Introduction of Androgen Receptor Targeting shRNA Inhibits Tumor Growth in Patient-Derived Prostate Cancer Xenografts

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation and Cultivation of Organoids from LuCaP35 and BM18 Xenografts
2.2. Lentiviral Transduction of Xenograft-Derived Organoids
2.3. Lentiviral Transduction of LuCaP Organoids to Knockdown AR
2.4. Establishment of LuCaP35-i-shAR Organoid Derived Xenografts
2.5. Passaging LuCaP35-i-shAR Xenografts
2.6. In Vivo Induction of AR Knockdown
2.7. In Vitro Culture of LuCaP35-i-shAR Xenograft and Induction of AR Knockdown
2.8. Histological Analysis of Xenograft Tissue and Organoids
3. Results
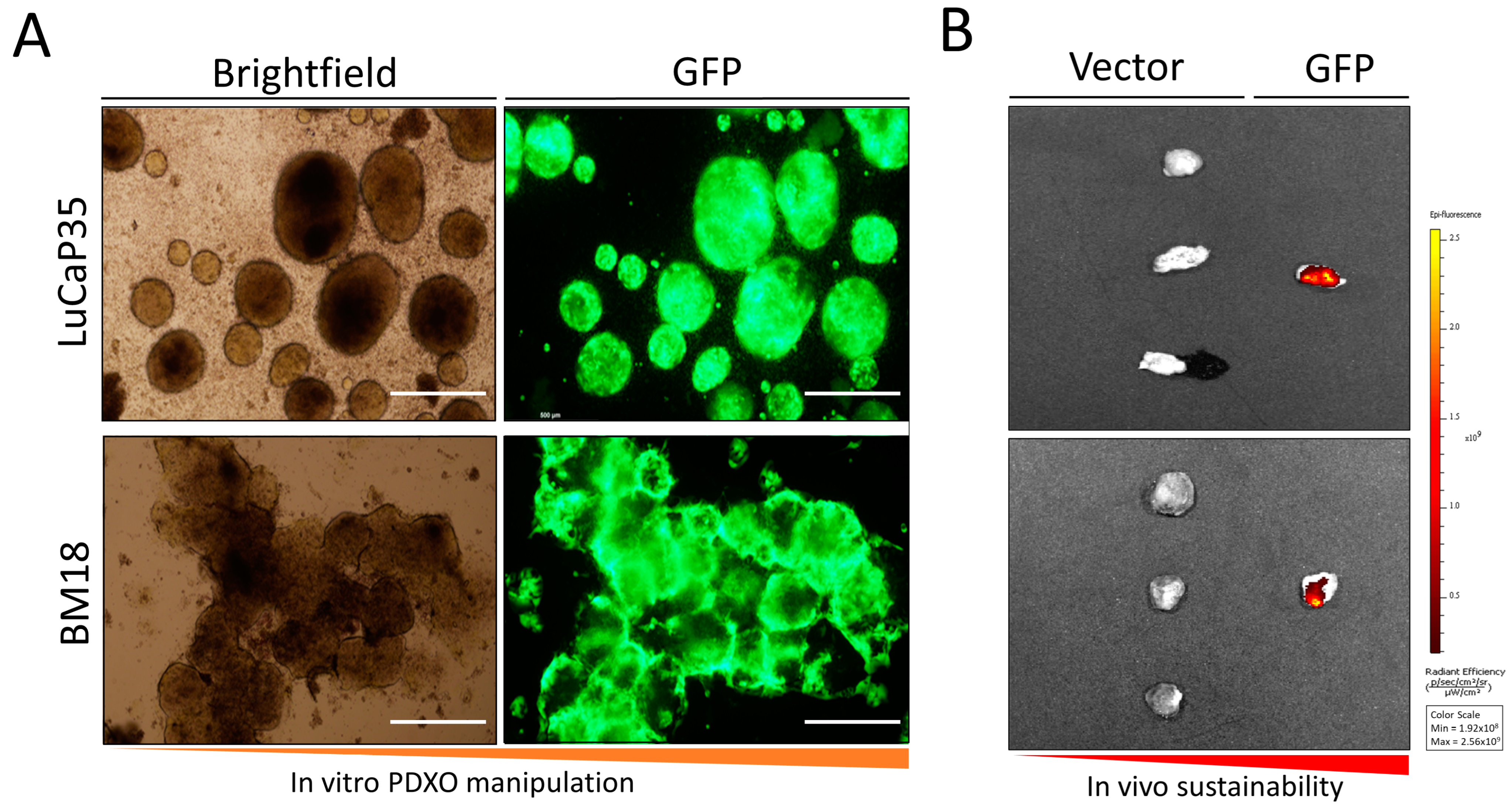
3.1. In Vitro Culture of LuCaP35 Xenografts and Lentiviral Transfection
3.2. Assessment of Tumor Formation of GFP-Labeled PDX Models In Vivo
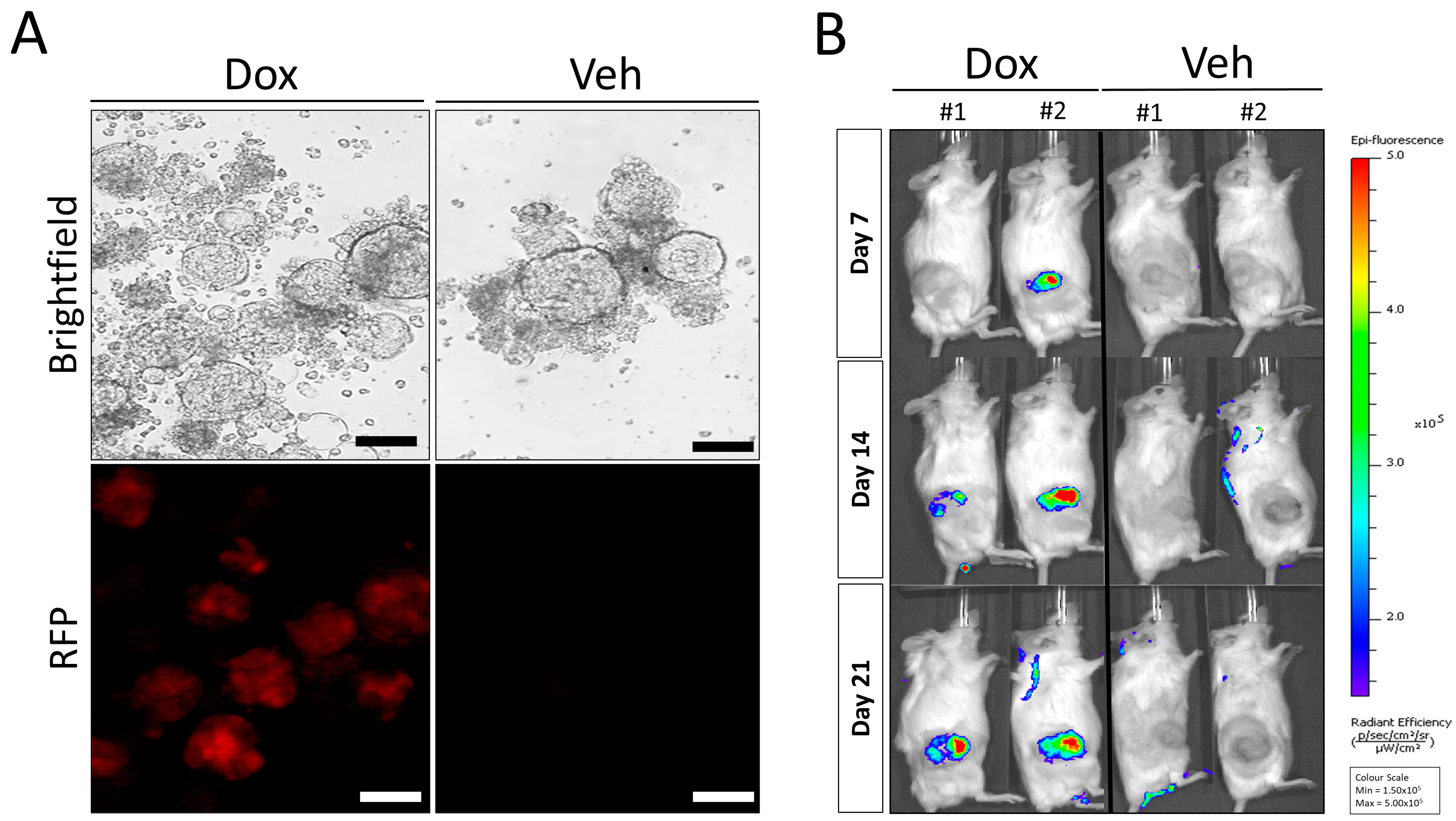
3.3. Doxycycline Inducible AR Knockdown Reduces In Vivo Tumor Growth in LuCaP35 PDX Model
3.4. Re-Implantation of LuCaP35-i-shAR Xenografts Retain Decreases in Tumor Growth
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2013. CA Cancer J. Clin. 2013, 63, 11–30. [Google Scholar] [CrossRef]
- Kongnyuy, M.; George, A.K.; Rastinehad, A.R.; Pinto, P.A. Magnetic resonance imaging-ultrasound fusion-guided prostate biopsy: Review of technology, techniques, and outcomes. Curr. Urol. Rep. 2016, 17, 32. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2015. CA Cancer J. Clin. 2015, 65, 5–29. [Google Scholar] [CrossRef]
- Valkenburg, K.C.; Pienta, K.J. Drug discovery in prostate cancer mouse models. Expert. Opin. Drug Discov. 2015, 10, 1011–1024. [Google Scholar] [CrossRef]
