endocrine 2017, 57, 409–417. [Google Scholar] [CrossRef]
- Rydzewska, M.; Jaromin, M.; Pasierowska, I.E.; Stożek, K.; Bossowski, A. Role of the T and B lymphocytes in pathogenesis of autoimmune thyroid diseases. Thyroid Res. 2018, 11, 1–11. [Google Scholar] [CrossRef]
- Viglietta, V.; Baecher-Allan, C.; Weiner, H.L.; Hafler, D.A. Loss of Functional Suppression by CD4+CD25+ Regulatory T Cells in Patients with Multiple Sclerosis. J. Exp. Med. 2004, 199, 971–979. [Google Scholar] [CrossRef]
- Jones, A.P.; Kermode, A.G.; Lucas, R.M.; Carroll, W.M.; Nolan, D.; Hart, P.H. Circulating immune cells in multiple sclerosis. Clin. Exp. Immunol. 2016, 187, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Carotenuto, A.; Scalia, G.; Ausiello, F.; Moccia, M.; Russo, C.V.; Saccà, F.; De Rosa, A.; Criscuolo, C.; Del Vecchio, L.; Morra, V.B.; et al. CD4/CD8 ratio during natalizumab treatment in multiple sclerosis patients. J. Neuroimmunol. 2017, 309, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Van Langelaar, J.; Van der Vuurst de Vries, R.M.; Janssen, M.; Wierenga-Wolf, A.F.; Spilt, I.M.; Siepman, T.A.; Dankers, W.; Verjans, G.M.G.M.; De Vries, H.E.; Lubberts, E.; et al. T helper 17.1 cells associate with multiple sclerosis disease activity: Perspectives for early intervention. Brain 2018, 141, 1334–1349. [Google Scholar] [CrossRef] [PubMed]
- Quirant-Sánchez, B.; Presas-Rodriguez, S.; Mansilla, M.J.; Serra, A.T.; Hervás-García, J.V.; Brieva, L.; Moral-Torres, E.; Cano, A.; Munteis, E.; Navarro-Barriuso, J.; et al. Th1Th17CM Lymphocyte Subpopulation as a Predictive Biomarker of Disease Activity in Multiple Sclerosis Patients under Dimethyl Fumarate or Fingolimod Treatment. Mediat. Inflamm. 2019, 2019, 8147803. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, J.; Xing, X.; Wan, L.; Li, M. Increased frequency of Th17 cells in systemic sclerosis is related to disease activity and collagen overproduction. Arthritis Res. Ther. 2014, 16, R4. [Google Scholar] [CrossRef] [PubMed]
- Rafael-Vidal, C.; Perez, S.G.; Altabás, I.; Garcia, S.; Pego-Reigosa, J.M. Blocking IL-17: A Promising Strategy in the Treatment of Systemic Rheumatic Diseases. Int. J. Mol. Sci. 2020, 21, 7100. [Google Scholar] [CrossRef] [PubMed]
- Leipe, J.; Grunke, M.; Dechant, C.; Reindl, C.; Kerzendorf, U.; Schulze-Koops, H.; Skapenko, A. Role of Th17 cells in human autoimmune arthritis. Arthritis Care Res. 2010, 62, 2876–2885. [Google Scholar] [CrossRef]
- Boniface, K.; Boniface, D.M.K. Role of Th17 cells in the pathogenesis of rheumatoid arthritis. World J. Rheumatol. 2013, 3, 25. [Google Scholar] [CrossRef]
- Dardalhon, V.; Korn, T.; Kuchroo, V.K.; Anderson, A.C. Role of Th1 and Th17 cells in organ-specific autoimmunity. J. Autoimmun. 2008, 31, 252–256. [Google Scholar] [CrossRef]
- Pandya, J.M.; Lundell, A.-C.; Hallström, M.; Andersson, K.; Nordström, I.; Rudin, A. Circulating T helper and T regulatory subsets in untreated early rheumatoid arthritis and healthy control subjects. J. Leukoc. Biol. 2016, 100, 823–833. [Google Scholar] [CrossRef]
- Rao, D.A.; Gurish, M.F.; Marshall, J.L.; Slowikowski, K.; Fonseka, C.Y.; Liu, Y.; Donlin, L.T.; Henderson, L.A.; Wei, W.; Mizoguchi, F.; et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature 2017, 542, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Kunwar, S.; Dahal, K.; Sharma, S. Anti-IL-17 therapy in treatment of rheumatoid arthritis: A systematic literature review and meta-analysis of randomized controlled trials. Rheumatol. Int. 2016, 36, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Luo, B.; Zhang, Y.; Xie, Z.; Ye, Z. Treatment of reactive arthritis with biological agents: A review. Biosci. Rep. 2020, 40. [Google Scholar] [CrossRef] [PubMed]
- Pacha, O.; Sallman, M.A.; Evans, S.E. COVID-19: A case for inhibiting IL-17? Nat. Rev. Immunol. 2020, 20, 345–346. [Google Scholar] [CrossRef] [PubMed]
- Vojdani, A.; Lambert, J. The Role of Th17 in Neuroimmune Disorders: Target for CAM Therapy. Part I. Evidence-Based Complement. Altern. Med. 2011, 2011, 927294. [Google Scholar] [CrossRef]
- Vojdani, A.; Lambert, J. The Role of Th17 in Neuroimmune Disorders: Target for CAM Therapy. Part II. Evidence-Based Complement. Altern. Med. 2011, 2011, 984965. [Google Scholar] [CrossRef]
- Vojdani, A.; Lambert, J.; Kellermann, G. The Role of Th17 in Neuroimmune Disorders: A Target for CAM Therapy. Part III. Evidence-Based Complement. Altern. Med. 2011, 2011, 548086. [Google Scholar] [CrossRef]
- Benham, H.; Norris, P.; Goodall, J.; Wechalekar, M.D.; FitzGerald, O.; Szentpetery, A.; Smith, M.; Thomas, R.; Gaston, H. Th17 and Th22 cells in psoriatic arthritis and psoriasis. Arthritis Res. Ther. 2013, 15, R136. [Google Scholar] [CrossRef]
- Kondělková, K.; Vokurková, D.; Krejsek, J.; Borská, L.; Fiala, Z.; Hamáková, K.; Andrýs, C. The Number of Immunoregulatory T Cells is Increased in Patients with Psoriasis after Goeckerman Therapy. Acta Med. (Hradec Kralove, Czech Republic) 2012, 55, 91–95. [Google Scholar] [CrossRef][Green Version]
- Owczarczyk-Saczonek, A.; Czerwińska, J.; Placek, W. The role of regulatory T cells and anti-inflammatory cytokines in psoriasis. Acta Dermatovenerol. Alp. Pannonica Adriat. 2018, 27. [Google Scholar] [CrossRef]
