Altered Gene Expression by Low-Dose Arsenic Exposure in Humans and Cultured Cardiomyocytes: Assessment by Real-Time PCR Arrays


Abstract
:1. Introduction
2. Experimental Section
2.1. Study Subjects
2.2. Water Collection and Analysis
2.3. Toenail and Urine Collection and Analysis
2.4. Blood Collection, RNA Isolation, and cDNA Synthesis
2.5. cDNA Synthesis
2.6. TaqMan Low-Density Array
2.7. In Vitro Studies
2.8. Arsenite Treatment and RNA Isolation
2.9. Data Analysis
3. Results
3.1. Study Subjects
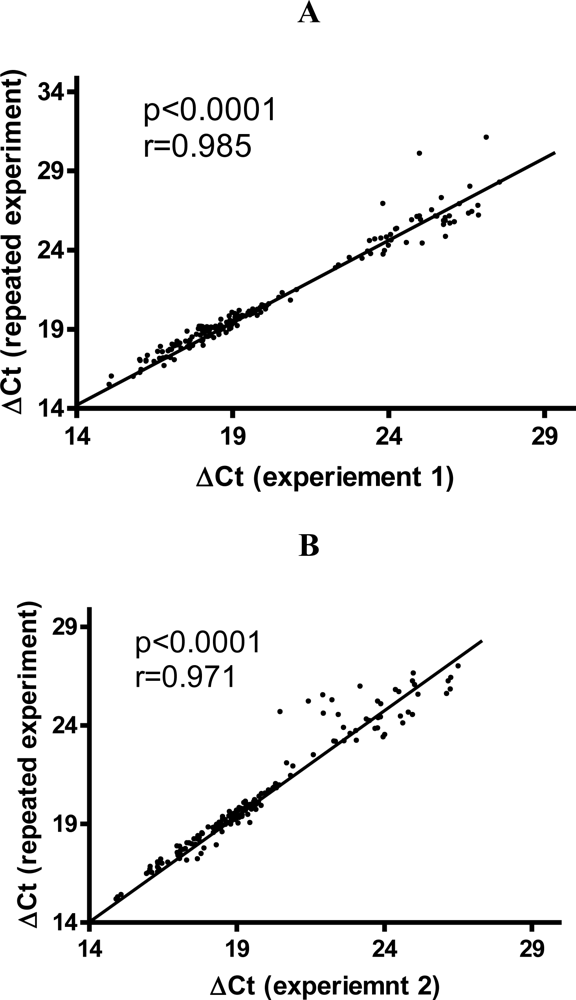
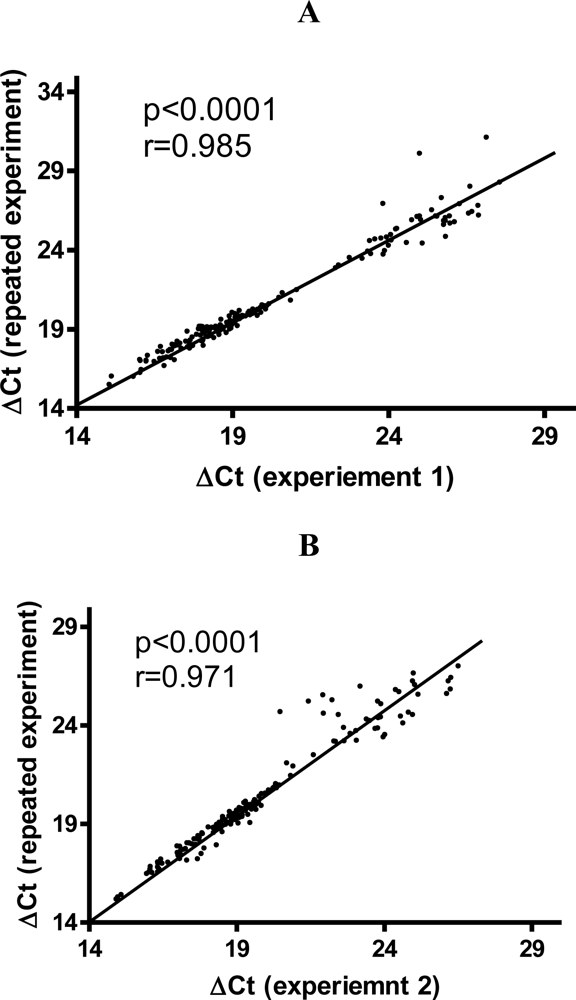
3.2. Variability of Gene Expression Data from TLDA
3.3. Arsenic-Altered Gene Expression in the Blood
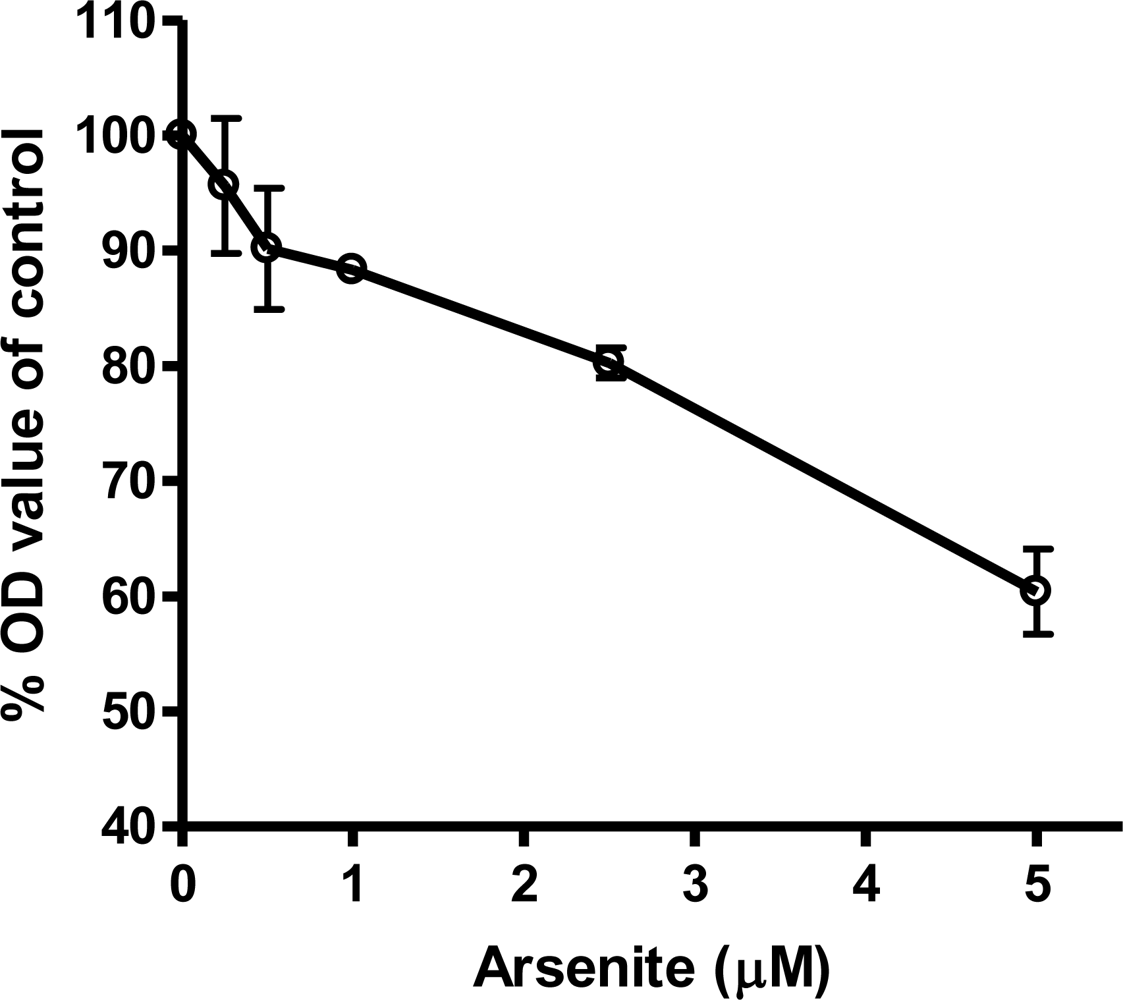
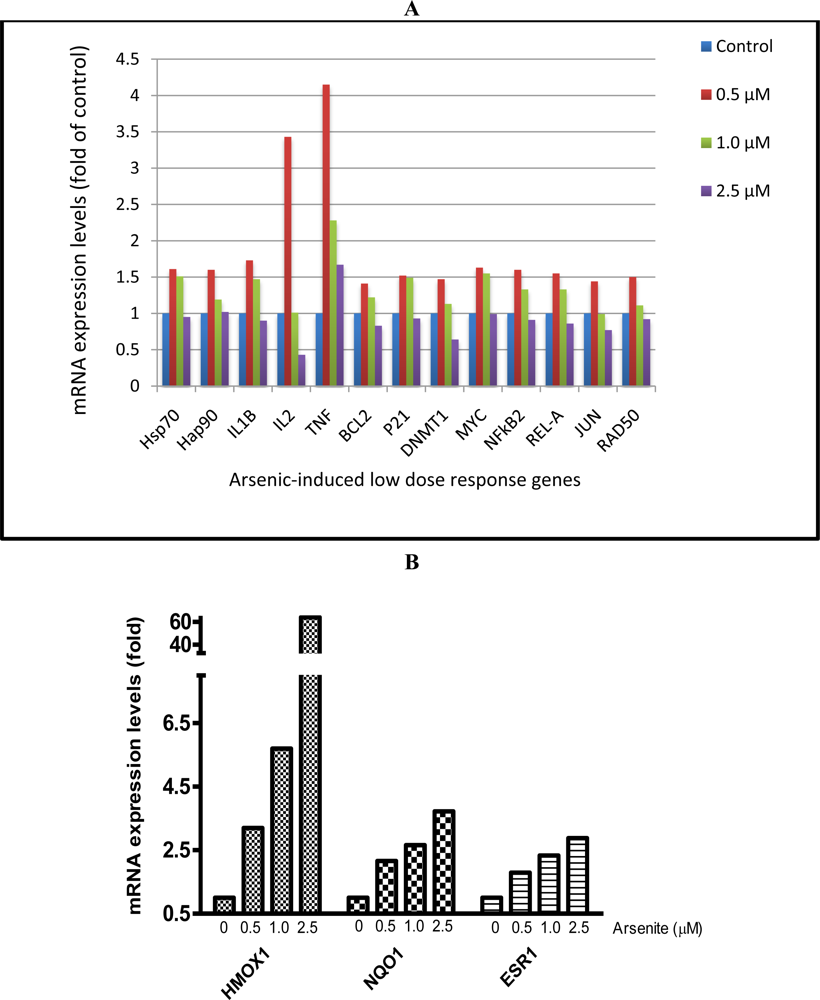
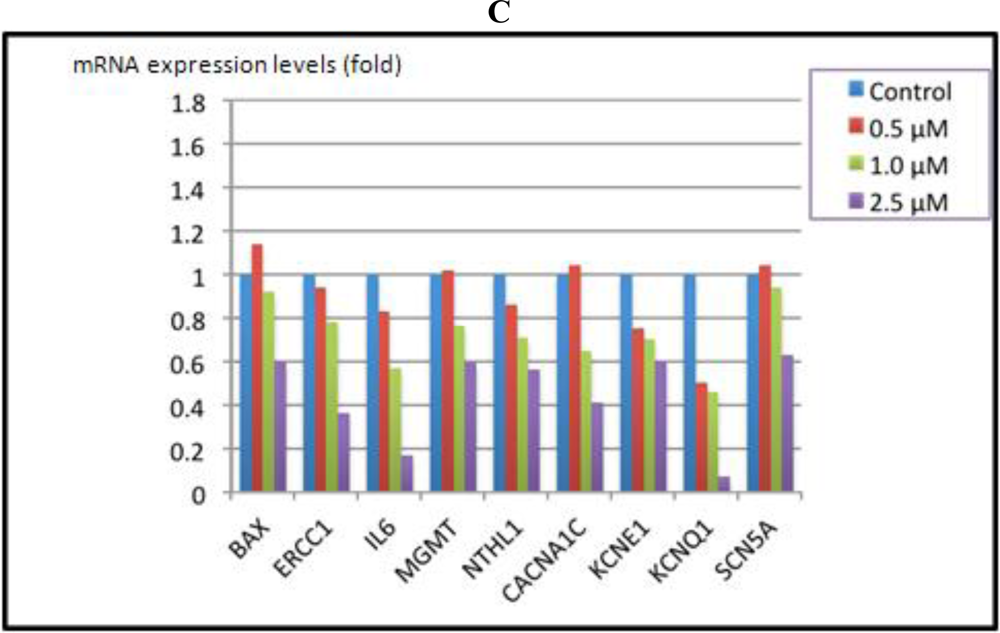
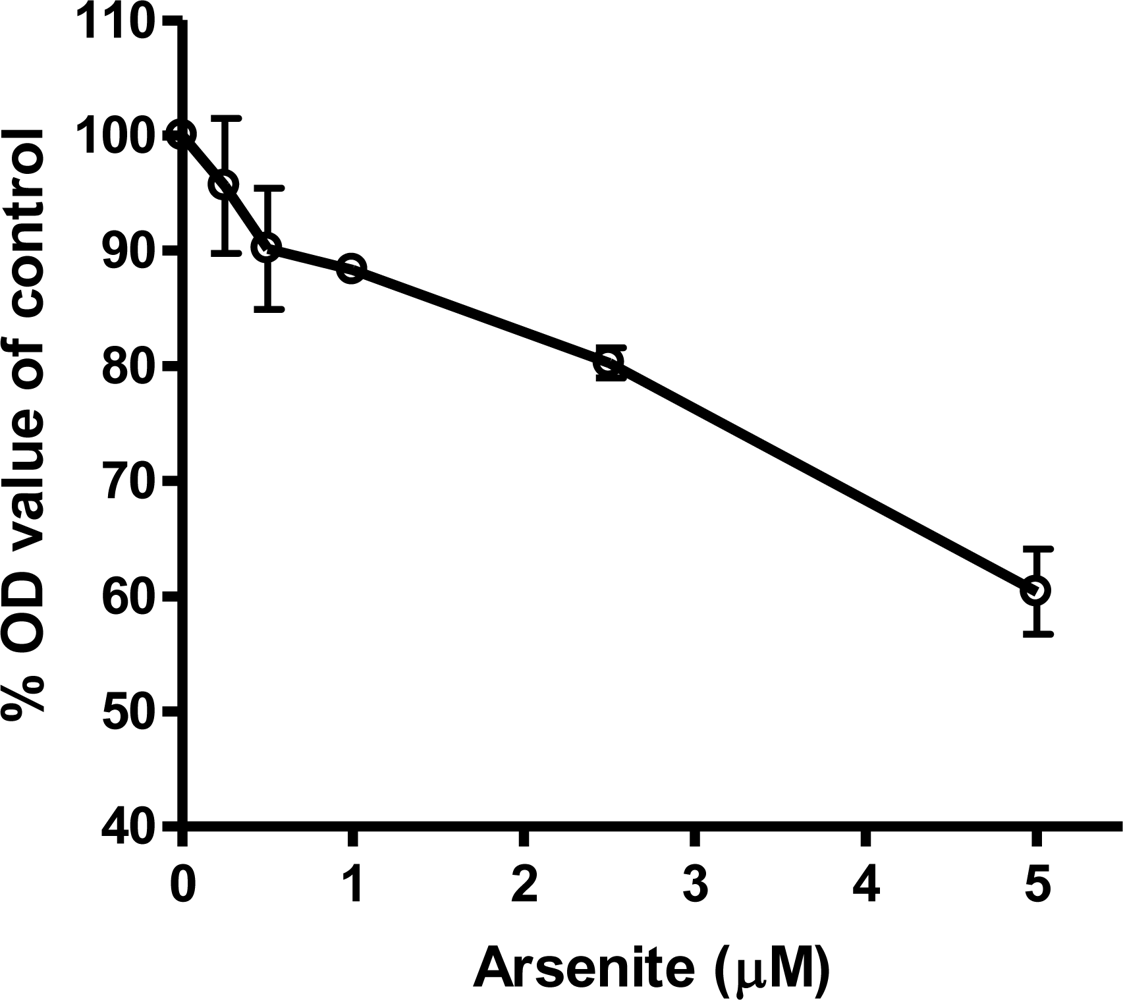
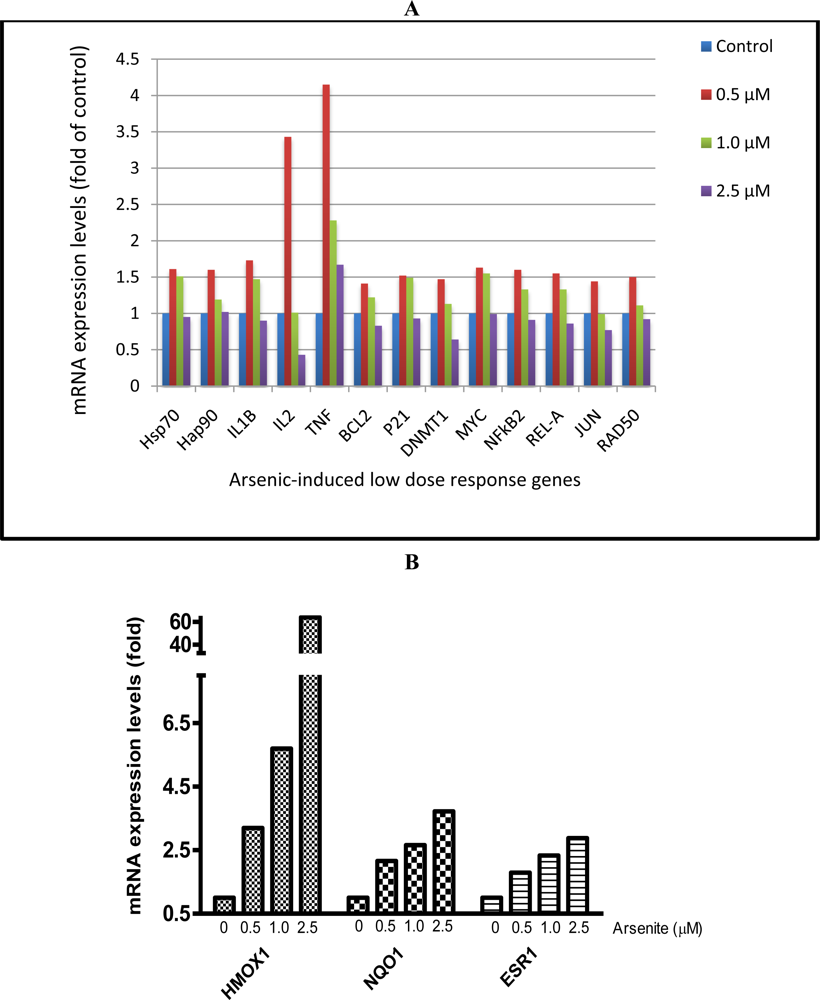
3.4. Cardiomyocytes Treated with Arsenite In Vitro
4. Discussion
4.1. Changes in Gene Expression in Blood Associated with Chronic Arsenic Exposure
4.2. Changes in Gene Expression in Arsenic-Exposed Cardiomyocytes
5. Conclusions
Acknowledgments
References
