Does α-Amino-β-methylaminopropionic Acid (BMAA) Play a Role in Neurodegeneration?

Abstract
:1. The Cycad Hypothesis
2. Proving the BMAA Link
3. Neurodegeneration is Caused by Excitotoxicity
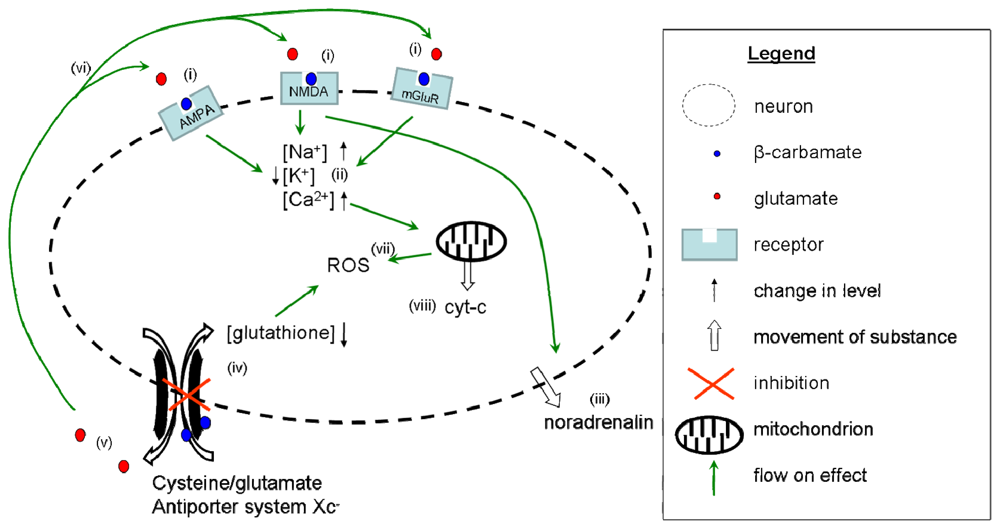
4. Summary of the Multiple Mechanisms of BMAA Activity
5. A Summary of the Mode of Action of BMAA based on the Current Literature
6. Concluding Remarks
References
- Arnold, A; Edgren, DC; Palladino, VS. Amyotrophic lateral sclerosis; fifty cases observed on Guam. J Nerv Ment Dis 1953, 117, 135–139. [Google Scholar]
- Kurland, LT; Mulder, DW. Epidemiologic investigations of amyotrophic lateral sclerosis. I. Preliminary report on geographic distribution and special reference to the Mariana Islands, including clinical and pathologic observations. Neurology 1954, 4, 438–448. [Google Scholar]
- Kurland, LT; Mulder, DW. Epidemiologic investigations of amyotrophic lateral sclerosis. I. Preliminary report on geographic distribution, with special reference to the Mariana Islands, including clinical and pathologic observations. Neurology 1954, 4, 355–378. [Google Scholar]
- Banack, SA; Murch, SJ; Cox, PA. Neurotoxic flying foxes as dietary items for the Chamorro people, Marianas Islands. J Ethnopharmacol 2006, 106, 97–104. [Google Scholar]
- Whiting, MG. Food Practices in Als Foci in Japan, the Marianas, and New Guinea. Fed Proc 1964, 23, 1343–1345. [Google Scholar]
- Whiting, M; Spatz, M; Matsumoto, H. Research progress on cycads. Econ Bot 1966, 20, 98–102. [Google Scholar]
- Borenstein, AR; Mortimer, JA; Schofield, E; Wu, Y; Salmon, DP; Gamst, A; Olichney, J; Thal, LJ; Silbert, L; Kaye, J; et al. Cycad exposure and risk of dementia, MCI, and PDC in the Chamorro population of Guam. Neurology 2007, 68, 1764–1771. [Google Scholar]
- Vega, A; Bell, EA. α-Amino-β-methylaminopropionic acid, a new amino acid from seeds of Cycas circinalis. Phytochemistry 1967, 6, 759–762. [Google Scholar]
- Vega, A; Bell, EA; Nunn, PB. The preparation of l- and d-α-amino-β-methylaminopropionic acids and the identification of the compound isolated from Cycas circinalis as the l-isomer. Phytochemistry 1968, 7, 1885–1887. [Google Scholar]
- Spencer, PS; Ohta, M; Palmer, VS. Cycad use and motor neurone disease in Kii peninsula of Japan. Lancet 1987, 2, 1462–1463. [Google Scholar]
- Spencer, PS; Palmer, VS; Herman, A; Asmedi, A. Cycad use and motor neurone disease in Irian Jaya. Lancet 1987, 2, 1273–1274. [Google Scholar]
- Spencer, PS; Nunn, PB; Hugon, J; Ludolph, A; Roy, DN. Motorneurone disease on Guam: Possible role of a food neurotoxin. Lancet 1986, 1, 965. [Google Scholar]
- Spencer, PS; Nunn, PB; Hugon, J; Ludolph, AC; Ross, SM; Roy, DN; Robertson, RC. Guam amyotrophic lateral sclerosis-parkinsonism-dementia linked to a plant excitant neurotoxin. Science 1987, 237, 517–522. [Google Scholar]
- Duncan, MW; Kopin, IJ; Garruto, RM; Lavine, L; Markey, SP. 2-Amino-3 (methylamino)-propionic acid in cycad-derived foods is an unlikely cause of amyotrophic lateral sclerosis/Parkinsonism. Lancet 1988, 2, 631–632. [Google Scholar]
- Garruto, R; Yanagihara, R; Gajdusek, DC. Cycads and amyotrophic lateral sclerosis/Parkinsonism dementia. Lancet 1988, 332, 1079–1079. [Google Scholar]
- Duncan, MW; Steele, JC; Kopin, IJ; Markey, SP. 2-Amino-3-(methylamino)-propanoic acid (BMAA) in cycad flour: An unlikely cause of amyotrophic lateral sclerosis and parkinsonism-dementia of Guam. Neurology 1990, 40, 767–772. [Google Scholar]
