Is an Immunosuppressive Microenvironment a Characteristic of Both Intra- and Extraparenchymal Central Nervous Tumors?

 ,
, 


 and
and 
Abstract
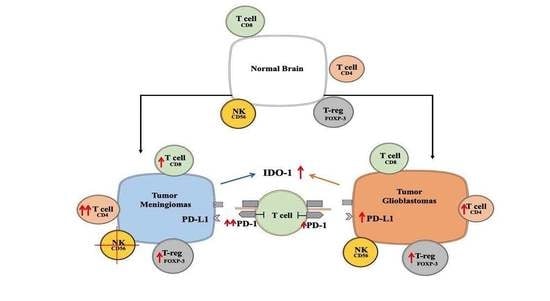
1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Patient Samples
2.3. RNA Isolation and Reverse Transcription
2.4. Real-Time Quantitative Polymerase Chain Reaction (qRT-PCR)
2.5. Hematoxylin-Eosin Staining
2.6. Immunohistochemistry
2.7. Immunofluorescent Staining
2.8. Statistical Analysis
3. Results
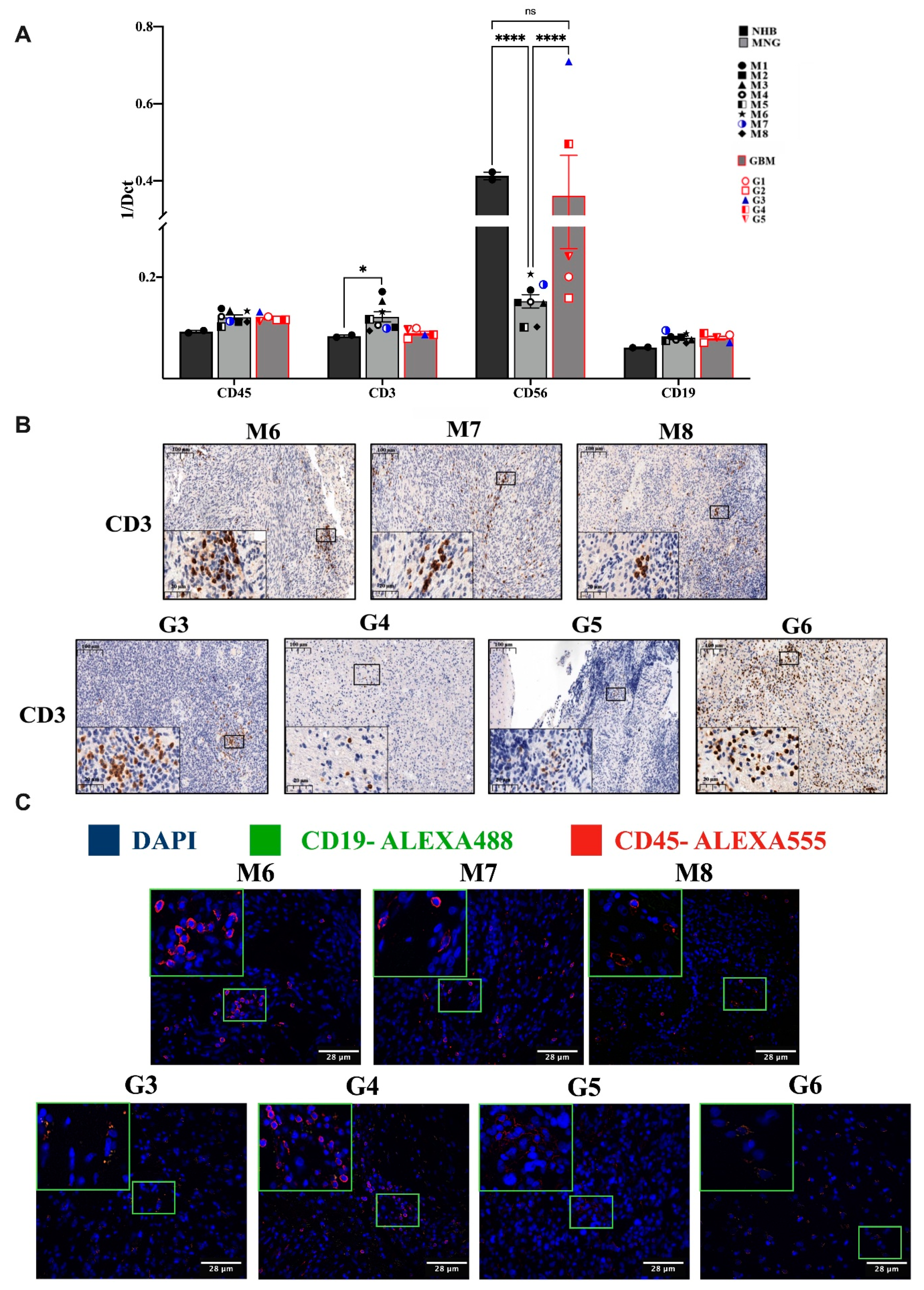
3.1. Variable Infiltration of T, B, NK Cells and Macrophages into MNG and GBM
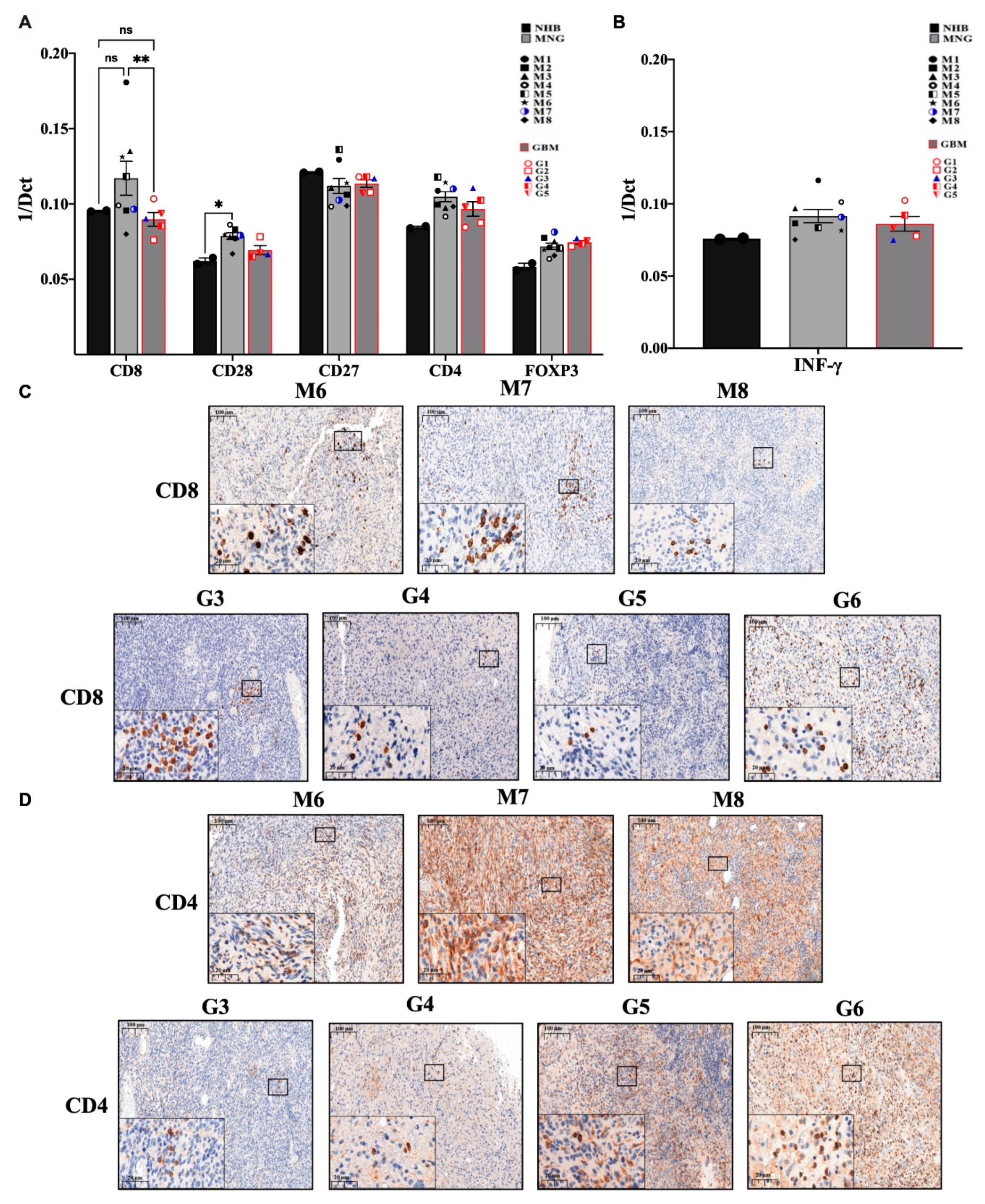
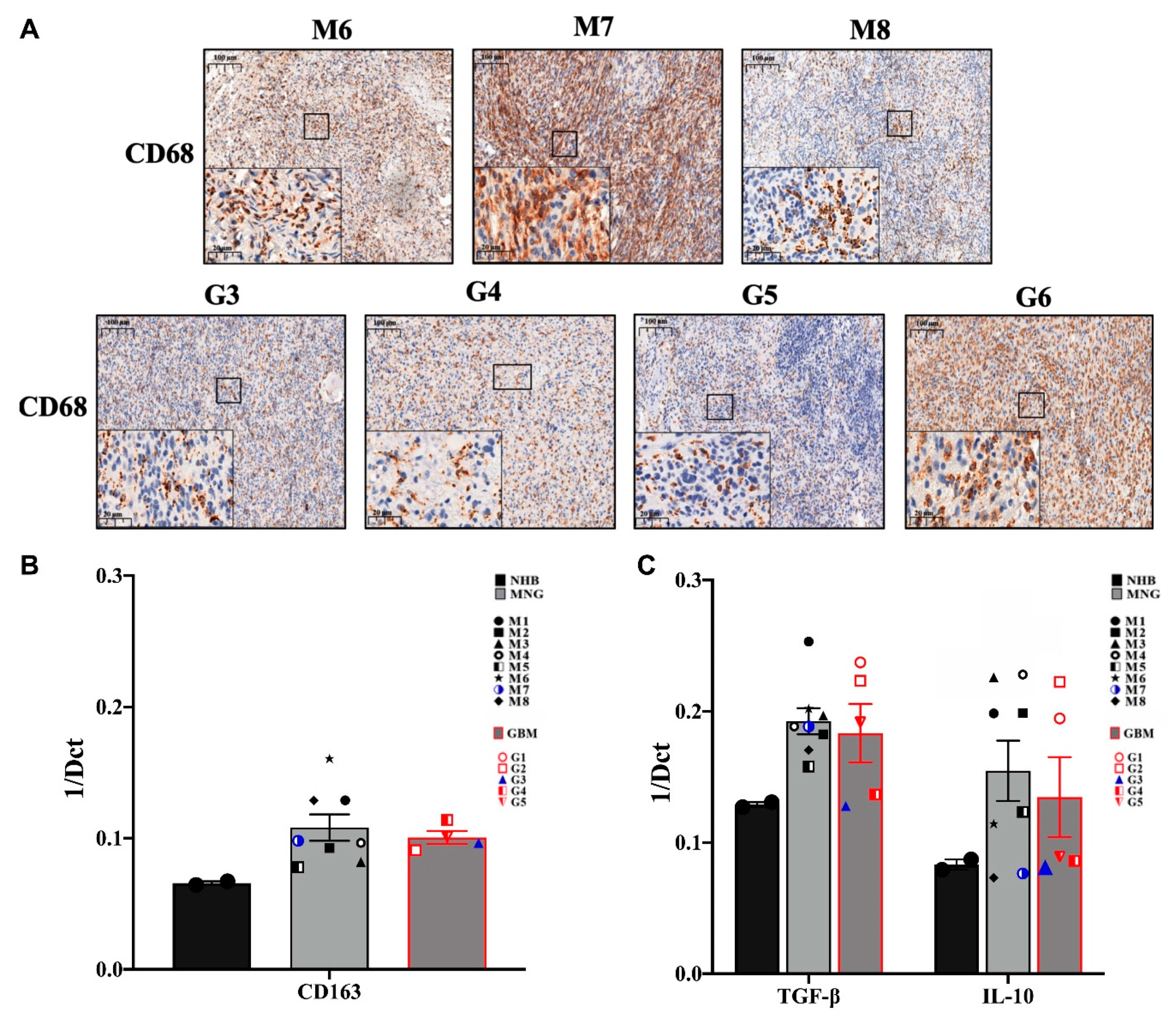
3.2. The Immune Microenvironment Is Actively Suppressive in Both MNG and GBM
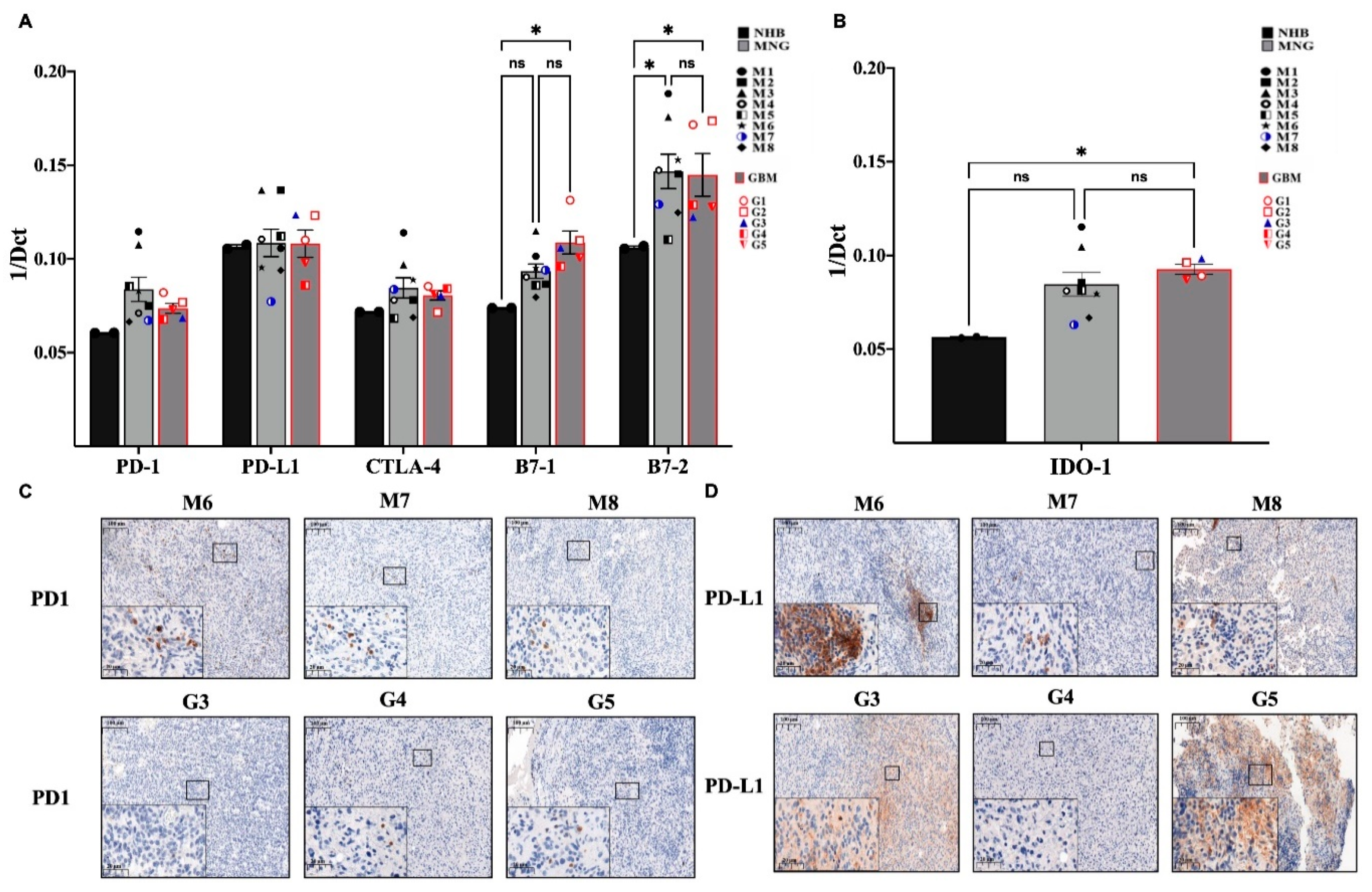
3.3. Immune Checkpoint Targets in MNG and GBM
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CNS | Central nervous system |
| GBM | Glioblastoma |
| MNG | Meningioma |
| Treg | Regulatory T cell |
| TAM | Tumor-associated macrophages |
| IDO1 | Indoleamine 2,3-dioxygenase 1 |
| LGG | Low-grade gliomas |
| TMZ | Temozolomide |
| NK | Natural killer |
| MDSSC | Myeloid-derived suppressor cells |
| BBB | Blood brain barrier |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| TBS | Tris Buffer Saline |
| BSA | Bovine serum albumin |
| INF-γ | Tumor interferon gamma |
| TGFβ | Transforming growth factor β |
| TNF | Tumor necrosis factor |
| PD-1 | Programmed cell death protein 1 |
| PD-L1 | Programmed death-ligand |
| CTLA-4 | Cytotoxic T-lymphocyte antigen 4 |
| CD80 | B7-1 |
| CD86 | B7-2 |
| TIL | Tumor infiltrating lymphocyte |
References
- Siegel, R.L.; Miller, K.D.M.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Ilkhanizadeh, S.; Lau, J.; Huang, M.; Foster, D.J.; Wong, R.; Frantz, A.; Wang, S.; Weiss, W.A.; Persson, A.I. Glial Progenitors as Targets for Transformation in Glioma. Adv. Cancer Res. 2014, 121, 1–65. [Google Scholar] [CrossRef]
- Gieryng, A.; Pszczolkowska, D.; Walentynowicz, K.A.; Rajan, W.D.; Kaminska, B. Immune microenvironment of gliomas. Lab. Investig. 2017, 97, 498–518. [Google Scholar] [CrossRef] [PubMed]