- International Agency for Research on Cancer (IARC). IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Some Drinking-Water Disinfectants and Contaminants, Including Arsenic; IARC: Lyon, France, 2004; Volume 84. [Google Scholar]
- Tapio, S; Grosche, B. Arsenic in the aetiology of cancer. Mutat. Res 2006, 612, 215–246. [Google Scholar]
- Straif, K; Benbrahim-Tallaa, L; Baan, R; Grosse, Y; Secretan, B; El Ghissassi, F; Bouvard, V; Guha, N; Freeman, C; Galichet, L; et al. A review of human carcinogens—Part C: Metals, arsenic, dusts, and fibres. Lancet Oncol 2009, 10, 453–454. [Google Scholar]
- Rossman, TG; Uddin, AN; Burns, FJ. Evidence that arsenite acts as a cocarcinogen in skin cancer. Toxicol. Appl. Pharmacol 2004, 198, 394–404. [Google Scholar]
- Mumford, JL; Wu, K; Xia, Y; Kwok, R; Yang, Z; Foster, J; Sanders, WE. Chronic arsenic exposure and cardiac repolarization abnormalities with QT interval prolongation in a population-based study. Environ. Health Perspect 2007, 115, 690–694. [Google Scholar]
- Westervelt, P; Brown, RA; Adkins, DR; Khoury, H; Curtin, P; Hurd, D; Luger, SM; Ma, MK; Ley, TJ; DiPersio, JF. Sudden death among patients with acute promyelocytic leukemia treated with arsenic trioxide. Blood 2001, 98, 266–271. [Google Scholar]