- Karamehic, J.; Zecevic, L.; Resic, H.; Jukic, M.; Jukic, T.; Ridjic, O.; Panjeta, M.; Coric, J. Immunophenotype Lymphocyte of Peripheral Blood in Patients with Psoriasis. Med. Arch. 2014, 68, 236–238. [Google Scholar] [CrossRef] [PubMed]
- Luan, L.; Han, S.; Wang, H.; Liu, X. Down-regulation of the Th1, Th17, and Th22 pathways due to anti-TNF-α treatment in psoriasis. Int. Immunopharmacol. 2015, 29, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Silfvast-Kaiser, A.; Paek, S.Y.; Menter, A. Anti-IL17 therapies for psoriasis. Expert Opin. Biol. Ther. 2018, 19, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, A.; Tahmasebi, S.; Mahmood, A.; Kuznetsova, M.; Valizadeh, H.; Taghizadieh, A.; Nazemiyeh, M.; Aghebati-Maleki, L.; Jadidi-Niaragh, F.; Abbaspour-Aghdam, S.; et al. Th17 and Treg cells function in SARS-CoV2 patients compared with healthy controls. J. Cell. Physiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Phetsouphanh, C.; Darley, D.R.; Wilson, D.B.; Howe, A.; Munier, C.M.L.; Patel, S.K.; Juno, J.A.; Burrell, L.M.; Kent, S.J.; Dore, G.J.; et al. Immunological dysfunction persists for 8 months following initial mild-to-moderate SARS-CoV-2 infection. Nat. Immunol. 2022, 23, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Vojdani, A. Molecular and immunological evidence for SARS-CoV-2 being theautoimmune virus. In Proceedings of the Abstract, Oral Presentation, 13th International Congress on Autoimmunity, Athens, Greece, 10–13 June 2022. [Google Scholar]
- Wentworth, B.B.; Alexander, E.R. Seroepidemiology of Infections Due to Members of the Herpesvirus Group. Am. J. Epidemiol. 1971, 94, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Arvin, A.M.; Prober, C.G. Herpes Simplex Viruses. In Manual of Clinical Microbiology, 6th ed.; Baron, E., Pfaller, M.A., Tenover, F.C., Yolken, R.H., Eds.; ASM Press: Washington, DC, USA, 1995; pp. 876–883. [Google Scholar]
- Ashley, R.L.; Militoni, J.; Lee, F.; Nahmias, A.; Corey, L. Comparison of Western blot (immunoblot) and glycoprotein G-specific immunodot enzyme assay for detecting antibodies to herpes simplex virus types 1 and 2 in human sera. J. Clin. Microbiol. 1988, 26, 662–667. [Google Scholar] [CrossRef]
- Bernstein, D.I.; Bryson, Y.J.; Lovett, M.A. Antibody response to type-common and type-unique epitopes of herpes simplex virus polypeptides. J. Med. Virol. 1985, 15, 251–263. [Google Scholar] [CrossRef]
- Münz, C.; Lünemann, J.D.; Getts, M.T.; Miller, S.D. Antiviral immune responses: Triggers of or triggered by autoimmunity? Nat. Rev. Immunol. 2009, 9, 246–258. [Google Scholar] [CrossRef]
- Olson, J.K.; Croxford, J.L.; Calenoff, M.A.; Canto, M.C.D.; Miller, S.D. A virus-induced molecular mimicry model of multiple sclerosis. J. Clin. Investig. 2001, 108, 311–318. [Google Scholar] [CrossRef]
- Sanchez-Ruiz, M.; Wilden, L.; Muller, W.; Stenzel, W.; Brunn, A.; Miletic, H.; Schlüter, D.; Deckert, M. Molecular Mimicry between Neurons and an Intracerebral Pathogen Induces a CD8 T Cell-Mediated Autoimmune Disease. J. Immunol. 2008, 180, 8421–8433. [Google Scholar] [CrossRef] [PubMed]
- Nahmias, A.J.; Dannenbarger, J.; Wickliffe, C. Clinical aspects of infection with herpes simplex viruses 1 and 2. In The Human Herpesviruses: An Interdisciplinary Perspective; Nahmias, A., Dowdle, W.R., Shinazai, R.F., Eds.; Elsevier: Amsterdam, The Netherlands, 1981. [Google Scholar]
- Virtanen, J.O.; Jacobson, S. Viruses and Multiple Sclerosis. CNS Neurol. Disord.-Drug Targets 2012, 11, 528–544. [Google Scholar] [CrossRef] [PubMed]
- Draborg, A.H.; Duus, K.; Houen, G. Epstein-Barr Virus and Systemic Lupus Erythematosus. Clin. Dev. Immunol. 2012, 2012, 370516. [Google Scholar] [CrossRef] [PubMed]
- Draborg, A.H.; Duus, K.; Houen, G. Epstein-Barr Virus in Systemic Autoimmune Diseases. Clin. Dev. Immunol. 2013, 2013, 535738. [Google Scholar] [CrossRef]
- Yu, S.-F.; Wu, H.-C.; Tsai, W.-C.; Yen, J.-H.; Chiang, W.; Yuo, C.-Y.; Lu, S.-N.; Chiang, L.-C.; Chen, C.-J. Detecting Epstein-Barr virus DNA from peripheral blood mononuclear cells in adult patients with systemic lupus erythematosus in Taiwan. Med. Microbiol. Immunol. 2004, 194, 115–120. [Google Scholar] [CrossRef]
- Moon, U.Y.; Park, S.J.; Oh, S.T.; Kim, W.-U.; Park, S.-H.; Lee, S.-H.; Cho, C.-S.; Kim, H.-Y.; Lee, W.-K.; Lee, S.K. Patients with systemic lupus erythematosus have abnormally elevated Epstein–Barr virus load in blood. Arthritis Res. Ther. 2004, 6, R295–R302. [Google Scholar] [CrossRef]
- Gross, A.J.; Hochberg, D.; Rand, W.M.; Thorley-Lawson, D.A. EBV and Systemic Lupus Erythematosus: A New Perspective. J. Immunol. 2005, 174, 6599–6607. [Google Scholar] [CrossRef]
- Draborg, A.H.; Jorgensen, J.M.; Müller, H.; Nielsen, C.T.; Jacobsen, S.; Iversen, L.V.; Theander, E.; Nielsen, L.P.; Houen, G.; Duus, K. Epstein–Barr virus early antigen diffuse (EBV-EA/D)-directed immunoglobulin A antibodies in systemic lupus erythematosus patients. Scand. J. Rheumatol. 2012, 41, 280–289. [Google Scholar] [CrossRef]
- Poole, B.D.; Scofield, R.H.; Harley, J.B.; James, J.A. Epstein-Barr virus and molecular mimicry in systemic lupus erythematosus. Autoimmunity 2006, 39, 63–70. [Google Scholar] [CrossRef]
- Iwakiri, D.; Zhou, L.; Samanta, M.; Matsumoto, M.; Ebihara, T.; Seya, T.; Imai, S.; Fujieda, M.; Kawa, K.; Takada, K. Epstein-Barr virus (EBV)–encoded small RNA is released from EBV-infected cells and activates signaling from toll-like receptor 3. J. Exp. Med. 2009, 206, 2091–2099. [Google Scholar] [CrossRef]
