Chelation in Metal Intoxication

Abstract
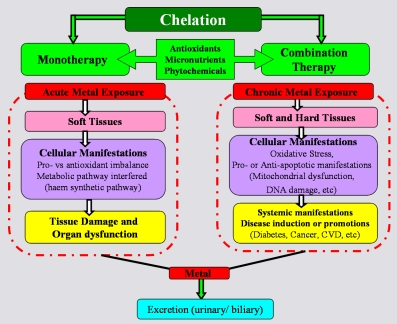
:
1. Introduction
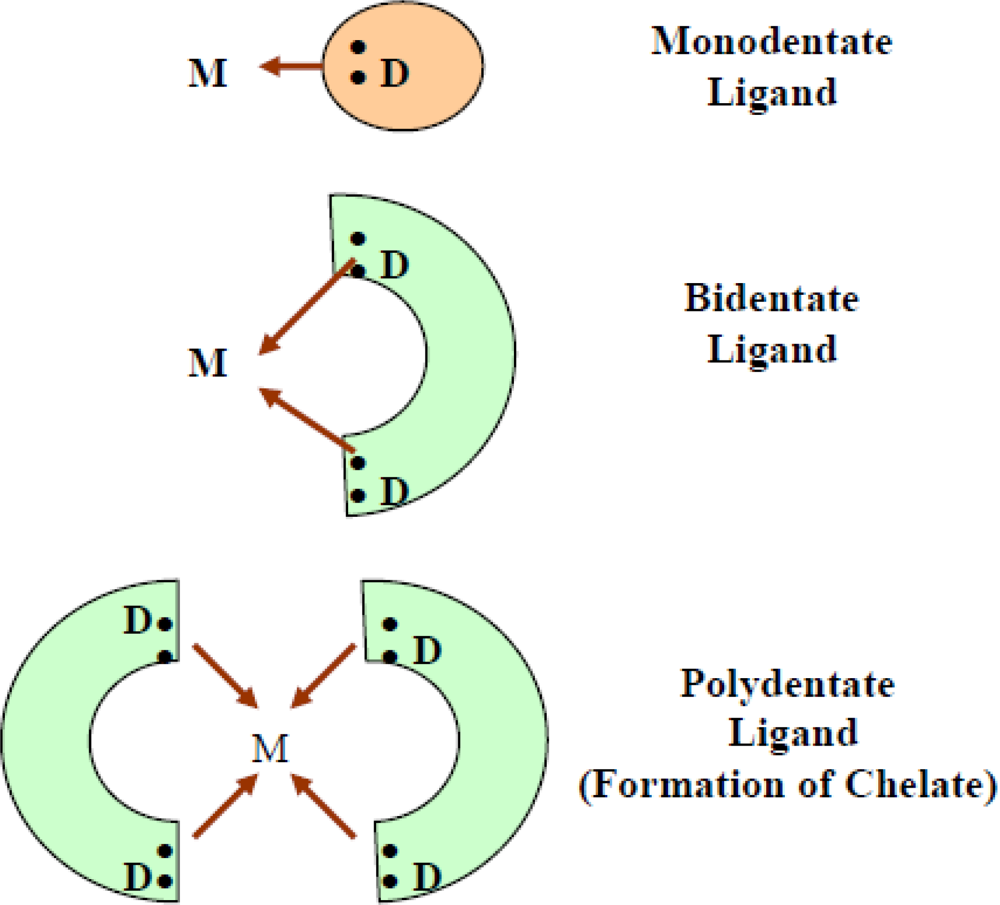
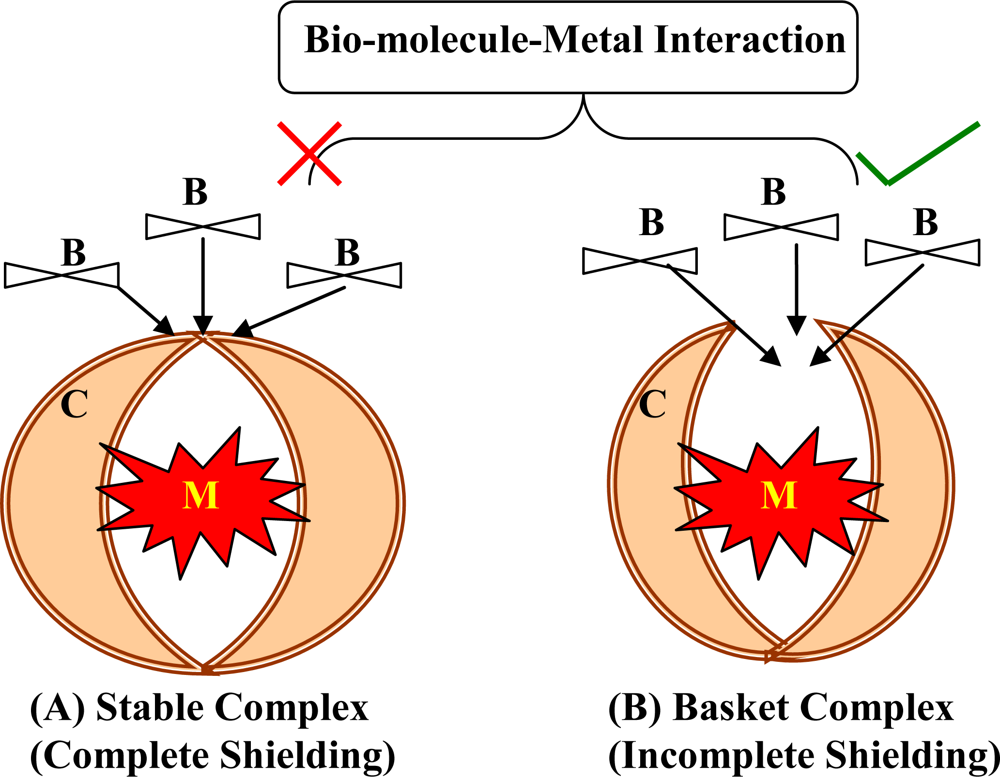
2. Chelation: Concept and Chemistry
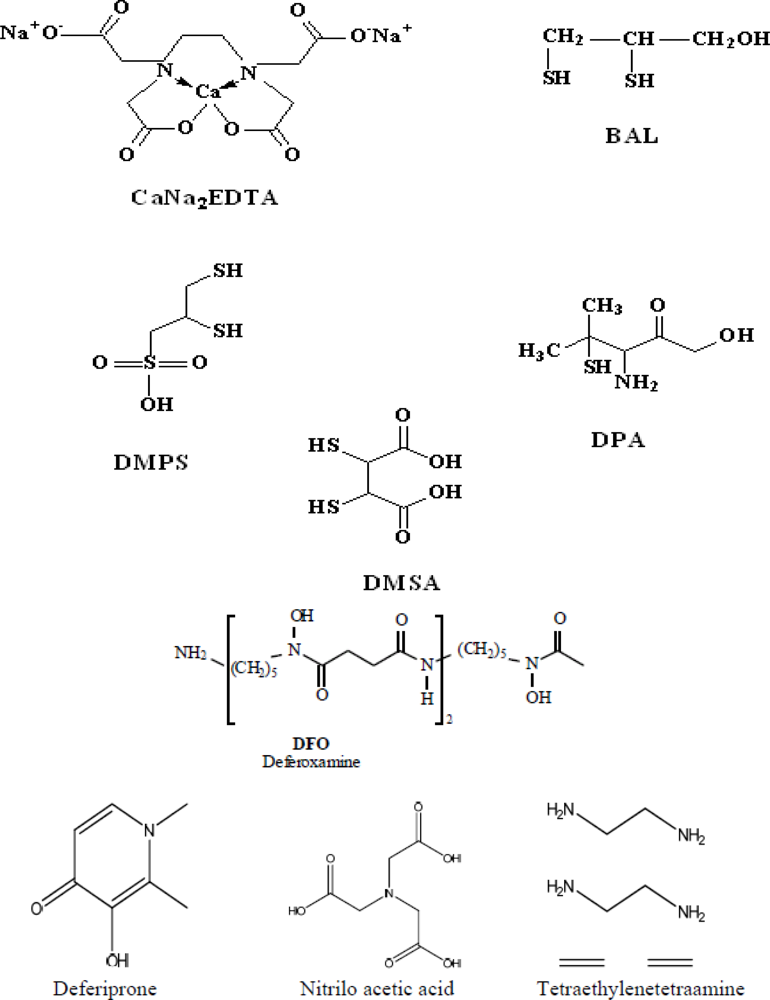