- Sanz, MA; Grimwade, D; Tallman, MS; Lowenberg, B; Fenaux, P; Estey, EH; Naoe, T; Lengfelder, E; Bűchner, T; Dőhner, H; Burnett, AK; Lo-Coco, F. Management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood 2009, 113, 1875–1891. [Google Scholar]
- States, JC; Srivastava, S; Chen, Y; Barchowsky, A. Arsenic and cardiovascular disease. Toxicol. Sci 2009, 107, 312–323. [Google Scholar]
- Wade, TJ; Xia, Y; Wu, K; Li, Y; Ning, Z; Le, XC; Lu, X; Feng, Y; He, X; Mumford, JL. Increased mortality associated with well-water arsenic exposure in Inner Mongolia, China. Int. J. Environ. Res. Public Health 2009, 6, 1107–1123. [Google Scholar]
- Wang, CH; Chen, CL; Hsiao, CK; Chiang, FT; Hsu, LI; Chiou, HY; Hsueh, YM; Wu, MM; Chen, CJ. Increased risk of QT prolongation associated with atherosclerotic diseases in arseniasis-endemic area in southwestern coast of Taiwan. Toxicol. Appl. Pharmacol 2009, 239, 320–324. [Google Scholar]
- Mordukhovich, I; Wright, RO; Amarasiriwardena, C; Baja, E; Baccarelli, A; Suh, H; Sparrow, D; Vokonas, P; Schwartz, J. Association between low-level environmental arsenic exposure and QT interval duration in a general population study. Am. J. Epidemiol 2009, 170, 739–746. [Google Scholar]