- Sanders, V.J.; Waddell, A.E.; Felisan, S.L.; Li, X.; Conrad, A.J.; Tourtellotte, W.W. Herpes Simplex Virus in Postmortem Multiple Sclerosis Brain Tissue. Arch. Neurol. 1996, 53, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Chucair-Elliott, A.J.; Conrady, C.; Zheng, M.; Kroll, C.M.; Lane, T.E.; Carr, D.J.J. Microglia-induced IL-6 protects against neuronal loss following HSV-1 infection of neural progenitor cells. Glia 2014, 62, 1418–1434. [Google Scholar] [CrossRef] [PubMed]
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L.; et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 2022, 375, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Ercolini, A.M.; Miller, S.D. The role of infections in autoimmune disease. Clin. Exp. Immunol. 2008, 155, 1–15. [Google Scholar] [CrossRef]
- Zhao, Z.-S.; Granucci, F.; Yeh, L.; Schaffer, P.A.; Cantor, H. Molecular Mimicry by Herpes Simplex Virus-Type 1: Autoimmune Disease After Viral Infection. Science 1998, 279, 1344–1347. [Google Scholar] [CrossRef]
- Verjans, G.M.; Remeijer, L.; Mooy, C.M.; Osterhaus, A.D. Herpes-simples virus-specific T cells infiltrate the cornea of patients with herpetic stromal keratitis: No evidence for autoreactive T cells. Investig. Opthalmol. Vis. Sci. 2000, 41, 2607–2612. [Google Scholar]
- Pak, C.; McArthur, R.; Eun, H.-M.; Yoon, J.-W. Association of Cytomegalovirus Infection with Autoimmune Type 1 Diabetes. Lancet 1988, 332, 1–4. [Google Scholar] [CrossRef]
- Nicoletti, F.; Scalia, G.; Lunetta, M.; Condorelli, F.; Di Mauro, M.; Barcellini, W.; Stracuzzi, S.; Pagano, M.; Meroni, P. Correlation between islet cell antibodies and anti-cytomegalovirus IgM and IgG antibodies in healthy first-degree relatives of type 1 (insulin-dependent) diabetic patients. Clin. Immunol. Immunopathol. 1990, 55, 139–147. [Google Scholar] [CrossRef]
- Kemppainen, K.M.; Lynch, K.F.; Liu, E.; Lönnrot, M.; Simell, V.; Briese, T.; Koletzko, S.; Hagopian, W.; Rewers, M.; She, J.-X.; et al. Factors That Increase Risk of Celiac Disease Autoimmunity After a Gastrointestinal Infection in Early Life. Clin. Gastroenterol. Hepatol. 2016, 15, 694–702.e5. [Google Scholar] [CrossRef]
- Lerner, A.; Arleevskaya, M.; Schmiedl, A.; Matthias, T. Microbes and Viruses Are Bugging the Gut in Celiac Disease. Are They Friends or Foes? Front. Microbiol. 2017, 8, 1392. [Google Scholar] [CrossRef]
- Geginat, J.; Paroni, M.; Pagani, M.; Galimberti, D.; De Francesco, R.; Scarpini, E.; Abrignani, S. The Enigmatic Role of Viruses in Multiple Sclerosis: Molecular Mimicry or Disturbed Immune Surveillance? Trends Immunol. 2017, 38, 498–512. [Google Scholar] [CrossRef] [PubMed]
- Arleevskaya, M.I.; Manukyan, G.; Inoue, R.; Aminov, R. Editorial: Microbial and Environmental Factors in Autoimmune and Inflammatory Diseases. Front. Immunol. 2017, 8, 243. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Kaistha, S.D.; Rouse, B.T. Viruses and autoimmunity. Autoimmunity 2006, 39, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Coppieters, K.T.; Wiberg, A.; Von Herrath, M.G. Viral infections and molecular mimicry in type 1 diabetes. APMIS 2012, 120, 941–949. [Google Scholar] [CrossRef]
- Gauntt, C.J.; Arizpe, H.M.; Higdon, A.L.; Wood, H.J.; Bowers, D.F.; Rozek, M.M.; Crawley, R. Molecular mimicry, an-ti-coxsackievirus B3 neutralizing monoclonal antibodies, and myocarditis. J. Immunol. 1995, 154, 2893–2995. [Google Scholar]
- Croxford, J.; Olson, J.K.; Miller, S.D. Epitope spreading and molecular mimicry as triggers of autoimmunity in the Theiler’s virus-induced demyelinating disease model of multiple sclerosis. Autoimmun. Rev. 2002, 1, 251–260. [Google Scholar] [CrossRef]
- Getts, D.R.; Chastain, E.M.L.; Terry, R.L.; Miller, S.D. Virus infection, antiviral immunity, and autoimmunity. Immunol. Rev. 2013, 255, 197–209. [Google Scholar] [CrossRef]
- Vojdani, A.; Monro, J.; Lanzisera, F.; Sadeghi, H. Serological cross-reactivity between viruses and their contribution to autoimmunity. Autoimmun. Rev. 2021, 20, 102840. [Google Scholar] [CrossRef]
- Fujinami, R.S.; von Herrath, M.G.; Christen, U.; Whitton, J.L. Molecular Mimicry, Bystander Activation, or Viral Persistence: Infections and Autoimmune Disease. Clin. Microbiol. Rev. 2006, 19, 80–94. [Google Scholar] [CrossRef]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). J. Cereb. Blood Flow Metab. 2011, 164, 1079–1106. [Google Scholar] [CrossRef]
- Leis, A.A.; Szatmary, G.; Ross, M.A.; Stokic, D. West nile virus infection and myasthenia gravis. Muscle Nerve 2013, 49, 26–29. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.D.; Vanderlugt, C.L.; Begolka, W.S.; Pao, W.; Yauch, R.L.; Neville, K.L.; Katz-Levy, Y.; Carrizosa, A.; Kim, B.S. Persistent infection with Theiler’s virus leads to CNS autoimmunity via epitope spreading. Nat. Med. 1997, 3, 1133–1136. [Google Scholar] [CrossRef] [PubMed]
- Nanbo, A.; Inoue, K.; Adachi-Takasawa, K.; Takada, K. Epstein-Barr virus RNA confers resistance to interferon-alpha-induced apoptosis in Burkitt’s lymphoma. EMBO J. 2002, 21, 954–965. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.J. Alzheimer’s Disease: A Pathogenetic Autoimmune Disorder Caused by Herpes Simplex in a Gene-Dependent Manner. Int. J. Alzheimer’s Dis. 2010, 2010, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, R.F.; Lathe, R.; Balin, B.J.; Ball, M.J.; Bearer, E.L.; Braak, H.; Bullido, M.J.; Carter, C.; Clerici, M.; Cosby, S.L.; et al. Microbes and Alzheimer’s Disease. J. Alzheimer’s Dis. JAD 2016, 5, 3–8. [Google Scholar] [CrossRef]