- Agarwala, S.S.; Kirkwood, J.M. Temozolomide, a Novel Alkylating Agent with Activity in the Central Nervous System, May Improve the Treatment of Advanced Metastatic Melanoma. Oncologist 2000, 5, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Harmancı, A.S.; Youngblood, M.W.; Clark, V.E.; Coşkun, S.; Henegariu, O.; Duran, D.; Erson-Omay, E.Z.; Kaulen, L.D.; Lee, T.I.; Abraham, B.J.; et al. Integrated genomic analyses of de novo pathways underlying atypical meningiomas. Nat. Commun. 2017, 8, 14433. [Google Scholar] [CrossRef] [PubMed]
- Aldape, K.D.; Zadeh, G.; Mansouri, S.; Reifenberger, G.; Von Deimling, A. Glioblastoma: Pathology, molecular mechanisms and markers. Acta Neuropathol. 2015, 129, 829–848. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Costa, A.; Osório, L.; Lago, R.C.; Linhares, P.; Carvalho, B.; Caeiro, C. Current Standards of Care in Glioblastoma Therapy; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2017. [Google Scholar]
- Kitange, G.J.; Carlson, B.L.; Schroeder, M.A.; Grogan, P.T.; Lamont, J.D.; Decker, P.A.; Wu, W.; James, C.D.; Sarkaria, J.N. Induction of MGMT expression is associated with temozolomide resistance in glioblastoma xenografts. Neuro Oncol. 2009, 11, 281–291. [Google Scholar] [CrossRef]
- Jiapaer, S.; Furuta, T.; Tanaka, S.; Kitabayashi, T.; Nakada, M. Potential Strategies Overcoming the Temozolomide Resistance for Glioblastoma. Neurol. Med. Chir. 2018, 58, 405–421. [Google Scholar] [CrossRef]
- van Genugten, J.A.B.; Leffers, P.; Baumert, B.G.; Tjon-A-Fat, H.; Twijnstra, A. Effectiveness of temozolomide for primary glio-blastoma multiforme in routine clinical practice. J. Neurooncol. 2010, 96, 249–257. [Google Scholar] [CrossRef]
- de Robles, P.; McIntyre, J.; Kalra, S.; Roldán, G.; Cairncross, G.; Forsyth, P.; Magliocco, T.; Hamilton, M.; Easaw, J. Methylation status of MGMT gene promoter in meningiomas. Cancer Genet. Cytogenet. 2008, 187, 25–27. [Google Scholar] [CrossRef]
- Sherman, W.; Raizer, J. Chemotherapy: What is its role in meningioma? Expert Rev. Neurother. 2012, 12, 1189–1196. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, G.C.; Malachowski, W.P.; DuHadaway, J.B.; Muller, A.J. Discovery of IDO1 Inhibitors: From Bench to Bedside. Cancer Res. 2017, 77, 6795–6811. [Google Scholar] [CrossRef]