- Liu, J; Kadiiska, MB; Liu, Y; Lu, T; Qu, W; Waalkes, MP. Stress-related gene expression in mice treated with inorganic arsenicals. Toxicol. Sci 2001, 61, 314–320. [Google Scholar]
- Rea, MA; Gregg, JP; Qin, Q; Phillips, MA; Rice, RH. Global alteration of gene expression in human keratinocytes by inorganic arsenic. Carcinogenesis 2003, 24, 747–756. [Google Scholar]
- Argos, M; Kibriya, MG; Parvez, F; Jasmine, F; Rakibuz-Zaman, M; Ahsan, H. Gene expression profiles in peripheral lymphocytes by arsenic exposure and skin lesion status in a Bangladeshi population. Cancer Epidemiol. Biomarkers Prev 2006, 15, 1367–1375. [Google Scholar]
- Fry, RC; Navasumrit, P; Valiathan, C; Svensson, JP; Hogan, BJ; Luo, M; Bhattacharya, S; Kandjanapa, K; Soontararuks, S; Nookabkaew, S; et al. Activation of inflammation/NF-κB signaling in infants born to arsenic-exposed mothers. PLoS Genet 2007, 3, e207. [Google Scholar]
- Ghosh, P; Banerjee, M; Giri, AK; Ray, K. Toxcogenomics of arsenic: Classical ideas and recent advances. Mutat. Res 2008, 659, 293–301. [Google Scholar]