- Smith, M.R.; Saad, F.; Chowdhury, S.; Oudard, S.; Hadaschik, B.A.; Graff, J.N.; Olmos, D.; Mainwaring, P.N.; Lee, J.Y.; Uemura, H.; et al. Apalutamide treatment and metastasis-free survival in prostate cancer. N. Engl. J. Med. 2018, 378, 1408–1418. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Zieren, R.C.; Xue, W.; de Reijke, T.M.; Pienta, K.J. Metastatic prostate cancer remains incurable, why? Asian J. Urol. 2019, 6, 26–41. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. The molecular taxonomy of primary prostate cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef]
- Kumar, A.; Coleman, I.; Morrissey, C.; Zhang, X.; True, L.D.; Gulati, R.; Etzioni, R.; Bolouri, H.; Montgomery, B.; White, T.; et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat. Med. 2016, 22, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J.; Tamura, S.; Buchdunger, E.; Ohno, S.; Segal, G.M.; Fanning, S.; Zimmermann, J.; Lydon, N.B. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat. Med. 1996, 2, 561–566. [Google Scholar] [CrossRef]
- Solit, D.B.; Garraway, L.A.; Pratilas, C.A.; Sawai, A.; Getz, G.; Basso, A.; Ye, Q.; Lobo, J.M.; She, Y.; Osman, I.; et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature 2006, 439, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Chen, Y. Organoid development in cancer genome discovery. Curr. Opin. Genet. Dev. 2015, 30, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Wilding, J.L.; Bodmer, W.F. Cancer cell lines for drug discovery and development. Cancer Res. 2014, 74, 2377–2384. [Google Scholar] [CrossRef] [PubMed]
- Namekawa, T.; Ikeda, K.; Horie-Inoue, K.; Inoue, S. Application of prostate cancer models for preclinical study: Advantages and limitations of cell lines, patient-derived xenografts, and three-dimensional culture of patient-derived cells. Cells 2019, 8, 74. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.M.; Vessella, R.L.; Morrissey, C.; Brown, L.G.; Coleman, I.M.; Higano, C.S.; Mostaghel, E.A.; Zhang, X.; True, L.D.; Lam, H.M.; et al. LuCaP prostate cancer patient-derived xenografts reflect the molecular heterogeneity of advanced disease and serve as models for evaluating cancer therapeutics. Prostate 2017, 77, 654–671. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.G.; Mathew, P.; Yang, J.; Starbuck, M.W.; Zurita, A.J.; Liu, J.; Sikes, C.; Multani, A.S.; Efstathiou, E.; Lopez, A.; et al. Androgen receptor-negative human prostate cancer cells induce osteogenesis in mice through FGF9-mediated mechanisms. J. Clin. Investig. 2008, 118, 2697–2710. [Google Scholar] [CrossRef] [PubMed]
- Moya, L.; Walpole, C.; Rae, F.; Srinivasan, S.; Seim, I.; Lai, J.; Nicol, D.; Williams, E.D.; Clements, J.A.; Batra, J. Characterisation of cell lines derived from prostate cancer patients with localized disease. Prostate Cancer Prostatic Dis. 2023, 26, 614–624. [Google Scholar] [CrossRef]
- Navone, N.M.; van Weerden, W.M.; Vessella, R.L.; Williams, E.D.; Wang, Y.; Isaacs, J.T.; Nguyen, H.M.; Culig, Z.; van der Pluijm, G.; Rentsch, C.A.; et al. Movember GAP1 PDX project: An international collection of serially transplantable prostate cancer patient-derived xenograft (PDX) models. Prostate 2018, 78, 1262–1282. [Google Scholar] [CrossRef]
- Corey, E.; Quinn, J.E.; Buhler, K.R.; Nelson, P.S.; Macoska, J.A.; True, L.D.; Vessella, R.L. LuCaP 35: A new model of prostate cancer progression to androgen independence. Prostate 2003, 55, 239–246. [Google Scholar] [CrossRef]
- Ellis, W.J.; Vessella, R.L.; Buhler, K.R.; Bladou, F.; True, L.D.; Bigler, S.A.; Curtis, D.; Lange, P.H. Characterization of a novel androgen-sensitive, prostate-specific antigen-producing prostatic carcinoma xenograft: LuCaP 23. Clin. Cancer Res. 1996, 2, 1039–1048. [Google Scholar]
- Young, S.R.; Saar, M.; Santos, J.; Nguyen, H.M.; Vessella, R.L.; Peehl, D.M. Establishment and serial passage of cell cultures derived from LuCaP xenografts. Prostate 2013, 73, 1251–1262. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, D.R.; Opeskin, K.; Thompson, E.W.; Williams, E.D. BM18: A novel androgen-dependent human prostate cancer xenograft model derived from a bone metastasis. Prostate 2005, 65, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Siolas, D.; Hannon, G.J. Patient-derived tumor xenografts: Transforming clinical samples into mouse models. Cancer Res. 2013, 73, 5315–5319. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Vela, I.; Sboner, A.; Iaquinta, P.J.; Karthaus, W.R.; Gopalan, A.; Dowling, C.; Wanjala, J.N.; Undvall, E.A.; Arora, V.K.; et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 2014, 159, 176–187. [Google Scholar] [CrossRef]
- Bose, S.; Clevers, H.; Shen, X. Promises and challenges of organoid-guided precision medicine. Med 2021, 2, 1011–1026. [Google Scholar] [CrossRef]
- Tomayko, M.M.; Reynolds, C.P. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother. Pharmacol. 1989, 24, 148–154. [Google Scholar] [CrossRef]
- Mehrara, E.; Forssell-Aronsson, E.; Ahlman, H.; Bernhardt, P. Specific growth rate versus doubling time for quantitative characterization of tumor growth rate. Cancer Res. 2007, 67, 3970–3975. [Google Scholar] [CrossRef] [PubMed]
- Tevz, G.; McGrath, S.; Demeter, R.; Magrini, V.; Jeet, V.; Rockstroh, A.; McPherson, S.; Lai, J.; Bartonicek, N.; An, J.; et al. Identification of a novel fusion transcript between human relaxin-1 (RLN1) and human relaxin-2 (RLN2) in prostate cancer. Mol. Cell Endocrinol. 2016, 420, 159–168. [Google Scholar] [CrossRef]
- Van Lidth de Jeude, J.F.; Vermeulen, J.L.M.; Montenegro-Miranda, P.S.; Van den Brink, G.R.; Heijmans, J. A protocol for lentiviral transduction and downstream analysis of intestinal organoids. J. Vis. Exp. 2015, 98, 52531. [Google Scholar]
- Lema, C.; Varela-Ramirez, A.; Aguilera, R.J. Differential nuclear staining assay for high-throughput screening to identify cytotoxic compounds. Curr. Cell Biochem. 2011, 1, 1–14. [Google Scholar] [PubMed]