- Cox, PA; Sacks, OW. Cycad neurotoxins, consumption of flying foxes, and ALS-PDC disease in Guam. Neurology 2002, 58, 956–959. [Google Scholar]
- Monson, CS; Banack, SA; Cox, PA. Conservation implications of chamorro consumption of flying foxes as a possible cause of amyotrophic lateral sclerosis/Parkinsonism dementia complex in guam. Conserv Biol 2003, 17, 678–686. [Google Scholar]
- Banack, SA; Cox, PA. Biomagnification of cycad neurotoxins in flying foxes: Implications for ALS-PDC in Guam. Neurology 2003, 61, 387–389. [Google Scholar]
- Cox, PA; Banack, SA; Murch, SJ. Biomagnification of cyanobacterial neurotoxins and neurodegenerative disease among the Chamorro people of Guam. Proc Natl Acad Sci USA 2003, 100, 13380–13383. [Google Scholar]
- Adams, D. The Ecology of Cyanobacteria; Kluwer Academic: New York, NY, USA, 2002; pp. 523–561. [Google Scholar]
- Cox, PA; Banack, SA; Murch, SJ; Rasmussen, U; Tien, G; Bidigare, RR; Metcalf, JS; Morrison, LF; Codd, GA; Bergman, B. Diverse taxa of cyanobacteria produce β-N-methylamino-l-alanine, a neurotoxic amino acid. Proc Natl Acad Sci USA 2005, 102, 5074–5078. [Google Scholar]
- Esterhuizen, M; Downing, TG. Beta-N-methylamino-l-alanine (BMAA) in novel South African cyanobacterial isolates. Ecotoxicol Environ Saf 2008, 71, 309–313. [Google Scholar]
- Marler, TE; Snyder, LR; Shaw, CA. Cycas micronesica (Cycadales) plants devoid of endophytic cyanobacteria increase in [beta]-methylamino-l-alanine. Toxicon 2010, 56, 563–568. [Google Scholar]
- Banack, SA; Cox, PA. Distribution of the neurotoxic nonprotein amino acid BMAA in Cycas micronesica. Bot J Linn Soc 2003, 143, 165–168. [Google Scholar]
- Murch, SJ; Cox, PA; Banack, SA; Steele, JC; Sacks, OW. Occurrence of β-methylamino-l-alanine (BMAA) in ALS/PDC patients from Guam. Acta Neurol Scand 2004, 110, 267–269. [Google Scholar]
- Murch, SJ; Cox, PA; Banack, SA. A mechanism for slow release of biomagnified cyanobacterial neurotoxins and neurodegenerative disease in Guam. Proc Natl Acad Sci USA 2004, 101, 12228–12231. [Google Scholar]
- Montine, TJ; Li, K; Perl, DP; Galasko, D. Lack of beta-methylamino-l-alanine in brain from controls, AD, or Chamorros with PDC. Neurology 2005, 65, 768–769. [Google Scholar]
- Snyder, LR; Cruz-Aguado, R; Sadilek, M; Galasko, D; Shaw, CA; Montine, TJ. Lack of cerebral BMAA in human cerebral cortex. Neurology 2009, 72, 1360–1361. [Google Scholar]
- Bradley, WG; Mash, DC. Beyond Guam: The cyanobacteria/BMAA hypothesis of the cause of ALS and other neurodegenerative diseases. Amyotroph Lateral Scler 2009, 10, 7–20. [Google Scholar]
- Karamyan, VT; Speth, RC. Animal models of BMAA neurotoxicity: A critical review. Life Sci 2008, 82, 233–246. [Google Scholar]
- Cohen, SA; de Antonis, KM. Applications of amino acid derivatization with 6-aminoquinolyl- N-hydroxysuccinimidyl carbamate. Analysis of feed grains, intravenous solutions and glycoproteins. J Chromatogr A 1994, 661, 25–34. [Google Scholar]
- Crimmins, DL; Cherian, R. Increasing the sensitivity of 6-aminoquinolyl-Nhydroxysuccinimidyl carbamate amino acid analysis: A Simple Solution. Anal Biochem 1997, 244, 407–410. [Google Scholar]
- Pablo, J; Banack, SA; Cox, PA; Johnson, TE; Papapetropoulos, S; Bradley, WG; Buck, A; Mash, DC. Cyanobacterial neurotoxin BMAA in ALS and Alzheimer’s disease. Acta Neurol Scand 2009, 120, 216–225. [Google Scholar]