- Abruzzo, LV; Lee, KY; Fuller, A; Silverman, A; Keating, MJ; Medeiros, LJ; Coombes, KR. Validation of oligonucleotide microarray data using microfluidic low-density arrays: A new statistical method to normalize real-time RT-PCR data. Biotechniques 2005, 38, 785–792. [Google Scholar]
- Goulter, AB; Harmer, DW; Clark, KLO. Evaluation of low density array technology for quantitative parallel measurement of multiple genes in human tissue. BMC Genomics 2006, 7, 34. [Google Scholar]
- Ma, HZ; Xia, YJ; Wu, KG; Sun, TZ; Mumford, JL. Human exposure to arsenic and health effects in Bayingnormen, Inner Mongolia. In Arsenic Exposure and Health Effects; Abernathy, WR, Calderon, CO, Chappell, R, Eds.; Elsevier: Amsterdam, The Netherlands, 1999; pp. 127–131. [Google Scholar]
- Mo, J; Xia, Y; Wade, TJ; Schmitt, M; Le, XC; Dang, R; Mumford, JL. Chronic arsenic exposure and oxidative stress: OGG1 expression and arsenic exposure, nail selenium, and skin hyperkeratosis in Inner Mongolia. Environ. Health Perspect 2006, 114, 835–841. [Google Scholar]
- World Medical Association. Declaration of Helsinki. 1989. Available online: http://www.wma.net/e/policy/pdf/17c.pdf (accessed on 23 March 2011).