3. Common Chelating Agents: Pharmacology and Toxicology
3.1. Calcium Disodium Ethylenediamine Tetraacetic Acid (CaNa2EDTA)
Pharmacological Profile
3.2. Calcium Trisodium DTPA
Pharmacological Profile
3.3. D-Penicillamine
Pharmacological Profile
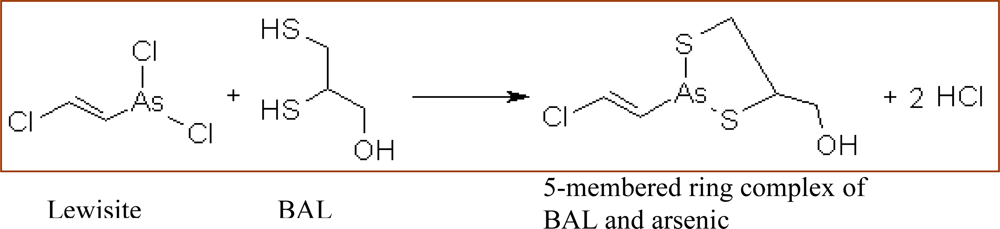
3.4. British Anti Lewisite (BAL)
Pharmacological Profile
- Low therapeutic index (small margin of safety)
- Tendency to redistribute arsenic to brain and testes
- Need for (painful) intramuscular injection