- Banack, SA; Johnson, HE; Cheng, R; Cox, PA. Production of the Neurotoxin BMAA by a Marine Cyanobacterium. Mar Drugs 2007, 5, 180–196. [Google Scholar]
- Jonasson, S; Eriksson, J; Berntzon, L; Spáčil, Z; Ilag, LL; Ronnevi, L-O; Rasmussen, U; Bergman, B. Transfer of a cyanobacterial neurotoxin within a temperate aquatic ecosystem suggests pathways for human exposure. Proc Natl Acad Sci USA 2010, 107, 9252–9257. [Google Scholar]
- Brand, LE; Pablo, J; Compton, A; Hammerschlag, N; Mash, DC. Cyanobacterial blooms and the occurrence of the neurotoxin, beta-N-methylamino-l-alanine (BMAA), in South Florida aquatic food webs. Harmful Algae 2010, 9, 620–635. [Google Scholar]
- Li, A; Tian, Z; Li, J; Yu, R; Banack, SA; Wang, Z. Detection of the neurotoxin BMAA within cyanobacteria isolated from freshwater in China. Toxicon 2010, 55, 947–953. [Google Scholar]
- Cox, PA; Richer, R; Metcalf, JS; Banack, SA; Codd, GA; Bradley, WG. Cyanobacteria and BMAA exposure from desert dust: A possible link to sporadic ALS among Gulf War veterans. Amyotroph Lateral Scler 2009, 10, 109–117. [Google Scholar]
- Esterhuizen, M; Pflugmacher, S; Downing, TG. [beta]-N-Methylamino-l-alanine (BMAA) uptake by the aquatic macrophyte Ceratophyllum demersum. Ecotoxicol Environ Saf 2011, 74, 74–77. [Google Scholar]
- Lürling, M; Faassen, EJ; van Eenennaam, JS. Effects of the cyanobacterial neurotoxin β-N-methylamino-l-alanine (BMAA) on the survival, mobility and reproduction of Daphnia magna. J Plankton Res 2011, 33, 333–342. [Google Scholar]
- Monaghan, DT; Bridges, RJ; Cotman, CW. The excitatory amino acid receptors: Their classes, pharmacology, and distinct properties in the function of the central nervous system. Annu Rev Pharmacol Toxicol 1989, 29, 365–402. [Google Scholar]
- Smith, PF; de Waele, C; Vidal, PP; Darlington, CL. Excitatory amino acid receptors in normal and abnormal vestibular function. Mol Neurobiol 1991, 5, 369–387. [Google Scholar]
- Curtis, DR; Watkins, JC. The excitation and depression of spinal neurones by structurally related amino acids. J Neurochem 1960, 6, 117–141. [Google Scholar]
- Doble, A. The role of excitotoxicity in neurodegenerative disease: Implications for therapy. Pharmacol Ther 1999, 81, 163–221. [Google Scholar]
- Shaw, PJ. Molecular and cellular pathways of neurodegeneration in motor neurone disease. J Neurol Neurosurg Psychiatry 2005, 76, 1046–1057. [Google Scholar]
- Strong, MJ; Kesavapany, S; Pant, HC. The pathobiology of amyotrophic lateral sclerosis: A proteinopathy? J Neuropathol Exp Neurol 2005, 64, 649–664. [Google Scholar]
- Cozzolino, M; Ferri, A; Teresa Carri, M. Amyotrophic lateral sclerosis: From current developments in the laboratory to clinical implications. Antioxid Redox Signal 2008, 10, 405–444. [Google Scholar]
- Boillee, S; Vande Velde, C; Cleveland, DW. ALS: A disease of motor neurons and their nonneuronal neighbors. Neuron 2006, 52, 39–59. [Google Scholar]
- Majoor-Krakauer, D; Willems, PJ; Hofman, A. Genetic epidemiology of amyotrophic lateral sclerosis. Clin Genet 2003, 63, 83–101. [Google Scholar]
- Dastur, DK. Cycad toxicity in monkeys: Clinical, pathological, and biochemical aspects. Fed Proc 1964, 23, 1368–1369. [Google Scholar]
- Bell, EA; Vega, A; Nunn, PB. Neurotoxic Effects of α-Amino-β-methyl-aminopropionic Acid. Proceedings of the Fifth Conference on Cycad Toxicity, Miami, FL, USA, 24–25 April 1967.