- Gong, Z; Lu, X; Watt, C; Wen, B; He, B; Mumford, J; Ning, Z; Xia, Y; Le, XC. Speciation analysis of arsenic in groundwater from Inner Mongolia with an emphasis on acid-leachable particulate arsenic. Anal. Chim. Acta 2006, 555, 181–187. [Google Scholar]
- Schmitt, MT; Schreinemachers, D; Wu, K; Ning, Z; Zhao, B; Le, XC; Mumford, JL. Human nails as a biomarker of arsenic exposure from well water in Inner Mongolia: Comparing atomic fluorescence spectrometry and neutron activation analysis. Biomarkers 2005, 10, 95–104. [Google Scholar]
- Heydorn, K. Neutron Activation Analysis for Clinical Trace Element Research; CRC Press: Boca Raton, FL, USA, 1984; Volume 1 and 2. [Google Scholar]
- Otto, D; Xia, Y; Li, Y; Wu, K; He, L; Telech, J; Hundell, H; Prah, J; Mumford, J; Wade, T. Neurosensory effects of chronic human exposure to arsenic associated with body burden and environmental measures. Hum. Exp. Toxicol 2007, 26, 169–177. [Google Scholar]
- Royston, P; Ambler, G; Sauerbrei, W. The use of fractional polynomials to model continuous risk variables in epidemiology. Int. J. Epidemiol 1999, 28, 964–974. [Google Scholar]
- Watanabe, C; Inaoka, T; Kadono, T; Nagano, M; Nakamura, S; Ushijima, K; Murayama, N; Miyazaki, K; Ohtsuka, R. Males in rural Bangladeshi communities are more susceptible to chronic arsenic poisoning than females: Analyses based on urinary arsenic. Environ. Health Perspect 2001, 109, 1265–1270. [Google Scholar]
- Ahsan, H; Perrin, M; Rahman, A; Parvez, F; Stute, M; Zheng, Y; Milton, AH; Brandt-Rauf, P; van Geen, A; Graziano, J. Associations between drinking water and urinary arsenic levels and skin lesions in Bangladesh. J. Occup. Environ. Med 2000, 42, 1195–1201. [Google Scholar]
- Maki-Paakkanen, J; Kurttio, P; Paldy, A; Pekkanen, J. Association between the clastogenic effect in peripheral lymphocytes and human exposure to arsenic through drinking water. Environ. Mol. Mutagen 1998, 32, 301–313. [Google Scholar]
- Bevnova, EE; Platoshyn, O; Zhang, S; Yuan, JX. Overexpression of human KCNA5 increases IK V and enhances apoptosis. Am. J. Physiol. Cell Physiol 2004, 287, C715–C722. [Google Scholar]
- Nattel, S; Bourne, G; Talajic, M. Insights into mechanisms of antiarrythmic drug action from experimental models of atrial fibrillation. J. Cardiovasc. Electrophysiol 1997, 8, 469–480. [Google Scholar]
- Fountain, SJ; Cheong, A; Li, J; Dondas, NY; Zeng, F; Wood, IC; Beech, DJ. K9v1.5 potassium channel gene regulation by Sp1 transcription factor and oxidative stress. Am. J. Physiol. Heart Circ. Physiol 2007, 293, H2719–H2725. [Google Scholar]
- Germolec, DR; Spalding, J; Boorman, GA; Wilmer, JL; Yoshida, T; Simeonova, PP; Bruccoleri, A; Kayama, F; Gaido, K; Tennant, R; et al. Arsenic can mediate skin neoplasia by chronic stimulation of keratinocyte-derived growth factors. Mutat. Res 1997, 386, 209–218. [Google Scholar]
- Yih, LH; Peck, K; Lee, TC. Changes in gene expression profiles of human fibroblasts in response to sodium arsenite treatment. Carcinogenesis 2002, 23, 867–876. [Google Scholar]
- Simeonova, PP; Luster, MI. Mechanisms of arsenic carcinogenicity: Genetic or epigenetic mechanisms? J. Environ. Pathol. Toxicol. Oncol 2000, 19, 281–286. [Google Scholar]
- Schoen, A; Beck, B; Sharma, R; Dube, E. Arsenic toxicity at low doses: Epidemiological and mode of action considerations. Toxicol. Appl. Pharmacol 2004, 198, 253–267. [Google Scholar]
- Itoh, K; Chiba, T; Takahashi, S; Ishii, T; Igarashi, K; Katoh, Y; Oyake, T; Hayashi, N; Satoh, K; Hatayama, I; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun 1997, 236, 313–322. [Google Scholar]
- Komissarova, EV; Li, P; Uddin, AN; Chen, X; Nadas, A; Rossman, TG. Gene expression levels in normal human lymphoblasts with variable sensitivities to arsenite: Identification of GGT1 and NFKBIE expression levels as possible biomarkers of susceptibility. Toxicol. Appl. Pharmacol 2008, 226, 199–205. [Google Scholar]
- Hu, Y; Jin, X; Snow, ET. Effect of arsenic on transcription factor AP-1 and NF-kappaB DNA binding activity and related gene expression. Toxicol. Lett 2002, 133, 33–45. [Google Scholar]