- Unpleasant odor (rotten eggs)
3.5. Meso-2,3-Dimercaptosuccinic Acid (DMSA)
Pharmacological Profile
3.6. Sodium 2,3 Dimercaptopropane-l-Sulphonate (DMPS)
Pharmacological Profile
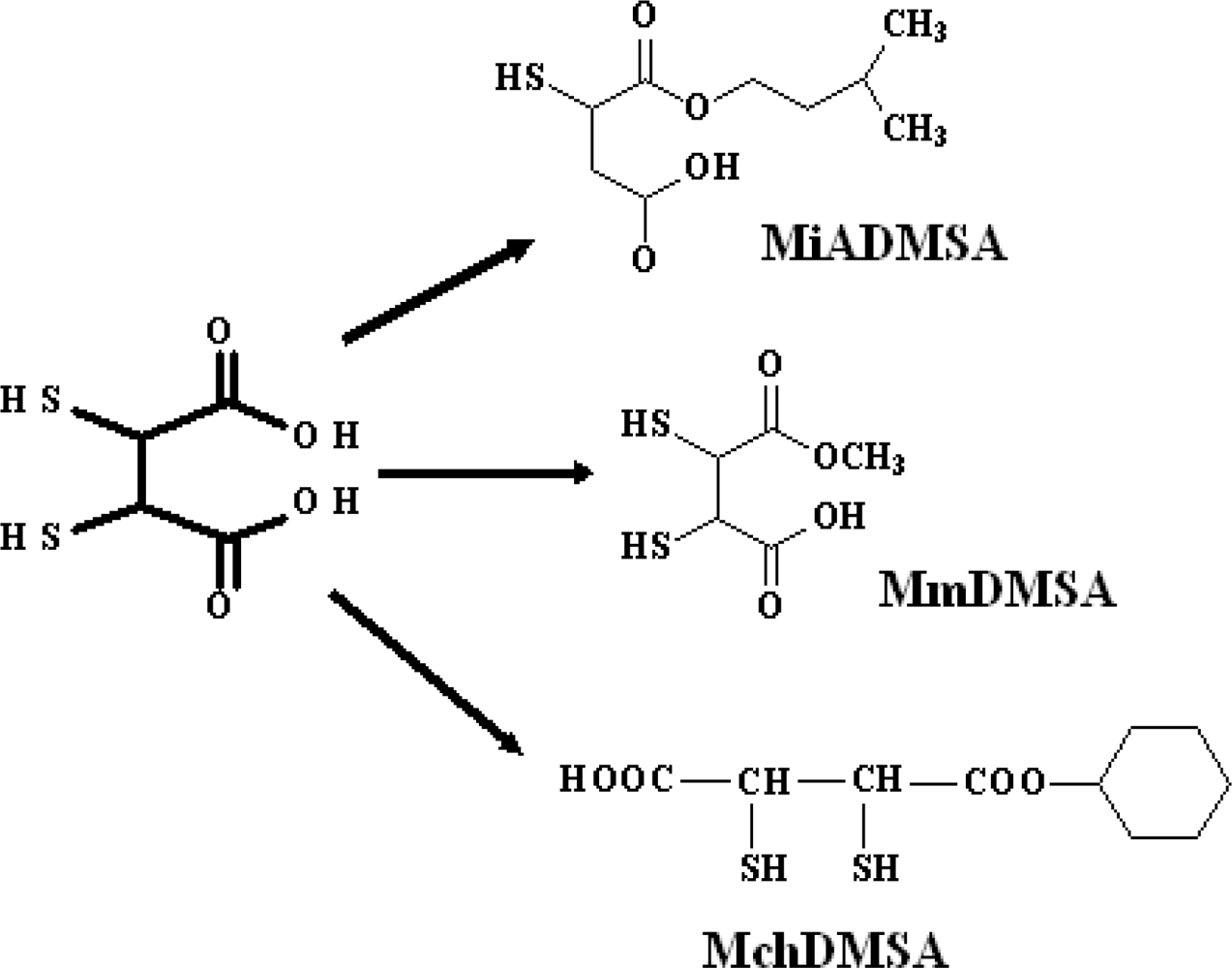
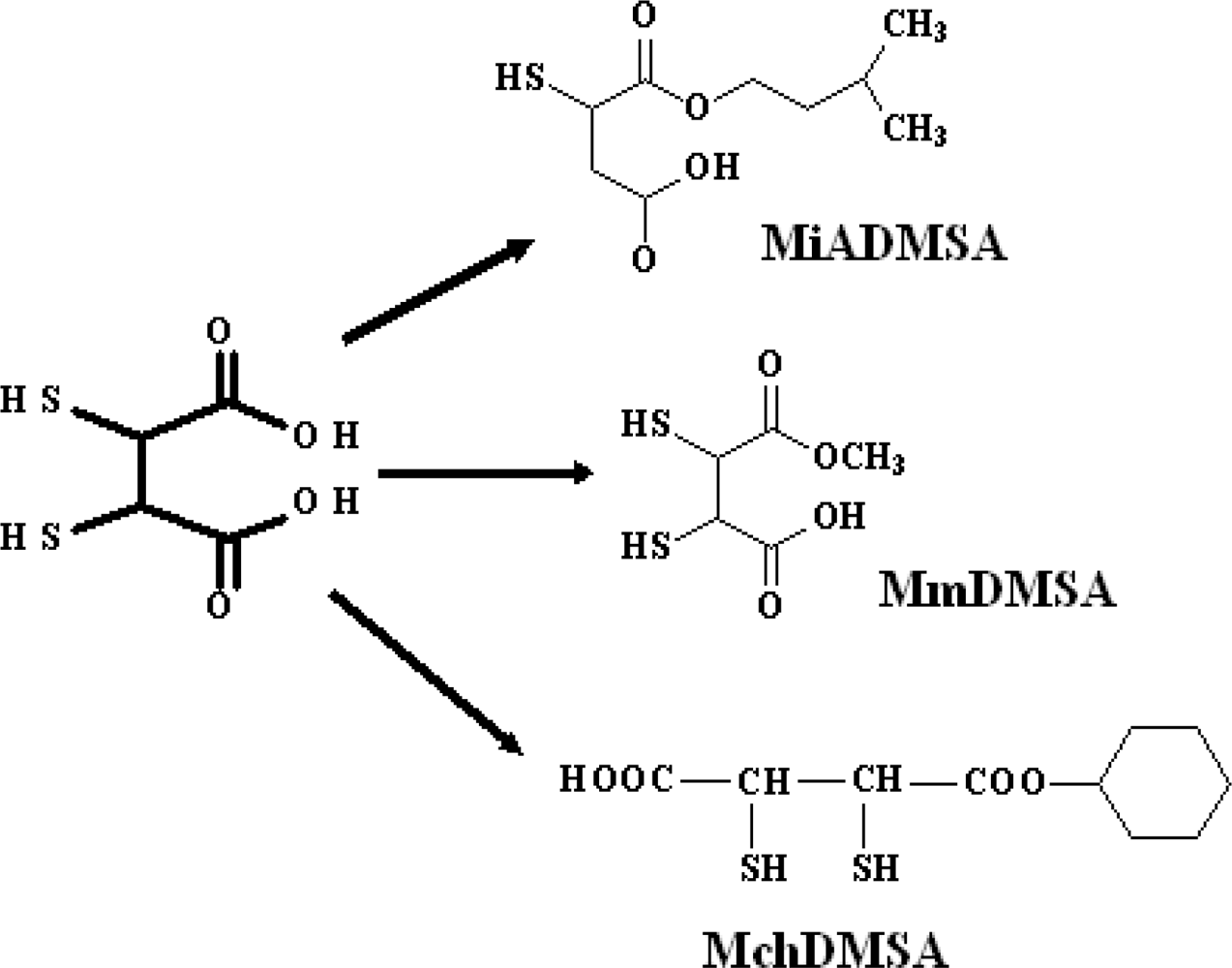