- Yousef, Z. Combined inhibition of the IDO and PD-1 pathways improves the response rate for patients with advanced melanoma. In Proceedings of the International Cancer Immunotherapy Conference, Mainz, Germany, 5–9 September 2017. [Google Scholar]
- Webb, E.S.; Liu, P.; Baleeiro, R.; Lemoine, N.R.; Yuan, M.; Wang, Y. Immune checkpoint inhibitors in cancer therapy. J. Biomed. Res. 2017, 32, 317–326. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Cao, Y.J. Engineered T Cell Therapy for Cancer in the Clinic. Front. Immunol. 2019, 10, 2250. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Kumar, A.B.; Finnes, H.; Markovic, S.N.; Park, S.; Dronca, R.S.; Dong, H. Combining Immune Checkpoint Inhibitors with Conventional Cancer Therapy. Front. Immunol. 2018, 9, 1739. [Google Scholar] [CrossRef] [PubMed]
- Costa, F.; Das, R.; Bailur, J.K.; Dhodapkar, K.; Dhodapkar, M.V. Checkpoint Inhibition in Myeloma: Opportunities and Challenges. Front. Immunol. 2018, 9, 2204. [Google Scholar] [CrossRef]
- Hu, W.; Wang, G.; Huang, D.; Sui, M.; Xu, Y. Cancer Immunotherapy Based on Natural Killer Cells: Current Progress and New Opportunities. Front. Immunol. 2019, 10, 1205. [Google Scholar] [CrossRef]
- Ameratunga, M.; Coleman, N.; Welsh, L.; Saran, F.; Lopez, J. CNS cancer immunity cycle and strategies to target this for glio-blastoma. Oncotarget 2018, 9, 22802–22816. [Google Scholar] [CrossRef]
- Fang, L.; Lowther, D.E.; Meizlish, M.L.; Anderson, R.C.E.; Bruce, J.N.; Devine, L.; Huttner, A.J.; Kleinstein, S.H.; Lee, J.-Y.; Stern, J.N.H.; et al. The immune cell infiltrate populating meningiomas is composed of mature, antigen-experienced T and B cells. Neuro Oncol. 2013, 15, 1479–1490. [Google Scholar] [CrossRef]
- Pinton, L.; Solito, S.; Masetto, E.; Vettore, M.; Canè, S.; Della Puppa, A.; Mandruzzato, S. Immunosuppressive activity of tumor-infiltrating myeloid cells in patients with meningioma. OncoImmunology 2018, 7, e1440931. [Google Scholar] [CrossRef] [PubMed]
- Kivisäkk, P.; Mahad, D.J.; Callahan, M.K.; Trebst, C.; Tucky, B.; Wei, T.; Wu, L.; Baekkevold, E.S.; Lassmann, H.; Staugaitis, S.M.; et al. Human cerebrospinal fluid central memory CD4+T cells: Evidence for trafficking through choroid plexus and meninges via P-selectin. Proc. Natl. Acad. Sci. USA 2003, 100, 8389–8394. [Google Scholar] [CrossRef] [PubMed]