- Liu, Q; Zhang, H; Smeester, L; Zou, F; Kesic, M; Jaspers, I; Pi, J; Fry, RC. The NRF2-mediated oxidative stress response pathway is associated with tumor cell resistance to arsenic trioxide across the NCI-60 panel. BMC Med. Genomics 2010, 3, 37. [Google Scholar]
- Navas-Acien, A; Sharrett, AR; Silbergeld, EK; Schwartz, BS; Nachman, KE; Burke, TA; Guallar, E. Arsenic exposure and cardiovascular disease: A systematic review of the epidemiologic evidence. Am. J. Epidemiol 2005, 162, 1037–1049. [Google Scholar]
- Kligerman, AD; Doerr, CL; Tennant, AH; Harrington-Brock, K; Allen, JW; Winkfield, E; Poorman-Allen, P; Kundu, B; Funasaka, K; Roop, BC; Mass, MJ; DeMarini, DM. Methylated trivalent arsenicals as candidate ultimate genotoxic forms of arsenic: induction of chromosomal mutations but not gene mutations. Environ. Mol. Mutagen 2003, 42, 192–205. [Google Scholar]
- Wang, CH; Hsiao, CK; Chen, CL; Hsu, LI; Chiou, HY; Chen, SY; Hsueh, YM; Wu, MM; Chen, CJ. A review of the epidemiologic literature on the role of environmental arsenic exposure and cardiovascular diseases. Toxicol. Appl. Pharmacol 2007, 222, 315–326. [Google Scholar]
- Sykora, P; Snow, ET. Modulation of DNA polymerase beta-dependent base excision repair in cultured human cells after low dose exposure to arsenite. Toxicol. Appl. Pharmacol 2008, 228, 385–394. [Google Scholar]
- Zhang, TC; Schmitt, MT; Mumford, JL. Effects of arsenic on telomerase and telomeres in relation to cell proliferation and apoptosis in human keratinocytes and leukemia cells in vitro. Carcinogenesis 2003, 24, 1811–1817. [Google Scholar]
- Pi, J; Yamauchi, H; Kumagai, Y; Sun, G; Yoshida, T; Aikawa, H; Hopenhayn-Rich, C; Shimojo, N. Evidence for induction of oxidative stress caused by chronic exposure of Chinese residents to arsenic contained in drinking water. Environ. Health Perspect 2002, 110, 331–336. [Google Scholar]
- Dixon, JE; McKinnon, D. Quantitative analysis of potassium channel mRNA expression in atrial and ventricular muscle of rats. Circ. Res 1994, 75, 252–260. [Google Scholar]
- Wan, X; Dennis, AT; Obejero-Paz, C; Overholt, JL; Heredia-Moya, J; Kirk, KL; Ficker, E. Oxidative inactivation of the lipid phosphatase phosphatase and tensin homolog on chromosome ten (PTEN) as a novel mechanism of acquired long QT syndrome. J. Biol. Chem 2011, 286, 2843–2852. [Google Scholar]
- Diaz-Villaseñor, A; Burns, AL; Hiriart, M; Cebrián, ME; Ostrosky-Wegman, P. Arsenic-induced alteration in the expression of genes related to type 2 diabetes mellitus. Toxicol. Appl. Pharmacol 2007, 225, 123–133. [Google Scholar]
- Diaz-Villaseñor, A; Hiriart, M; Cebrián, ME; Zacarías-Castillo, R; Ostrosky-Wegman, P. The activity of calpains in lymphocytes is glucose-dependent and is decreased in diabetic patients. Blood Cells Mol. Dis 2008, 40, 414–419. [Google Scholar]
- Smeester, L; Rager, JE; Bailey, KA; Guan, X; Smith, N; García-Vargas, G; Del Razo, LM; Drobná, Z; Kelkar, H; Stýblo, M; Fry, RC. Epigenetic changes in individuals with arsenicosis. Chem. Res. Toxicol 2011, 24, 165–167. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene symbol and assay ID | Functional group | Gene description |
|---|---|---|
| ACTB-Hs00242273_m1 | Endogenous control | beta actin |
| 18S-Hs99999901_s1 | 18S Ribosomal RNA | |
| HMOX1-Hs00157965_m1 | Nrf2 pathway | |
| KEAP1-Hs00202227_m1 | kelch-like ECH-associated protein 1 | |
| MAF-Hs00193519_m1 | ||
| MT1A-Hs00831826_s1 | ||
| NQO1-Hs00168547_m1 | ||
| HSPA1A-Hs00359163_s1 | Heat shock protein | |
| HSP90AA1-Hs00743767_sH | ||
| BAX-Hs00180269_m1 | Apoptosis | |
| BCL2-Hs00153350_m1 | ||
| IL1B-Hs00174097_m1 | Inflammation | |
| IL2-Hs00174114_m1 | ||
| IL6-Hs00174131_m1 | ||
| TNF-Hs00174128_m1 | ||
| CHUK-Hs00175141_m1 | NF-κB pathway | |
| NFE2L2-Hs00232352_m1 | nuclear factor (erythroid-derived 2)-like 2 | |
| NFKB1-Hs00231653_m1 | nuclear factor in B-cells 1 (p105) | |
| NFKB2-Hs00174517_m1 | nuclear factor in B-cells 2 (p49/p100) | |
| RELB-Hs00232399_m1 | ||
| RELA-Hs00153294_m1 | ||
| CDKN1A-Hs00355782_m1 | Cell proliferation | |
| CCND1-Hs00277039_m1 | ||
| ESR1-Hs00174860_m1 | ||
| FOS-Hs00170630_m1 | ||
| JUN-Hs99999141_s1 | ||
| MYC-Hs00153408_m1 | ||