- Vojdani, A.; Vojdani, E.; Saidara, E.; Kharrazian, D. Reaction of Amyloid-β Peptide Antibody with Different Infectious Agents Involved in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 63, 847–860. [Google Scholar] [CrossRef]
- Remington, J.S.; Klein, J.O.; Wilson, C.B.; Baker, C.J. Infectious Diseases of the Fetus and Newborn Infant; Saunders Company: Philadelphia, PA, USA, 1976; pp. 521–583. [Google Scholar] [CrossRef]
- Williamson, A.P. Clinical Review: The Varicella-Zoster Virus in the Etiology of Severe Congenital Defects. Clin. Pediatr. 1975, 14, 553–555. [Google Scholar] [CrossRef]
- Gilden, D.; White, T.; Khmeleva, N.; Heintzman, A.; Choe, A.; Boyer, P.J.; Grose, C.; Carpenter, J.E.; Rempel, A.; Bos, N.; et al. Prevalence and distribution of VZV in temporal arteries of patients with giant cell arteritis. Neurology 2015, 84, 1948–1955. [Google Scholar] [CrossRef]
- Franco-Paredes, C.; Bellehemeur, T.; Merchant, A.; Sanghi, P.; DiazGranados, C.; Rimland, D. Aspetic Meningitis and Optic Neuritis Preceding Varicella-Zoster Progressive Outer Retinal Necrosis in a Patient with AIDS. AIDS 2002, 16, 1045–1049. [Google Scholar] [CrossRef]
- Lennette, E.T.; Henle, W. Epstein-Barr virus infections: Clinical and serological features. Lab Manag. 1987, 25, 23–28. [Google Scholar]
- Henle, G.; Henle, W.; Horwitz, C.A. Antibodies to Epstein-Barr Virus-Associated Nuclear Antigen in Infectious Mononucleosis. J. Infect. Dis. 1974, 130, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Farina, A.; Cirone, M.; York, M.; Lenna, S.; Padilla, C.; Mclaughlin, S.; Faggioni, A.; Lafyatis, R.; Trojanowska, M.; Farina, G. Epstein–Barr Virus Infection Induces Aberrant TLR Activation Pathway and Fibroblast–Myofibroblast Conversion in Scleroderma. J. Investig. Dermatol. 2014, 134, 954–964. [Google Scholar] [CrossRef] [PubMed]
- Poole, B.D.; Gross, T.; Maier, S.; Harley, J.B.; James, J.A. Lupus-like autoantibody development in rabbits and mice after immunization with EBNA-1 fragments. J. Autoimmun. 2008, 31, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Sospedra, M.; Martin, R. Molecular mimicry in multiple sclerosis. Autoimmunity 2006, 39, 3–8. [Google Scholar] [CrossRef]
- Paroni, M.; Maltese, V.; De Simone, M.; Ranzani, V.; Larghi, P.; Fenoglio, C.; Pietroboni, A.M.; De Riz, M.A.; Crosti, M.C.; Maglie, S.; et al. Recognition of viral and self-antigens by T H 1 and T H 1/T H 17 central memory cells in patients with multiple sclerosis reveals distinct roles in immune surveillance and relapses. J. Allergy Clin. Immunol. 2017, 140, 797–808. [Google Scholar] [CrossRef]
- Donati, D.; Martinelli, E.; Cassiani-Ingoni, R.; Ahlqvist, J.; Hou, J.; Major, E.O.; Jacobson, S. Variant-Specific Tropism of Human Herpesvirus 6 in Human Astrocytes. J. Virol. 2005, 79, 9439–9448. [Google Scholar] [CrossRef][Green Version]
- Krueger, G.R.; Sander, C.; Hoffmann, A.; Barth, A.; Koch, B.; Braun, M. Isolation of human herpesvirus-6 (HHV-6) from patients with collagen vascular diseases. Vivo 1991, 5, 217–225. [Google Scholar]
- Broccolo, F.; Drago, F.; Paolino, S.; Cassina, G.; Gatto, F.; Fusetti, L.; Matteoli, B.; Zaccaria, E.; Parodi, A.; Lusso, P.; et al. Reactivation of human herpesvirus 6 (HHV-6) infection in patients with connective tissue diseases. J. Clin. Virol. 2009, 46, 43–46. [Google Scholar] [CrossRef]
- Rogez, S.; Vidal, E.; Liozon, F.; Denis, F. Primary Sjogren’s Syndrome and Antibodies to Human Herpesvirus Type 6. Clin. Infect. Dis. 1994, 19, 1159–1160. [Google Scholar] [CrossRef]
- Yagasaki, H.; Kato, M.; Shimizu, N.; Shichino, H.; Chin, M.; Mugishima, H. Autoimmune hemolytic anemia and autoimmune neutropenia in a child with erythroblastopenia of childhood (TEC) caused by human herpesvirus-6 (HHV-6). Ann. Hematol. 2010, 90, 851–852. [Google Scholar] [CrossRef]
- Cirone, M.; Cuomo, L.; Zompetta, C.; Ruggieri, S.; Frati, L.; Faggioni, A.; Ragona, G. Human herpesvirus 6 and multiple sclerosis: A study of t cell cross-reactivity to viral and myelin basic protein antigens. J. Med. Virol. 2002, 68, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Liakos, V.; Michou, V.; Kapranos, N.; Kaltsas, G.; Tsilivakos, V.; Tsatsoulis, A. Detection of Herpes Virus DNA in Post-operative Thyroid Tissue Specimens of Patients with Autoimmune Thyroid Disease. Exp. Clin. Endocrinol. Diabetes 2008, 116, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Sekigawa, I.; Nawata, M.; Seta, N.; Yamada, M.; Iida, N.; Hashimoto, H. Cytomegalovirus infection in patients with systemic lupus erythematosus. Clin. Exp. Rheumatol. 2002, 20, 559–564. [Google Scholar]
- Cappel, R.; de Cuyper, F.; de Braekeleer, J. Rapid detection of IgG and IgM antibodies for cytomegalovirus by the enzyme linked immunosorbent assay (ELISA). Arch. Virol. 1978, 58, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Lunardi, C.; Bason, C.; Corrocher, R.; Puccetti, A. Induction of endothelial cell damage by hCMV molecular mimicry. Trends Immunol. 2005, 26, 19–24. [Google Scholar] [CrossRef]
- Cytomegalovirus (CMV) and Congenital CMV Infection. Available online: https://www.cdc.gov/cmv/overview.html (accessed on 1 April 2022).