- Norris, G.T.; Kipnis, J. Immune cells and CNS physiology: Microglia and beyond. J. Exp. Med. 2018, 216, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Fortin, D. The blood-brain barrier: Its influence in the treatment of brain tumors metastases. Curr. Cancer Drug Targets 2012, 12, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Filley, A.C.; Henriquez, M.; Dey, M. Recurrent glioma clinical trial, CheckMate-143: The game is not over yet. Oncotarget 2017, 8, 91779–91794. [Google Scholar] [CrossRef]
- Galstyan, A.; Markman, J.L.; Shatalova, E.S.; Chiechi, A.; Korman, A.J.; Patil, R.; Klymyshyn, D.; Tourtellotte, W.G.; Israel, L.L.; Braubach, O.; et al. Blood–brain barrier permeable nano immunoconjugates induce local immune responses for glioma therapy. Nat. Commun. 2019, 10, 3850. [Google Scholar] [CrossRef]
- Asai, J.; Suzuki, R.; Fujimoto, T.; Suzuki, T.; Nakagawa, N.; Nagashima, G.; Miyo, T.; Hokaku, H.; Takei, A. Fluorescence automatic cell sorter and immunohistochemical investigation of CD68-positive cells in meningioma. Clin. Neurol. Neurosurg. 1999, 101, 229–234. [Google Scholar] [CrossRef]
- Denning, T.L.; Wang, Y.C.; Patel, S.R.; Williams, I.R.; Pulendran, B. Lamina propria macrophages and dendritic cells differential-ly induce regulatory and interleukin 17-producing T cell responses. Nat. Immunol. 2007, 8, 1086–1094. [Google Scholar] [CrossRef]
- Shima, T.; Shimoda, M.; Shigenobu, T.; Ohtsuka, T.; Nishimura, T.; Emoto, K.; Hayashi, Y.; Iwasaki, T.; Abe, T.; Asamura, H.; et al. Infiltration of tumor-associated macrophages is involved in tumor programmed death-ligand 1 expression in early lung adenocarcinoma. Cancer Sci. 2020, 111, 727–738. [Google Scholar] [CrossRef]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef]
- Qazi, M.; Bakhshinyan, D.; Singh, S. Deciphering brain tumor heterogeneity, one cell at a time. Nat. Med. 2019, 25, 1474–1476. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef]
- Garber, S.T.; Hashimoto, Y.; Weathers, S.P.; Xiu, J.; Gatalica, Z.; Verhaak, R.G.; Zhou, S.; Fuller, G.N.; Khasraw, M.; de Groot, J.; et al. Immune checkpoint blockade as a potential therapeutic target: Surveying CNS malignancies. Neuro Oncol. 2016, 18, 1357–1366. [Google Scholar] [CrossRef]