- Nunn, PB; Vega, A; Bell, EA. Neurotoxic effects of alpha-amino-beta-methylaminopropionic acid. Biochem J 1968, 106, 15. [Google Scholar]
- Polsky, FI; Nunn, PB; Bell, EA. Distribution and toxicity of alpha-amino-beta-methylaminopropionic acid. Fed Proc 1972, 31, 1473–1475. [Google Scholar]
- Rakonczay, Z; Matsuoka, Y; Giacobini, E. Effects of l-beta-N-methylamino-l-alanine (l-BMAA) on the cortical cholinergic and glutamatergic systems of the rat. J Neurosci Res 1991, 29, 121–126. [Google Scholar]
- Matsuoka, Y; Rakonczay, Z; Giacobini, E; Naritoku, D. l-beta-methylaminoalanine-induced behavioral changes in rats. Pharmacol Biochem Behav 1993, 44, 727–734. [Google Scholar]
- Perry, TL; Bergeron, C; Biro, AJ; Hansen, S. Beta-N-methylamino-l-alanine. Chronic oral administration is not neurotoxic to mice. J Neurol Sci 1989, 94, 173–180. [Google Scholar]
- Cruz-Aguado, R; Winkler, D; Shaw, CA. Lack of behavioral and neuropathological effects of dietary [beta]-methylamino-l-alanine (BMAA) in mice. Pharmacol Biochem Behav 2006, 84, 294–299. [Google Scholar]
- Banack, SA; Caller, TA; Stommel, EW. The cyanobacteria derived toxin beta-N-methylamino-l-alanine and amyotrophic lateral sclerosis. Toxins 2010, 2, 2837–2850. [Google Scholar]
- Weiss, JH; Choi, DW. Beta-N-methylamino-l-alanine neurotoxicity: Requirement for bicarbonate as a cofactor. Science 1988, 241, 973–975. [Google Scholar]
- Richter, KE; Mena, EE. l-beta-Methylaminoalanine inhibits [3H]glutamate binding in the presence of bicarbonate ions. Brain Res 1989, 492, 385–388. [Google Scholar]
- Weiss, JH; Koh, JY; Choi, DW. Neurotoxicity of beta-N-methylamino-l-alanine (BMAA) and beta-N-oxalylamino-l-alanine (BOAA) on cultured cortical neurons. Brain Res 1989, 497, 64–71. [Google Scholar]
- Myers, TG; Nelson, SD. Neuroactive carbamate adducts of beta-N-methylamino-l-alanine and ethylenediamine. Detection and quantitation under physiological conditions by 13C NMR. J Biol Chem 1990, 265, 10193–10195. [Google Scholar]
- Lindstrom, H; Luthman, J; Mouton, P; Spencer, P; Olson, L. Plant-derived neurotoxic amino acids (beta-N-oxalylamino-l-alanine and beta-N-methylamino-l-alanine): Effects on central monoamine neurons. J Neurochem 1990, 55, 941–949. [Google Scholar]
- Copani, A; Canonico, PL; Catania, MV; Aronica, E; Bruno, V; Ratti, E; van Amsterdam, FT; Gaviraghi, G; Nicoletti, F. Interaction between beta-N-methylamino-l-alanine and excitatory amino acid receptors in brain slices and neuronal cultures. Brain Res 1991, 558, 79–86. [Google Scholar]
- Duncan, MW; Markey, SP; Weick, BG; Pearson, PG; Ziffer, H; Hu, Y; Kopin, IJ. 2-Amino-3-(methylamino)propanoic acid (BMAA) bioavailability in the primate. Neurobio Aging 1992, 13, 333–337. [Google Scholar]
- Kisby, GE; Roy, DN; Spencer, PS. Determination of beta-N-methylamino-l-alanine (BMAA) in plant (Cycas circinalis L.) and animal tissue by precolumn derivatization with 9-fluorenylmethyl chloroformate (FMOC) and reversed-phase high-performance liquid chromatography. J Neurosci Methods 1988, 26, 45–54. [Google Scholar]