- Paul, P.J.; Raghu, D.; Chan, A.-L.; Gulati, T.; Lambeth, L.; Takano, E.; Herold, M.J.; Hagekyriakou, J.; Vessella, R.L.; Fedele, C.; et al. Restoration of tumor suppression in prostate cancer by targeting the E3 ligase E6AP. Oncogene 2016, 35, 6235–6245. [Google Scholar] [CrossRef] [PubMed]
- Hulton, C.H.; Costa, E.A.; Shah, N.S.; Quintanal-Villalonga, A.; Heller, G.; de Stanchina, E.; Rudin, C.M.; Poirier, J.T. Direct genome editing of patient-derived xenografts using CRISPR-Cas9 enables rapid in vivo functional genomics. Nat. Cancer 2020, 1, 359–369. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thomas, P.B.; Alinezhad, S.; Joshi, A.; Sweeney, K.; Tse, B.W.C.; Tevz, G.; McPherson, S.; Nelson, C.C.; Williams, E.D.; Vela, I. Introduction of Androgen Receptor Targeting shRNA Inhibits Tumor Growth in Patient-Derived Prostate Cancer Xenografts. Curr. Oncol. 2023, 30, 9437-9447. https://doi.org/10.3390/curroncol30110683
Thomas PB, Alinezhad S, Joshi A, Sweeney K, Tse BWC, Tevz G, McPherson S, Nelson CC, Williams ED, Vela I. Introduction of Androgen Receptor Targeting shRNA Inhibits Tumor Growth in Patient-Derived Prostate Cancer Xenografts. Current Oncology. 2023; 30(11):9437-9447. https://doi.org/10.3390/curroncol30110683
Chicago/Turabian StyleThomas, Patrick B., Saeid Alinezhad, Andre Joshi, Katrina Sweeney, Brian W. C. Tse, Gregor Tevz, Stephen McPherson, Colleen C. Nelson, Elizabeth D. Williams, and Ian Vela. 2023. "Introduction of Androgen Receptor Targeting shRNA Inhibits Tumor Growth in Patient-Derived Prostate Cancer Xenografts" Current Oncology 30, no. 11: 9437-9447. https://doi.org/10.3390/curroncol30110683
APA StyleThomas, P. B., Alinezhad, S., Joshi, A., Sweeney, K., Tse, B. W. C., Tevz, G., McPherson, S., Nelson, C. C., Williams, E. D., & Vela, I. (2023). Introduction of Androgen Receptor Targeting shRNA Inhibits Tumor Growth in Patient-Derived Prostate Cancer Xenografts. Current Oncology, 30(11), 9437-9447. https://doi.org/10.3390/curroncol30110683