- Martinez-Lage, M.; Lynch, T.M.; Bi, Y.; Cocito, C.; Way, G.P.; Pal, S.; Haller, J.; Yan, R.E.; Ziober, A.; Nguyen, A.; et al. Immune landscapes associated with different glioblastoma molecular subtypes. Acta Neuropathol. Commun. 2019, 7, 203. [Google Scholar] [CrossRef]
- Gibellini, L.; De Biasi, S.; Porta, C.; Lo Tartaro, D.; Depenni, R.; Pellacani, G.; Sabbatini, R.; Cossarizza, A. Single-Cell Approaches to Profile the Response to Immune Checkpoint Inhibitors. Front. Immunol. 2020, 11, 490. [Google Scholar] [CrossRef]
- Tang, F.; Du, X.; Liu, M.; Zheng, P.; Liu, Y. Anti-CTLA-4 antibodies in cancer immunotherapy: Selective depletion of intra-tumoral regulatory T cells or checkpoint blockade? Cell Biosci. 2018, 8, 30. [Google Scholar] [CrossRef]
- Le Mercier, I.; Lines, J.L.; Noelle, R.J. Beyond CTLA-4 and PD-1, the Generation Z of Negative Checkpoint Regulators. Front Immunol. 2015, 6, 418. [Google Scholar] [CrossRef]
- Engelhardt, J.J.; Sullivan, T.J.; Allison, J.P. CTLA-4 overexpression inhibits T cell responses through a CD28-B7-dependent mechanism. J. Immunol. 2006, 177, 1052–1061. [Google Scholar] [CrossRef]
- Riella, L.V.; Paterson, A.M.; Sharpe, A.H.; Chandraker, A. Role of the PD-1 pathway in the immune response. Am. J. Transplant. 2012, 12, 2575–2587. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, F.; Jiang, F.; Lv, X.; Zhang, R.; Lu, A.; Zhang, G. A Mini-Review for Cancer Immunotherapy: Molecular Understanding of PD-1/PD-L1 Pathway & Translational Blockade of Immune Checkpoints. Int. J. Mol. Sci. 2016, 17, 1151. [Google Scholar] [CrossRef]
- Terrén, I.; Orrantia, A.; Vitallé, J.; Zenarruzabeitia, O.; Borrego, F. NK Cell Metabolism and Tumor Microenvironment. Front. Immunol. 2019, 10, 2278. [Google Scholar] [CrossRef] [PubMed]
- Mushtaq, M.U.; Papadas, A.; Pagenkopf, A.; Flietner, E.; Morrow, Z.; Chaudhary, S.G.; Asimakopoulos, F. Tumor matrix remodeling and novel immunotherapies: The promise of matrix-derived immune biomarkers. J. Immunother. Cancer 2018, 6, 65. [Google Scholar] [CrossRef] [PubMed]
- Vitale, M.; Cantoni, C.; Pietra, G.; Mingari, M.C.; Moretta, L. Effect of tumor cells and tumor microenvironment on NK-cell function. Eur. J. Immunol. 2014, 44, 1582–1592. [Google Scholar] [CrossRef] [PubMed]