- Duncan, MW; Villacreses, NE; Pearson, PG; Wyatt, L; Rapoport, SI; Kopin, IJ; Markey, SP; Smith, QR. 2-amino-3-(methylamino)-propanoic acid (BMAA) pharmacokinetics and blood-brain barrier permeability in the rat. J Pharmacol Exp Ther 1991, 258, 27–35. [Google Scholar]
- Smith, QR; Nagura, H; Takada, Y; Duncan, MW. Facilitated transport of the neurotoxin, beta-N-methylamino-l-alanine, across the blood-brain barrier. J Neurochem 1992, 58, 1330–1337. [Google Scholar]
- Brownson, DM; Mabry, TJ; Leslie, SW. The cycad neurotoxic amino acid, β-N-methylamino-l-alanine (BMAA), elevates intracellular calcium levels in dissociated rat brain cells. J Ethnopharmacol 2002, 82, 159–167. [Google Scholar]
- Choi, DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron 1988, 1, 623–634. [Google Scholar]
- Meldrum, B; Garthwaite, J. Excitatory amino acid neurotoxicity and neurodegenerative disease. Trends Pharmacol Sci 1990, 11, 379–387. [Google Scholar]
- Nedeljkov, V; Lopicic, S; Pavlovic, D; Cemerikic, D. Electrophysiological Effect of β-N-Methylamino-l-Alanine on Retzius Nerve Cells of the Leech Haemopis sanguisuga. Ann N Y Acad Sci 2005, 1048, 349–351. [Google Scholar]
- Buenz, EJ; Howe, CL. Beta-methylamino-alanine (BMAA) injures hippocampal neurons in vivo. Neuro Toxicol 2007, 28, 702–704. [Google Scholar]
- Lobner, D; Piana, PMT; Salous, AK; Peoples, RW. [beta]-N-methylamino-l-alanine enhances neurotoxicity through multiple mechanisms. Neurobiol Dis 2007, 25, 360–366. [Google Scholar]
- Rao, SD; Banack, SA; Cox, PA; Weiss, JH. BMAA selectively injures motor neurons via AMPA/kainate receptor activation. Exp Neurol 2006, 201, 244–252. [Google Scholar]
- Liu, X; Rush, T; Zapata, J; Lobner, D. [beta]-N-methylamino-l-alanine induces oxidative stress and glutamate release through action on system Xc−. Exp Neurol 2009, 217, 429–433. [Google Scholar]
- Nunn, PB; Ponnusamy, M. [beta]-N-Methylaminoalanine (BMAA): Metabolism and metabolic effects in model systems and in neural and other tissues of the rat in vitro. Toxicon 2009, 54, 85–94. [Google Scholar]
- Deng, Y; Boomsma, F; Yu, PH. Deamination of methylamine and aminoacetone increases aldehydes and oxidative stress in rats. Life Sciences 1998, 63, 2049–2058. [Google Scholar]
- Karlsson, O; Berg, C; Brittebo, EB; Lindquist, NG. Retention of the cyanobacterial neurotoxin β-N-methylamino-l-alanine in melanin and neuromelanin-containing cells—A possible link between Parkinson-dementia complex and pigmentary retinopathy. Pigment Cell Melanoma Res 2009, 22, 120–130. [Google Scholar]
- Lopicic, S; Nedeljkov, V; Cemerikic, D. Augmentation and ionic mechanism of effect of [beta]-N-methylamino-l-alanine in presence of bicarbonate on membrane potential of Retzius nerve cells of the leech Haemopis sanguisuga. Comp Biochem Physiol Part A 2009, 153, 284–292. [Google Scholar]