- Bradshaw, M.J.; Pawate, S.; Lennon, V.A.; Bloch, K.C.; Brown, K.M. Herpes simplex virus 1 encephalitis associated with voltage-gated calcium channel autoimmunity. Neurology 2015, 85, 2176–2177. [Google Scholar] [CrossRef]
- Armangue, T.; Leypoldt, F.; Málaga, I.; Raspall-Chaure, M.; Marti, I.; Nichter, C.; Pugh, J.; Vicente-Rasoamalala, M.; Lafuente-Hidalgo, M.; Macaya, A.; et al. Herpes simplex virus encephalitis is a trigger of brain autoimmunity. Ann. Neurol. 2013, 75, 317–323. [Google Scholar] [CrossRef]
- Kothur, K.; Gill, D.; Wong, M.; Mohammad, S.S.; Bandodkar, S.; Arbunckle, S.; Wienholt, L.; Dale, R.C. Cerebrospinal fluid cyto-/chemokine profile during acute herpes simplex virus induced anti-N-methyl-d-aspartate receptor encephalitis and in chronic neurological sequelae. Dev. Med. Child Neurol. 2017, 59, 806–814. [Google Scholar] [CrossRef]
- Deshpande, S.; Zheng, M.; Lee, S.; Banerjee, K.; Gangappa, S.; Kumaraguru, U.; Rouse, B.T. Bystander Activation Involving T Lymphocytes in Herpetic Stromal Keratitis. J. Immunol. 2001, 167, 2902–2910. [Google Scholar] [CrossRef]
- Itzhaki, R. Overwhelming Evidence for a Major Role for Herpes Simplex Virus Type 1 (HSV1) in Alzheimer’s Disease (AD); Underwhelming Evidence against. Vaccines 2021, 9, 679. [Google Scholar] [CrossRef]
- Sotelo, J.; Corona, T. Varicella Zoster Virus and Relapsing Remitting Multiple Sclerosis. Mult. Scler. Int. 2011, 2011, 214763. [Google Scholar] [CrossRef] [PubMed]
- Cabibi, D. Autoimmune hepatitis following Epstein-Barr virus infection. BMJ Case Rep. 2008, 2008. [Google Scholar] [CrossRef] [PubMed]
- Pender, M.P. Infection of autoreactive B lymphocytes with EBV, causing chronic autoimmune diseases. Trends Immunol. 2003, 24, 584–588. [Google Scholar] [CrossRef] [PubMed]
- Nagata, K.; Kumata, K.; Nakayama, Y.; Satoh, Y.; Sugihara, H.; Hara, S.; Matsushita, M.; Kuwamoto, S.; Kato, M.; Murakami, I.; et al. Epstein–Barr Virus Lytic Reactivation Activates B Cells Polyclonally and Induces Activation-Induced Cytidine Deaminase Expression: A Mechanism Underlying Autoimmunity and Its Contribution to Graves’ Disease. Viral Immunol. 2017, 30, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Janegova, A.; Janega, P.; Rychly, B.; Kuracinova, K.; Babal, P. Rola infekcji wirusem Epstein-Barr’a w rozwoju autoimmunologicznych chorób tarczycy. Endokrynol. Polska 2015, 66, 132–136. [Google Scholar] [CrossRef]
- Zivadinov, R.; Guan, Y.; Jakimovski, D.; Ramanathan, M.; Weinstock-Guttman, B. The role of Epstein-Barr virus in multiple sclerosis: From molecular pathophysiology to in vivo imaging. Neural Regen. Res. 2019, 14, 373–386. [Google Scholar] [CrossRef]
- Robinson, W.H.; Steinman, L. Epstein-Barr virus and multiple sclerosis. Science 2022, 375, 264–265. [Google Scholar] [CrossRef]
- Svendsen, A.J.; Westergaard, M.C.W.; Draborg, A.H.; Holst, R.; Kyvik, K.O.; Jakobsen, M.A.; Junker, P.; Houen, G. Altered Antibody Response to Epstein-Barr Virus in Patients With Rheumatoid Arthritis and Healthy Subjects Predisposed to the Disease. A Twin Study. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef]
- Neidhart, M.; Kuchen, S.; Distler, O.; Michel, B.A.; Gay, R.E.; Gay, S. Increased serum levels of antibodies against human cytomegalovirus and prevalence of autoantibodies in systemic sclerosis. Arthritis Care Res. 1999, 42, 389–392. [Google Scholar] [CrossRef]
- Farina, A.; Rosato, E.; York, M.; Gewurz, B.E.; Trojanowska, M.; Farina, G.A. Innate Immune Modulation Induced by EBV Lytic Infection Promotes Endothelial Cell Inflammation and Vascular Injury in Scleroderma. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef]
- Davignon, J.-L.; Combe, B.; Cantagrel, A. Cytomegalovirus infection: Friend or foe in rheumatoid arthritis? Arthritis Res. Ther. 2021, 23, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Dolcino, M.; Puccetti, A.; Barbieri, A.; Bason, C.; Tinazzi, E.; Ottria, A.; Patuzzo, G.; Martinelli, N.; Lunardi, C. Infections and autoimmunity: Role of human cytomegalovirus in autoimmune endothelial cell damage. Lupus 2015, 24, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Caselli, E.; D’Accolti, M.; Soffritti, I.; Zatelli, M.C.; Rossi, R.; Degli Uberti, E.; Di Luca, D. HHV-6A in vitro infection of thyrocytes and T cells alters the expression of miRNA associated to autoimmune thyroiditis. Virol. J. 2017, 14, 3. [Google Scholar] [CrossRef] [PubMed]
- Akhyani, N.; Berti, R.; Brennan, M.B.; Soldan, S.S.; Eaton, J.M.; McFarland, H.F.; Jacobson, S. Tissue Distribution and Variant Characterization of Human Herpesvirus (HHV)–6: Increased Prevalence of HHV-6A in Patients with Multiple Sclerosis. J. Infect. Dis. 2000, 182, 1321–1325. [Google Scholar] [CrossRef] [PubMed]