| MSH2-Hs00179887_m1 | ||
| NRAS-Hs00180035_m1 | ||
| TERT-Hs00162669_m1 | ||
| DNMT1-Hs00154749_m1 | DNA methylation | DNA (cytosine-5-)-methyltransferase 1 |
| DNMT3A-Hs00173377_m1 | ||
| ERCC1-Hs00157415_m1 | DNA repair | |
| MGMT-Hs00172470_m1 | ||
| NTHL1-Hs00267385_m1 | ||
| OGG1-Hs00213454_m1 | ||
| POLB-Hs00160263_m1 | ||
| DNMT1-Hs00154749_m1 | DNA methylation | DNA (cytosine-5-)-methyltransferase 1 |
| RAD50-Hs00194871_m1 | ||
| TP53-Hs00153349_m1 | ||
| CACNA1C-Hs00167681_m1 | Ion channel | calcium channel, voltage-dependent, L type, alpha 1C subunit |
| SCN5A-Hs00165693_m1 | sodium channel, voltage-gated, type V, alpha (long QT syndrome 3) | |
| KCNJ2-Hs00265315_m1 | potassium inwardly-rectifying channel, subfamily J, member 2 | |
| KCNA5-Hs00266898_s1 | potassium voltage-gated channel, shaker-related subfamily, member 5 | |
| KCND3-Hs00542597_m1 | potassium voltage-gated channel, Shal-related subfamily, member 3 | |
| KCNE1-Hs00264799_s1 | potassium voltage-gated channel, Isk-related family, member 1 | |
| KCNQ1-Hs00165003_m1 | potassium voltage-gated channel, KQT-like subfamily, member 1 | |
| KCNH2-Hs00165120_m1 | potassium voltage-gated channel, subfamily H (eag-related), member 2 | |
| KCNE2-Hs00270822_s1 | potassium voltage-gated channel, Isk-related family, member 2 | |
| Variables | Number of subjects | Percent |
|---|---|---|
| Gender | ||
| Male | 48 | 30.2 |
| Female | 111 | 69.8 |
| Age | ||
| 11–18 | 49 | 30.8 |
| 19–49 | 93 | 58.5 |
| >50 | 17 | 10.7 |
| Occupation | ||
| Farmer | 110 | 69.6 |
| Other | 48 | 30.4 |
| Education | ||
| None | 24 | 15.2 |
| Elementary | 50 | 31.6 |
| Jr high school | 76 | 48.1 |
| High school | 8 | 5.1 |
| Meat frequency | ||
| Often | 159 | 100 |
| Vegetable frequency | ||
| Occasionally | 1 | 0.6 |
| Often | 158 | 99.4 |
| Use of pesticides in past 5 years | ||
| Yes | 45 | 28.3 |
| No | 114 | 71.7 |
| Alcohol use 2 x/week | ||
| No | 114 | 95.6 |
| Yes | 7 | 4.4 |
| Eat fish frequency | ||
| Never | 1 | 0.6 |
| Occasionally | 146 | 91.8 |
| Often | 12 | 7.5 |
| Gene | Function group | Water arsenic | Nail arsenic | Urine arsenic | |||
|---|---|---|---|---|---|---|---|
| Slope | p value b | Slope | p value | Slope | p value | ||
| KCNA5 | Ion channel | 0.343 | 0.031 | 0.547 | 0.043 | 0.189 | 0.078 |
| TNF-α | Inflammation | 0.178 | 0.043 | 0.146 | 0.013 | ||
| POLB | DNA repair | 0.306 | 0.049 | 0.135 | 0.028 | ||
| MAF | Nrf2 pathway | 0.137 | 0.038 | ||||
| BCL2 | Apoptosis | 0.131 | 0.033 | ||||
| BAX | Apoptosis | 0.117 | 0.038 | ||||
| DNMT1 | DNA methylation | 0.122 | 0.031 | ||||
| HSP1 | Heat shock protein | 0.105 | 0.054 | ||||
| JUN | Signal transduction | 0.149 | 0.023 | ||||
| NTHL1 | DNA repair | 0.132 | 0.034 | ||||
| RAD50 | DNA repair | 0.113 | 0.050 | ||||
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mo, J.; Xia, Y.; Wade, T.J.; DeMarini, D.M.; Davidson, M.; Mumford, J. Altered Gene Expression by Low-Dose Arsenic Exposure in Humans and Cultured Cardiomyocytes: Assessment by Real-Time PCR Arrays. Int. J. Environ. Res. Public Health 2011, 8, 2090-2108. https://doi.org/10.3390/ijerph8062090
Mo J, Xia Y, Wade TJ, DeMarini DM, Davidson M, Mumford J. Altered Gene Expression by Low-Dose Arsenic Exposure in Humans and Cultured Cardiomyocytes: Assessment by Real-Time PCR Arrays. International Journal of Environmental Research and Public Health. 2011; 8(6):2090-2108. https://doi.org/10.3390/ijerph8062090
Chicago/Turabian StyleMo, Jinyao, Yajuan Xia, Timothy J. Wade, David M. DeMarini, Mercy Davidson, and Judy Mumford. 2011. "Altered Gene Expression by Low-Dose Arsenic Exposure in Humans and Cultured Cardiomyocytes: Assessment by Real-Time PCR Arrays" International Journal of Environmental Research and Public Health 8, no. 6: 2090-2108. https://doi.org/10.3390/ijerph8062090
APA StyleMo, J., Xia, Y., Wade, T. J., DeMarini, D. M., Davidson, M., & Mumford, J. (2011). Altered Gene Expression by Low-Dose Arsenic Exposure in Humans and Cultured Cardiomyocytes: Assessment by Real-Time PCR Arrays. International Journal of Environmental Research and Public Health, 8(6), 2090-2108. https://doi.org/10.3390/ijerph8062090