- Holmgaard, R.B.; Zamarin, D.; Munn, D.H.; Wolchok, J.D.; Allison, J.P. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J. Exp. Med. 2013, 210, 1389–1402. [Google Scholar] [CrossRef] [PubMed]
- Hornyák, L.; Dobos, N.; Koncz, G.; Karányi, Z.; Páll, D.; Szabó, Z.; Halmos, G.; Székvölgyi, L. The Role of Indoleamine-2,3-Dioxygenase in Cancer Development, Diagnostics, and Therapy. Front. Immunol. 2018, 9, 151. [Google Scholar] [CrossRef]
- Bilir, C.; Sarisozen, C. Indoleamine 2,3-dioxygenase (IDO): Only an enzyme or a checkpoint controller? J. Oncol. Sci. 2017, 3, 52–56. [Google Scholar] [CrossRef]
- Wainwright, D.A.; Lesniak, M.S. Ménage à trois: Sustained therapeutic anti-tumor immunity requires multiple partners in malignant glioma. Oncoimmunology 2014, 3, e28927. [Google Scholar] [CrossRef]
- Penter, L.; Dietze, K.; Ritter, J.; Lammoglia Cobo, M.F.; Garmshausen, J.; Aigner, F.; Bullinger, L.; Hackstein, H.; Wienzek-Lischka, S.; Blankenstein, T.; et al. Localization-associated immune phenotypes of clonally expanded tumor-infiltrating T cells and distribution of their target antigens in rectal cancer. Oncoimmunology 2017, 6, e1252894. [Google Scholar] [CrossRef]
- Quattrocchi, K.B.; Miller, C.H.; Cush, S.; Bernard, S.A.; Dull, S.T.; Smith, M.; Gudeman, S.; Varia, M.A. Pilot Study of Local Autologous Tumor Infiltrating Lymphocytes for the Treatment of Recurrent Malignant Gliomas. J. Neuro-Oncol. 1999, 45, 141–157. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor Type | N° | Code | Age Range | IDH | Diagnosis | Radio- or Other Therapy before Surgery |
|---|---|---|---|---|---|---|
| Meningioma | 1 | M1 | 50–60 | N/A | Grade I meningioma | - |
| 2 | M2 | 70–80 | N/A | Grade I meningioma | - | |
| 3 | M3 | 40–50 | N/A | Grade I meningioma | - | |
| 4 | M4 | 70–80 | N/A | Grade I transitional meningioma | - | |
| 5 | M5 | 60–70 | N/A | Grade I meningioma brain invasion | - | |
| 6 | M6 | 40–50 | N/A | Grade I meningioma | - | |
| 7 | M7 | 40–50 | N/A | Grade I meningioma | + | |
| 8 | M8 | 70–80 | N/A | Grade I meningioma | - | |
| Glioblastoma | 1 | G1 | 60–70 | (-) | Grade IV Glioblastoma | - |
| 2 | G2 | 70–80 | (-) | Grade IV Glioblastoma | - | |
| 3 | G3 | 60–70 | (-) | Grade IV Glioblastoma | + | |
| 4 | G4 | 40–50 | (-) | Grade IV Glioblastoma | - | |