- Tejada-Simon, M.V.; Zang, Y.C.Q.; Hong, J.; Rivera, V.M.; Zhang, J.Z. Cross-reactivity with myelin basic protein and human herpesvirus-6 in multiple sclerosis. Ann. Neurol. 2003, 53, 189–197. [Google Scholar] [CrossRef]
- Broccolo, F.; Drago, F.; Cassina, G.; Fava, A.; Fusetti, L.; Matteoli, B.; Ceccherini-Nelli, L.; Sabbadini, M.G.; Lusso, P.; Parodi, A.; et al. Selective reactivation of human herpesvirus 6 in patients with autoimmune connective tissue diseases. J. Med. Virol. 2013, 85, 1925–1934. [Google Scholar] [CrossRef]
- Nagler, C.R. Modern World Influences on the Microbiome and Their Consequences for Immune-Mediated Disease. J. Immunol. 2021, 207, 1695–1696. [Google Scholar] [CrossRef]
- Blaser, M. The theory of disappearing microbiota and the epidemics of chronic diseases. Nat. Rev. Immunol. 2017, 17, 461–463. [Google Scholar] [CrossRef]
- Khan, M.F.; Wang, G. Environmental agents, oxidative stress and autoimmunity. Curr. Opin. Toxicol. 2017, 7, 22–27. [Google Scholar] [CrossRef]
- Dehner, C.; Fine, R.; Kriegel, M. The microbiome in systemic autoimmune disease: Mechanistic insights from recent studies. Curr. Opin. Rheumatol. 2019, 31, 201–207. [Google Scholar] [CrossRef]
- De Luca, F.; Shoenfeld, Y. The microbiome in autoimmune diseases. Clin. Exp. Immunol. 2018, 195, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Selmi, C.; Tang, R.; Gershwin, M.E.; Ma, X. The microbiome and autoimmunity: A paradigm from the gut–liver axis. Cell. Mol. Immunol. 2018, 15, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Gianchecchi, E.; Fierabracci, A. Recent Advances on Microbiota Involvement in the Pathogenesis of Autoimmunity. Int. J. Mol. Sci. 2019, 20, 283. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.M.; Edwards, M.R.; Mu, Q.; Yu, Y.; Vieson, M.D.; Reilly, C.; Ahmed, S.A.; Bankole, A.A. Gut Microbiota in Human Systemic Lupus Erythematosus and a Mouse Model of Lupus. Appl. Environ. Microbiol. 2018, 84. [Google Scholar] [CrossRef] [PubMed]
- Kirby, T.O.; Ochoa-Repáraz, J. The Gut Microbiome in Multiple Sclerosis: A Potential Therapeutic Avenue. Med. Sci. 2018, 6, 69. [Google Scholar] [CrossRef] [PubMed]
- Maeda, Y.; Kurakawa, T.; Umemoto, E.; Motooka, D.; Ito, Y.; Gotoh, K.; Hirota, K.; Matsushita, M.; Furuta, Y.; Narazaki, M.; et al. Dysbiosis Contributes to Arthritis Development via Activation of Autoreactive T Cells in the Intestine. Arthritis Rheumatol. 2016, 68, 2646–2661. [Google Scholar] [CrossRef]
- Bellocchi, C.; Volkmann, E.R. Update on the Gastrointestinal Microbiome in Systemic Sclerosis. Curr. Rheumatol. Rep. 2018, 20, 49. [Google Scholar] [CrossRef]
- Clemente, J.C.; Manasson, J.; Scher, J.U. The role of the gut microbiome in systemic inflammatory disease. BMJ 2018, 360, j5145. [Google Scholar] [CrossRef]
- Moulton, V.R.; Suárez-Fueyo, A.; Meidan, E.; Li, H.; Mizui, M.; Tsokos, G.C. Pathogenesis of Human Systemic Lupus Erythematosus: A Cellular Perspective. Trends Mol. Med. 2017, 23, 615–635. [Google Scholar] [CrossRef]
- De Oliveira, G.L.V.; Leite, A.Z.; Higuchi, B.S.; Gonzaga, M.I.; Mariano, V.S. Intestinal dysbiosis and probiotic applications in autoimmune diseases. Immunology 2017, 152, 1–12. [Google Scholar] [CrossRef]
- Mu, Q.; Zhang, H.; Liao, X.; Lin, K.; Liu, H.; Edwards, M.R.; Ahmed, S.A.; Yuan, R.; Li, L.; Cecere, T.E.; et al. Control of lupus nephritis by changes of gut microbiota. Microbiome 2017, 5, 1–12. [Google Scholar] [CrossRef]
- Hevia, A.; Milani, C.; López, P.; Cuervo, A.; Arboleya, S.; Duranti, S.; Turroni, F.; González, S.; Suárez, A.; Gueimonde, M.; et al. Intestinal Dysbiosis Associated with Systemic Lupus Erythematosus. mBio 2014, 5, e01548-14. [Google Scholar] [CrossRef] [PubMed]
- Katz, U.; Zandman-Goddard, G. Drug-induced lupus: An update. Autoimmun. Rev. 2010, 10, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Azzouz, D.; Omarbekova, A.; Heguy, A.; Schwudke, D.; Gisch, N.; Rovin, B.H.; Caricchio, R.; Buyon, J.P.; Alekseyenko, A.V.; Silverman, G.J. Lupus nephritis is linked to disease-activity associated expansions and immunity to a gut commensal. Ann. Rheum. Dis. 2019, 78, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Zhang, Z.; Yu, A.; Wang, W.; Wei, Z.; Akhter, E.; Maurer, K.; Reis, P.C.; Song, L.; Petri, M.; et al. The SLE Transcriptome Exhibits Evidence of Chronic Endotoxin Exposure and Has Widespread Dysregulation of Non-Coding and Coding RNAs. PLoS ONE 2014, 9, e93846. [Google Scholar] [CrossRef] [PubMed]
- Ogunrinde, E.; Zhou, Z.; Luo, Z.; Alekseyenko, A.; Li, Q.; Macedo, D.; Kamen, D.L.; Oates, J.C.; Gilkeson, G.S.; Jiang, W. A Link Between Plasma Microbial Translocation, Microbiome, and Autoantibody Development in First-Degree Relatives of Systemic Lupus Erythematosus Patients. Arthritis Rheumatol. 2019, 71, 1858–1868. [Google Scholar] [CrossRef]