- Santucci, S; Zsürger, N; Chabry, J. β-N-methylamino-l-alanine induced in vivo retinal cell death. J Neurochem 2009, 109, 819–825. [Google Scholar]
- Purdie, EL; Samsudin, S; Eddy, FB; Codd, GA. Effects of the cyanobacterial neurotoxin [beta]-N-methylamino-l-alanine on the early-life stage development of zebrafish (Danio rerio). Aquat Toxicol 2009, 95, 279–284. [Google Scholar]
- Cucchiaroni, ML; Viscomi, MT; Bernardi, G; Molinari, M; Guatteo, E; Mercuri, NB. Metabotropic glutamate receptor 1 mediates the electrophysiological and toxic actions of the cycad derivative {beta}-N-Methylamino-l-alanine on substantia nigra pars compacta DAergic neurons. J Neurosci 2010, 30, 5176–5188. [Google Scholar]
- Karlsson, O; Roman, E; Berg, A-L; Brittebo, EB. Early hippocampal cell death, and late learning and memory deficits in rats exposed to the environmental toxin BMAA ([beta]-N-methylamino-l-alanine) during the neonatal period. Behav Brain Res 2011, 219, 310–320. [Google Scholar]
- Lee, M; McGeer, PL. Weak BMAA toxicity compares with that of the dietary supplement beta-alanine. Neurobiol Aging 2011, in press. [Google Scholar]
- Steele, JC; McGeer, PL. The ALS/PDC syndrome of Guam and the cycad hypothesis. Neurology 2008, 70, 1984–1990. [Google Scholar]
- Cheng, R; Banack, SA. Previous studies underestimate BMAA concentrations in cycad flour. Amyotroph Lateral Scler 2009, 10, 41–43. [Google Scholar]
- Khabazian, I; Bains, JS; Williams, DE; Cheung, J; Wilson, JM; Pasqualotto, BA; Pelech, SL; Andersen, RJ; Wang, YT; Liu, L; et al. Isolation of various forms of sterol beta-d-glucoside from the seed of Cycas circinalis: Neurotoxicity and implications for ALS-parkinsonism dementia complex. J Neurochem 2002, 82, 516–528. [Google Scholar]
- Ly, PTT; Singh, S; Shaw, CA. Novel environmental toxins: Steryl glycosides as a potential etiological factor for age-related neurodegenerative diseases. J Neurosci Res 2007, 85, 231–237. [Google Scholar]
- Tabata, RC; Wilson, JM; Ly, P; Zwiegers, P; Kwok, D; van Kampen, JM; Cashman, N; Shaw, CA. Chronic exposure to dietary sterol glucosides is neurotoxic to motor neurons and induces an ALS-PDC phenotype. Neuromol Med 2008, 10, 24–39. [Google Scholar]
- Metcalf, JS; Banack, SA; Lindsay, J; Morrison, LF; Cox, PA; Codd, GA. Co-occurrence of beta-N-methylamino-l-alanine, a neurotoxic amino acid with other cyanobacterial toxins in British waterbodies, 1990–2004. Environ Microbiol 2008, 10, 702–708. [Google Scholar]
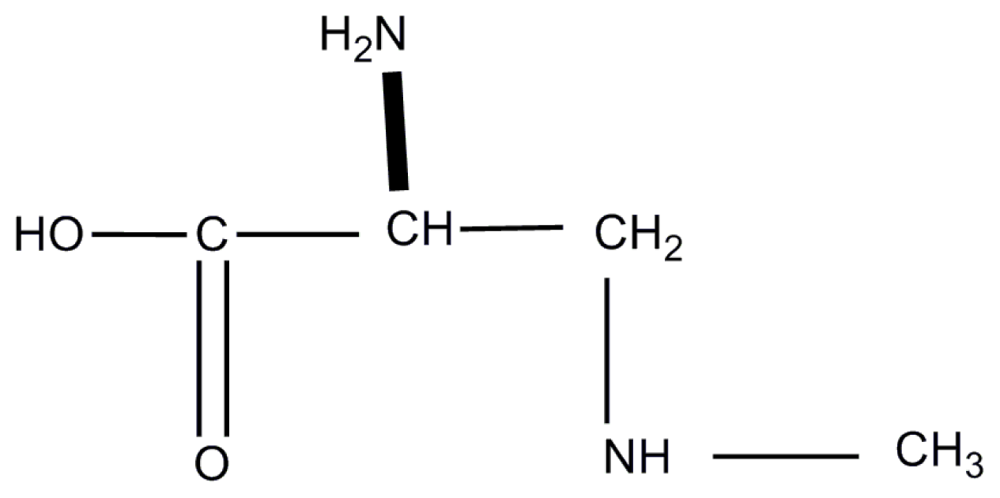


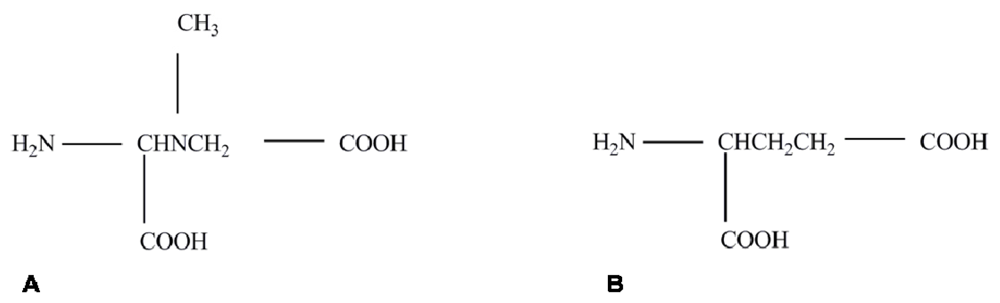
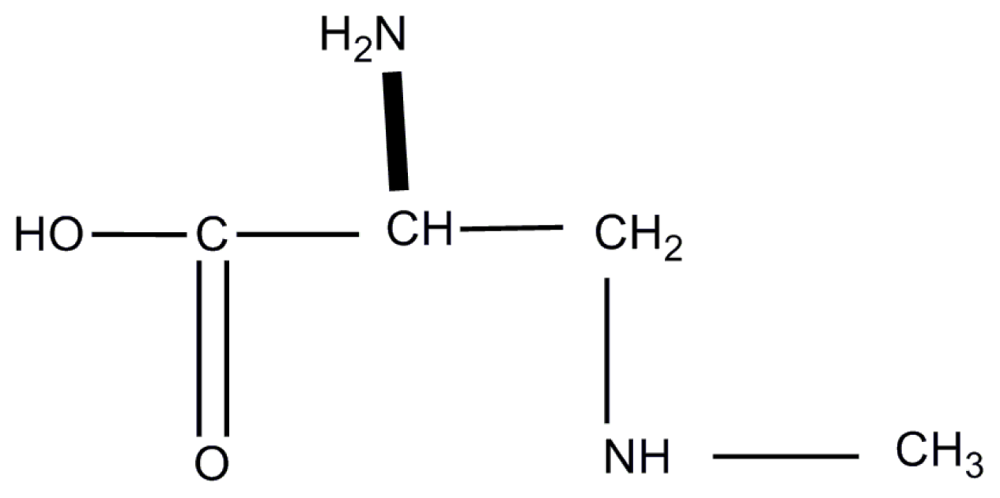{kind=link}
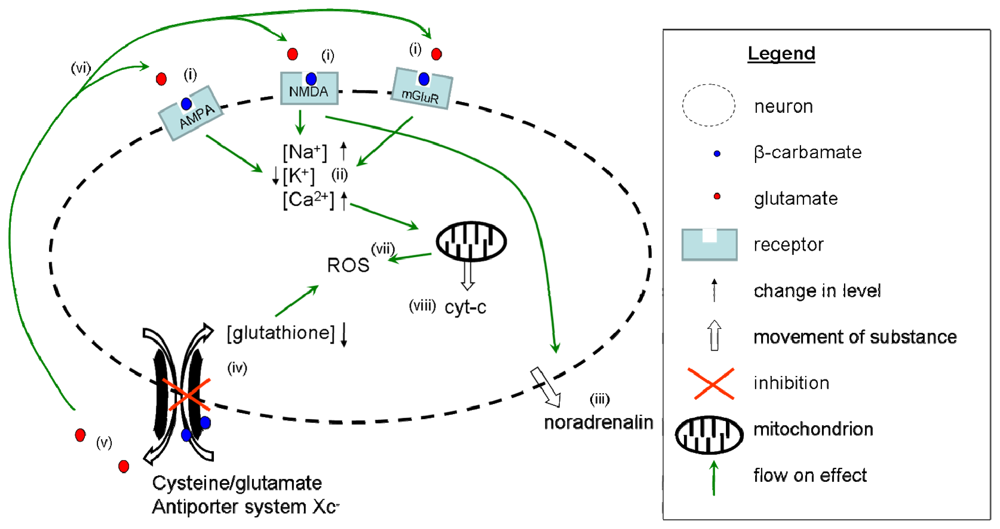
{kind=link}
{kind=link}
| Routez of exposure | Species | Dose level, exposure time | Research group and date | Observations |
|---|---|---|---|---|
| Intraperitoneal injections | Rat Chicken | 6–14 μmoles/g body weight 3–7 μmoles/g body weight | Vega and Bell. 1967 | Weakness, convulsions and uncoordination |