| 5 | G5 | 60–70 | (-) | Grade IV Glioblastoma | - |
| Target Genes | Accession Number | Primers Sequences |
|---|---|---|
| beta-Actin | NM_001101.5 | |
| Forward | 5′-GCGCGGCTACAGCTTCA-3′ | |
| Reverse | 5′-CTTAATGTCACGCACGATTTCC-3′ | |
| GAPDH | NM_002046.7 | |
| Forward | 5′-ATCCCTCCAAAATCAAGTGA-3′ | |
| Reverse | 5′-GGCTGTTGTCATACTTCTCA-3′ | |
| FOXP3 | XM_006724533.2 | |
| Forward | 5′-AAGGACAGGTCAGTGGACAG-3′ | |
| Reverse | 5′-CGAAGACCTTCTCACATCCG-3′ | |
| IDO1 | NM_002164.5 | |
| Forward | 5′-CCAAGAAACTGGAACTGCCT -3′ | |
| Reverse | 5′-CTGCAGTCTCCATCACGAAA-3′ | |
| IL-10 | NM_000572.3 | |
| Forward | 5′-CCTGCCTAACATGCTTCGAG-3′ | |
| Reverse | 5′-GGTCTTGGTTCTCAGCTTGG-3′ | |
| INF- gamma | NM_000619.2 | |
| Forward | 5′-GAATGTCCAACGCAAAGCAA-3′ | |
| Reverse | 5′-ACCTCGAAACAGCATCTGAC-3′ | |
| CD27 | >NM_001242.4 | |
| Forward | 5′-TGCAGAGCCTTGTCGTTACAG-3′ | |
| Reverse | 5′-GCTCCGGTTTTCGGTAATCCT-3′ | |
| CD163 | XM_024449278.1 | |
| Forward | 5′-GGACAGGGTTAGGGAGTCAT-3′ | |
| Reverse | 5′-TAAGCTGCTGGCAAAGAACA-3′ | |
| CTLA4 | NM_005214.5 | |
| Forward | 5′-ATGTACCCACCGCCATACTA-3′ | |
| Reverse | 5′-CGAACTAACTGCTGCAAGGA-3′ | |
| CD28 | XM_011512194.2 | |
| Forward | 5′-GCCTTGGCAGGAAACAAGAT-3′ | |
| Reverse | 5′-AGTCCTTTGTGAAGGGATGC-3′ | |
| TGF-beta | NM_000660.6 | |
| Forward | 5′-GACATCAACGGGTTCACTACC-3′ | |
| Reverse | 5′-CGTGGAGCTGAAGCAATAGTT-3′ | |
| CD4 | NM_001195014.2 | |
| Forward | 5′-TGCACCCTCATCTTCCTATCT-3′ | |
| Reverse | 5′-AGGAGAACTCCACCTGTTCC-3′ | |
| PD-1 | NM_005018.3 | |
| Forward | 5′-CAGTTCCAAACCCTGGTGGT-3′ | |
| Reverse | 5′-GGCTCCTATTGTCCCTCGTG-3′ | |
| PD-L1 | NM_014143.4 | |
| Forward | 5′-ATGGTGGTGCCGACTACAAG-3′ | |
| Reverse | 5′-GGAATTGGTGGTGGTGGTCT-3′ | |
| CD19 | XM_006721103.3 | |
| Forward | 5′-CAGGGTCCCAGTCCTATGAG-3′ | |
| Reverse | 5′-TCTGGCCCATCGGGATTAT-3′ | |
| CD56 | NM_001242608.1 | |
| Forward | 5′-TAGTTCCCAGCTGACCATCA-3′ | |
| Reverse | 5′-TGGCAGTCTGGTTCTCTACA-3′ | |
| CD3 | NM_000733.3 | |
| Forward | 5′-ATGTCTGCTACCCCAGAGGA-3′ | |
| Reverse | 5′-GTTTTGTCCCCTTTGCCTGC-3′ | |
| CD8 | NM_001145873.1 | |
| Forward | 5′-ACCCTTTACTGCAACCAC-3′ | |
| Reverse | 5′-TTGTCTCCCGATTTGACCAC-3′ | |
| PAX5 | NM_016734.3 | |
| Forward | 5′-GTAGTCCGCCAGAGGATAGT-3′ | |
| Reverse | 5′-TCCAATTACCCCAGGCTTGA-3′ | |
| CD70 | NM_001330332.2 | |