- Tursi, S.A.; Lee, E.Y.; Medeiros, N.J.; Lee, M.; Nicastro, L.K.; Buttaro, B.; Gallucci, S.; Wilson, R.P.; Wong, G.C.L.; Tükel, C. Bacterial amyloid curli acts as a carrier for DNA to elicit an autoimmune response via TLR2 and TLR9. PLoS Pathog. 2017, 13, e1006315. [Google Scholar] [CrossRef] [PubMed]
- Gallo, P.M.; Rapsinski, G.J.; Wilson, R.P.; Oppong, G.O.; Sriram, U.; Goulian, M.; Buttaro, B.; Caricchio, R.; Gallucci, S.; Tükel, C. Amyloid-DNA Composites of Bacterial Biofilms Stimulate Autoimmunity. Immunity 2015, 42, 1171–1184. [Google Scholar] [CrossRef]
- Glassner, K.L.; Abraham, B.P.; Quigley, E.M. The microbiome and inflammatory bowel disease. J. Allergy Clin. Immunol. 2020, 145, 16–27. [Google Scholar] [CrossRef]
- Boziki, M.K.; Kesidou, E.; Theotokis, P.; Mentis, A.-F.A.; Karafoulidou, E.; Melnikov, M.; Sviridova, A.; Rogovski, V.; Boyko, A.; Grigoriadis, N. Microbiome in Multiple Sclerosis: Where Are We, What We Know and Do Not Know. Brain Sci. 2020, 10, 234. [Google Scholar] [CrossRef]
- Zhang, H.; Liao, X.; Sparks, J.B.; Luo, X.M. Dynamics of Gut Microbiota in Autoimmune Lupus. Appl. Environ. Microbiol. 2014, 80, 7551–7560. [Google Scholar] [CrossRef] [PubMed]
- Abdelhamid, L.; Luo, X.M. Retinoic Acid, Leaky Gut, and Autoimmune Diseases. Nutrients 2018, 10, 1016. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.Y.; Ramirez, Z.E.; Surana, N.K. A Modern-World View of Host–Microbiota–Pathogen Interactions. J. Immunol. 2021, 207, 1710–1718. [Google Scholar] [CrossRef]
- Gupta, S.; Allen-Vercoe, E.; Petrof, E.O. Fecal microbiota transplantation: In perspective. Ther. Adv. Gastroenterol. 2015, 9, 229–239. [Google Scholar] [CrossRef]
- Britton, G.J.; Contijoch, E.J.; Spindler, M.P.; Aggarwala, V.; Dogan, B.; Bongers, G.; Mateo, L.S.; Baltus, A.; Das, A.; Gevers, D.; et al. Defined microbiota transplant restores Th17/RORγt + regulatory T cell balance in mice colonized with inflammatory bowel disease microbiotas. Proc. Natl. Acad. Sci. USA 2020, 117, 21536–21545. [Google Scholar] [CrossRef] [PubMed]
- Shamriz, O.; Mizrahi, H.; Werbner, M.; Shoenfeld, Y.; Avni, O.; Koren, O. Microbiota at the crossroads of autoimmunity. Autoimmun. Rev. 2016, 15, 859–869. [Google Scholar] [CrossRef] [PubMed]
- Klag, K.A.; Round, J.L. Microbiota-Immune Interactions Regulate Metabolic Disease. J. Immunol. 2021, 207, 1719–1724. [Google Scholar] [CrossRef]
- Bishai, J.D.; Palm, N.W. Small Molecule Metabolites at the Host–Microbiota Interface. J. Immunol. 2021, 207, 1725–1733. [Google Scholar] [CrossRef]
- Dotan, A.; Mahroum, N.; Bogdanos, D.P.; Shoenfeld, Y. COVID-19 as an infectome paradigm of autoimmunity. J. Allergy Clin. Immunol. 2022, 149, 63–64. [Google Scholar] [CrossRef]
- Wang, H.; Wang, H.; Sun, Y.; Ren, Z.; Zhu, W.; Li, A.; Cui, G. Potential Associations Between Microbiome and COVID-19. Front. Med. 2021, 8. [Google Scholar] [CrossRef]
- Ascher, S.; Wilms, E.; Pontarollo, G.; Formes, H.; Bayer, F.; Müller, M.; Malinarich, F.; Grill, A.; Bosmann, M.; Saffarzadeh, M.; et al. Gut Microbiota Restricts NETosis in Acute Mesenteric Ischemia-Reperfusion Injury. Arter. Thromb. Vasc. Biol. 2020, 40, 2279–2292. [Google Scholar] [CrossRef] [PubMed]
- Chamardani, T.M.; Amiritavassoli, S. Inhibition of NETosis for treatment purposes: Friend or foe? Mol. Cell. Biochem. 2022, 477, 673–688. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Chen, X.; Liu, X. NETosis and Neutrophil Extracellular Traps in COVID-19: Immunothrombosis and Beyond. Front. Immunol. 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Saito, M.; Tamura, A.; Prawisuda, D.; Mizutani, T.; Yotsuyanagi, H. The human microbiome and COVID-19: A systematic review. PLoS ONE 2021, 16, e0253293. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Song, Z.-G.; Liu, C.; Tan, S.; Lin, S.; Zhu, J.; Dai, F.-H.; Gao, J.; She, J.-L.; Mei, Z.; et al. Gut microbiome alterations and gut barrier dysfunction are associated with host immune homeostasis in COVID-19 patients. BMC Med. 2022, 20, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Cheng, X.; Jiang, G.; Tang, H.; Ming, S.; Tang, L.; Lu, J.; Guo, C.; Shan, H.; Huang, X. Altered oral and gut microbiota and its association with SARS-CoV-2 viral load in COVID-19 patients during hospitalization. npj Biofilms Microbiomes 2021, 7. [Google Scholar] [CrossRef] [PubMed]