| Intraperitoneal injections | Rat Chicken Mouse | 6–14 μmoles/g body weight 3–7 μmoles/g body weight 6–14 μmoles/g body weight | Polski et al. 1972 | Weakness, convulsions and uncoordination |
| Perorally Intraperitoneal injections | Monkey Rat | 100–350 mg/kg, 12 months 500 mg/kg daily, 14 days | Kisby et al. 1988 | BMAA can cross from gut to blood BMAA can cross the blood brain barrier |
| Gavage | Monkey | 100–350 mg/kg daily, up to 10 weeks | Spencer et al. 1987 | Corticomotoneuronal dysfunction, Parkinsonian features and behavioural abnormalities |
| Gavage | Cynomologous monkey | 500 mg/kg daily, 18 days, then 500 mg/kg 2 daily, 28 days, then 100mg/kg 2 daily, 30 days | Perry et al. 1989 | No behavioral or physiological effects observed |
| Intracerebral injections | Rat | 10 μg or 400 μg/150–200 g rat | Lindström et al. 1990 | Activation of NMDA receptor, release noradrenalin from cells |
| Intracerebroventricular injections | Rat | 500 μg/day 200–250 g body weight, 10–60 days | Rakonczay et al. 1990 Matsuoka et al. 1993 | Agonistic effects on NMDA, EAA and AMPA receptors in synapse Physical impairment. Mixed agonistic receptor activity |
| Gavage and intravenous injections | Cynomologous monkey, rat | 2 mg/kg gavage; 1 mg/kg iv 100 mg/kg gavage; 24–400 mg/kg iv | Duncan et al. 1991–1992 | 80% of ingested BMAA enters systemic circulation. BMAA can cross the blood brain barrier. BMAA is transported by neutral amino acid carriers so uptake can be influenced by diet, metabolism, disease and age |
| Dosed feed pellets | Mouse | 28 mg/kg daily, 30 days | Cruz-Aguado et al. 2006 | No motor, cognitive or neuropathological effect observed |
| Intracranial injections | Mouse | 10 μL of 100 mM, 24 h | Buenz and Howe. 2007 | Injury to hippocampal neurons |
| Intravenous and subcutaneous injections | Mouse and frog | 7.3 μg/kg, 30 min, 1 h, 3 h, 24 h, 12 days | Karlsson et al. 2009 | BMAA interacts/binds melanin, particularly during synthesis, and accumulates in melanin and neuromelanin containing cells increasingly over time |
| Ocular injections | Mouse | 5–10 nmol, 4, 8 and 24 h | Santucci et al. 2009 | Retinal neuron death and production of ROS |
| Experimental model | Species | Dose level, exposure time | Research group and date | Conclusion |
|---|---|---|---|---|
| Primary cortical neurons | Mouse | 3 mM, 1 h With and without 10–24 mM HCO3− | Weiss and Choi, 1988 | BMAA activity is dependent on bicarbonate at a min. of 20mM |
| Primary cortical neurons | Mouse | 300 μM–3 mM, 24 h | Weiss et al. 1989 | BMAA has activity on NMDA and non-NMDA receptors |
| Primary cortical neurons | Rat | 1 mM | Richter and Mena, 1989 | Inhibition of glutamate binding in synapse, impaired neuron function |
| Chemical assay | - | Myers and Nelson, 1990 | Formation of bicarbonate adduct with structural similarity to glutamate | |
| Brain slices | Rat | 1 mM, acute | Copani et al. 1991 | BMAA acts as a mixed agonist of metabotropic and NMDA receptors |