| Forward | 5′-GGCATCTACATGGTACACATCC-3′ | |
| Reverse | 5′-ACTTGACTTTGAGTCCCCAG-3′ | |
| B7-1 | NM_005191.4 | |
| Forward | 5′-CAGGTGTTATCCACGTGACC-3′ | |
| Reverse | 5′-CCTTTTGCCAGTAGATGCGA-3′ | |
| B7-2 | NM_175862.5 | |
| Forward | 5′-CACAGCAGAAGCAGCCAAAATG-3′ | |
| Reverse | 5′-CTTCAGAGGAGCAGCACCAGA-3′ |
| Antibody | Clone | Source | Isotype | Source | Dilution |
|---|---|---|---|---|---|
| Anti-CD4 | 4B12 | Mouse | IgG1, kappa | Thermo Fisher Scientific | 1:20 |
| Anti-CD8 | C8/114B | Mouse | IgG1, kappa | Thermo Fisher Scientific | 1:50 |
| Anti-CD3 | Polyclonal | Rabbit | / | Dako | 1:400 |
| Anti-CD45 | 2B11 + PD7/26 | Mouse | IgG1, kappa | Dako | 1:400 |
| Anti-CD19 | EPR5906 | Rabbit | IgG | Abcam | 1:500–1:1000 |
| Anti-CD68 | PGM1 | Mouse | IgG3, kappa | Dako | 1:200 |
| Anti-PD1 | NAT105 | Mouse | IgG1, kappa | Abcam | 1:50 |
| Anti-PDL1 | 22C3 | Mouse | IgG1 | Dako | 1:50 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soltani, A.; Kajtar, B.; Abdelwahab, E.H.M.M.; Steib, A.; Horvath, Z.; Mangel, L.; Jaromi, L.; Pongracz, J.E. Is an Immunosuppressive Microenvironment a Characteristic of Both Intra- and Extraparenchymal Central Nervous Tumors? Pathophysiology 2021, 28, 34-49. https://doi.org/10.3390/pathophysiology28010004
Soltani A, Kajtar B, Abdelwahab EHMM, Steib A, Horvath Z, Mangel L, Jaromi L, Pongracz JE. Is an Immunosuppressive Microenvironment a Characteristic of Both Intra- and Extraparenchymal Central Nervous Tumors? Pathophysiology. 2021; 28(1):34-49. https://doi.org/10.3390/pathophysiology28010004
Chicago/Turabian StyleSoltani, Amina, Bela Kajtar, El Husseiny Mohamed Mahmud Abdelwahab, Anita Steib, Zsolt Horvath, Laszlo Mangel, Luca Jaromi, and Judit E. Pongracz. 2021. "Is an Immunosuppressive Microenvironment a Characteristic of Both Intra- and Extraparenchymal Central Nervous Tumors?" Pathophysiology 28, no. 1: 34-49. https://doi.org/10.3390/pathophysiology28010004
APA StyleSoltani, A., Kajtar, B., Abdelwahab, E. H. M. M., Steib, A., Horvath, Z., Mangel, L., Jaromi, L., & Pongracz, J. E. (2021). Is an Immunosuppressive Microenvironment a Characteristic of Both Intra- and Extraparenchymal Central Nervous Tumors? Pathophysiology, 28(1), 34-49. https://doi.org/10.3390/pathophysiology28010004