- Finlay, B.B.; Amato, K.R.; Azad, M.; Blaser, M.J.; Bosch, T.C.G.; Chu, H.; Dominguez-Bello, M.G.; Ehrlich, S.D.; Elinav, E.; Geva-Zatorsky, N.; et al. The hygiene hypothesis, the COVID pandemic, and consequences for the human microbiome. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.F.; Wang, H. Environmental Exposures and Autoimmune Diseases: Contribution of Gut Microbiome. Front. Immunol. 2020, 10, 3094. [Google Scholar] [CrossRef]
- Wallace, M.A.G.; Kormos, T.M.; Pleil, J.D. Blood-borne biomarkers and bioindicators for linking exposure to health effects in environmental health science. J. Toxicol. Environ. Heal. Part B 2016, 19, 380–409. [Google Scholar] [CrossRef]
















| Pathogen Antigen | Cross-Reactive Self-Antigens | Autoimmune Disease |
|---|---|---|
| Herpes simplex virus | Corneal antigen | Stromal keratitis |
| Campylobacter jejuni | Ganglioside in peripheral nerve | Guillain-Barré syndrome |
| Coxsackievirus | Glutamic acid decarboxylase | Type 1 diabetes |
| Theiler’s murine encephalomyelitis virus | Proteolipid protein | Multiple sclerosis |
| Yersinia enterocolitica | Thyrotropin receptor | Thyroid autoimmunity |
| Borrelia burgdorferi | Leukocyte function associated antigen | Lyme arthritis |
| Salmonella typhi and Yersinia enterocolitica | HLA-B27 | Reactive arthritis |
| HHV-6, EBV, Rubeolla, influenza virus, and HPV | Myelin basic protein | Multiple sclerosis |
| Streptococcal M protein | Myosin and other heart valve proteins | Rheumatic fever |
| Porphyromonas gingivalis | Heat-shock proteins | Atherosclerosis |
| Trypanosoma cruzi | Cardiac myosis | Chagas heart disease |
| SARS-CoV-2 | More than 20 tissue antigens | More than 20 ADs |
| Shared Heptapeptide | Human Proteins Sharing Heptapeptides with SARS-CoV-2 |
|---|---|
| SSRSSSR | Corneal antigen |
| ALALLLL | Ganglioside in peripheral nerve |
| ALALLLL | Glutamic acid decarboxylase |
| ALALLLL | Proteolipid protein |
| IGAGICA | Thyrotropin receptor |
| TGRLQSL | Leukocyte function associated antigen |
| NASVVNI | HLA-B27 |
| AEGSRGG | More than 20 tissue antigens |
| Other Viral Antigen | Other Viral Sequence | Mapped Start to End | HHV-6 Sequence | ID (%) |
|---|---|---|---|---|
| Crystal Structure of NendoU (Uridylate-specific endoribonuclease, nsp15) of SARS-CoV-2 | SHHHHHHSSG | 4–13 | SHHHHHHSSG | 100 |
| Peptide-bound SARS-CoV-2 Nsp9 RNA-replicase | HHHHHHSAAL | 3–12 | HHHHHHSSGL | 80 |
| HSV-1 portal vertex-adjacent capsid/CATC, asymmetric unit | DPPSAIPPPPPS | 347–358 | DPPRT---PPPS | 58 |
| Crystal Structure of a gE-gI/Fc complex of HSV-1 | TPPPTPADYDE | 148–158 | TPPPS---YSE | 55 |
| Atomic structure of the herpes simplex virus type 2 B-capsid | ATIAAVRGAFE | 609–619 | ATIGMVRGLFD | 64 |
| Structure of the Herpes simplex virus type 2 C-capsid with capsid-vertex-specific component | DPRPSPPTPS | 2634–2643 | DPPRTPP-PS | 60 |
| An atomic structure of the HCMV capsid with its securing layer of pp150 | KL-LVKELRMC-LS | 233–244 | KLQLDKQL—CGLS | 57 |
| Human Cytomegalovirus protease | VYVGGFLARYDQSPDE | 14–29 | VWVGGFLCVYGEEPSE | 56 |
| Epstein-Barr virus protease | GKLSFFDHVSIC | 132–143 | GK-PFFHHVSVC | 67 |
| EBV major envelope glycoprotein | SKKL-PINITAGEE | 108–120 | SKTLFPIPRSA-EE | 57 |
| Structure of Varicella-zoster virus protease | DGN-FFTHVALC | 123–133 | DGKPFFHHVSVC | 58 |
| gHgL of Varicella-zoster virus in complex with human neutralizing antibodies | TG-AI-MDIIII | 737–746 | TGLAIAM-ILFI | 58 |
| Crystal structure of measles N0-P complex | LKAEPIGS-LA | 408–417 | LTTEP-GSELA | 64 |
| Crystal structure of the prefusion form of measles virus fusion protein | DLIGQKLGLKL | 84–94 | DLL—KLNKKL | 55 |
| B. burgdorferi BmpD nucleoside binding protein bound to adenosine | LNINIIEKASTG | 78–89 | LNINHNEKATIG | 67 |
| Structure of DNA gyrase A C-terminal domain [Borrelia burgdorferi] | VIKLNDKDFV | 144–153 | VI—NDTSFV | 60 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vojdani, A.; Vojdani, E.; Rosenberg, A.Z.; Shoenfeld, Y. The Role of Exposomes in the Pathophysiology of Autoimmune Diseases II: Pathogens. Pathophysiology 2022, 29, 243-280. https://doi.org/10.3390/pathophysiology29020020
Vojdani A, Vojdani E, Rosenberg AZ, Shoenfeld Y. The Role of Exposomes in the Pathophysiology of Autoimmune Diseases II: Pathogens. Pathophysiology. 2022; 29(2):243-280. https://doi.org/10.3390/pathophysiology29020020
Chicago/Turabian StyleVojdani, Aristo, Elroy Vojdani, Avi Z. Rosenberg, and Yehuda Shoenfeld. 2022. "The Role of Exposomes in the Pathophysiology of Autoimmune Diseases II: Pathogens" Pathophysiology 29, no. 2: 243-280. https://doi.org/10.3390/pathophysiology29020020
APA StyleVojdani, A., Vojdani, E., Rosenberg, A. Z., & Shoenfeld, Y. (2022). The Role of Exposomes in the Pathophysiology of Autoimmune Diseases II: Pathogens. Pathophysiology, 29(2), 243-280. https://doi.org/10.3390/pathophysiology29020020