| Minced brain | Rat | 5 mM, acute | Brownson et al. 2002 | Impairment of intracellular calcium ion homeostasis. Possible neuronal death. Effects on calcium dependent cascades |
| Primary nerve cells | Leech | 1–10 mM, acute | Nedeljkov et al. 2005 | Depolarisation of cell, impaired nerve function. Membrane permeabilisation. Activity via glutamate receptors |
| Primary embryonic spinal cord culture | Mouse | 30–1000 μM, 20–24 h | Rao et al. 2006 | Increase on calcium ion concentration and ROS. Selective damage to motor neurons |
| Primary mixed cortical cells | Mouse | 0.1–10 mM, 24 h 3 mM, 3 h (DCFDA) | Lobner et al. 2007 | Potentiation of other insults, makes cells more sensitive to other compounds. Increase in ROS |
| NSC-34 cells | Mouse | 50–1000 μM, 18 h | Buenz and Howe 2007 | Dose dependent death of NSC-34 cells |
| Primary mixed cortical cell cultures | Mouse | 3 mM, 3 h | Liu et al. 2009 | Induction of oxidative stress is through inhibition of the cystine/glutamate antiporter system Xc− |
| Brain slices. Brain, liver, kidney homogenates | Rat | 10 mM, 30 min for slices 1 h for homogenates | Nunn and Ponnusamy, 2009 | The dimethylated product of BMAA, 2,3-diaminopropionic acid was formed in liver and kidney (but not brain) preparations |
| Nerve cells | Leech | 100–3000 μM, acute | Lopicic et al. 2009 | Action on non-NMDA ionotropic glutamate receptors, with a concomitant increase in cell membrane input conductance, as well as an increase in Na+ activity and a decrease in K+ activity. Possible initiation of excitotoxicity through activation of non-NMDA ionotropic glutamate receptors |
| Brain slices | Rat | 100–10000 μM, acute | Cucchiaroni et al. 2010 | BMAA activates mGluR1 receptors to cause neuronal degeneration Massive release of cyt-c into cytosol |
© 2011 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chiu, A.S.; Gehringer, M.M.; Welch, J.H.; Neilan, B.A. Does α-Amino-β-methylaminopropionic Acid (BMAA) Play a Role in Neurodegeneration? Int. J. Environ. Res. Public Health 2011, 8, 3728-3746. https://doi.org/10.3390/ijerph8093728
Chiu AS, Gehringer MM, Welch JH, Neilan BA. Does α-Amino-β-methylaminopropionic Acid (BMAA) Play a Role in Neurodegeneration? International Journal of Environmental Research and Public Health. 2011; 8(9):3728-3746. https://doi.org/10.3390/ijerph8093728
Chicago/Turabian StyleChiu, Alexander S., Michelle M. Gehringer, Jeffrey H. Welch, and Brett A. Neilan. 2011. "Does α-Amino-β-methylaminopropionic Acid (BMAA) Play a Role in Neurodegeneration?" International Journal of Environmental Research and Public Health 8, no. 9: 3728-3746. https://doi.org/10.3390/ijerph8093728
APA StyleChiu, A. S., Gehringer, M. M., Welch, J. H., & Neilan, B. A. (2011). Does α-Amino-β-methylaminopropionic Acid (BMAA) Play a Role in Neurodegeneration? International Journal of Environmental Research and Public Health, 8(9), 3728-3746. https://doi.org/10.3390/ijerph8093728