3.7. New DMSA Analogues
3.7.1. Monoisoamyl DMSA (MiADMSA)
Pharmacological Profile
3.7.2. Monomethyl DMSA (MmDMSA) and Monocyclohexyl DMSA (MchDMSA)
3.8. Deferoxamine (DFO)
Pharmacological Profile
3.9. Deferiprone (L1)
Pharmacological Profile
3.10. TETA
Pharmacological Profile
3.11. Nitrilotriacetic Acid (NTA)
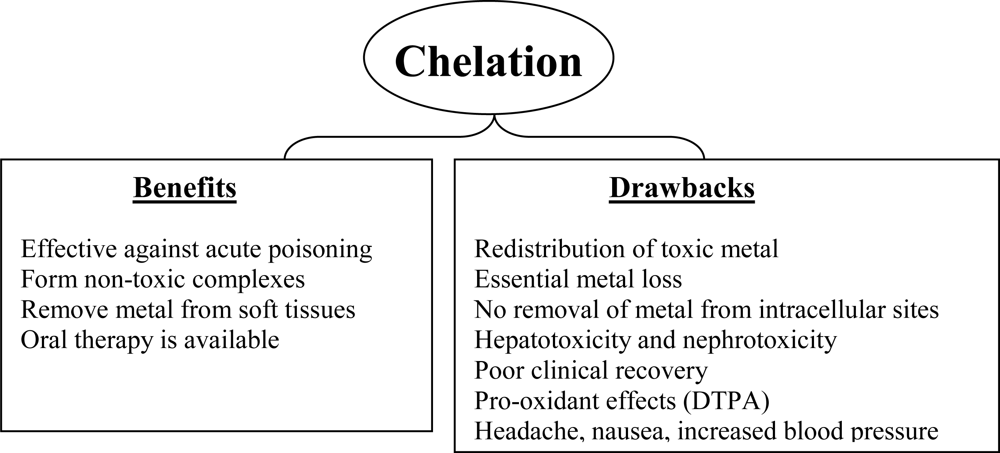
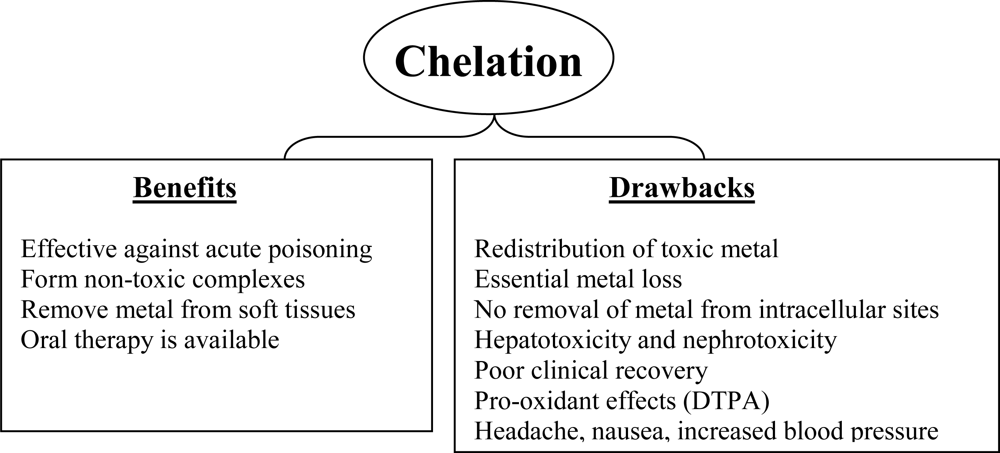
4. Limitations of Current Chelation Therapy
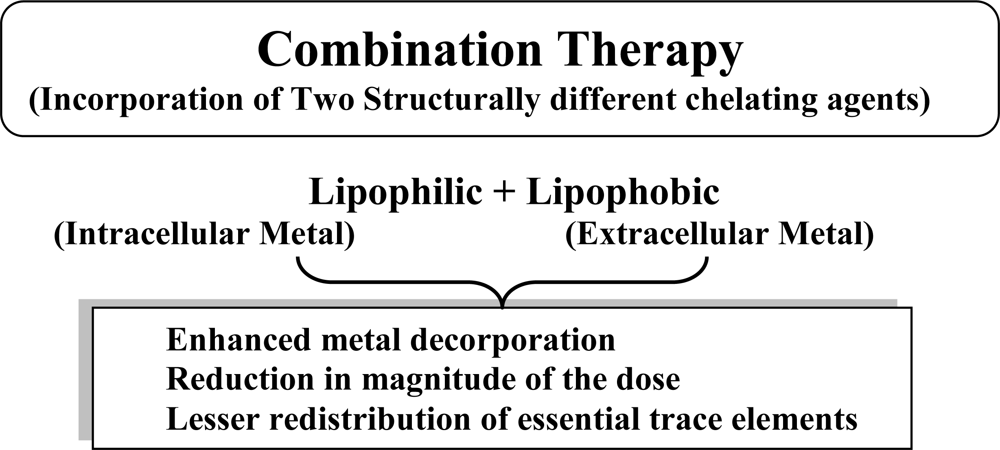
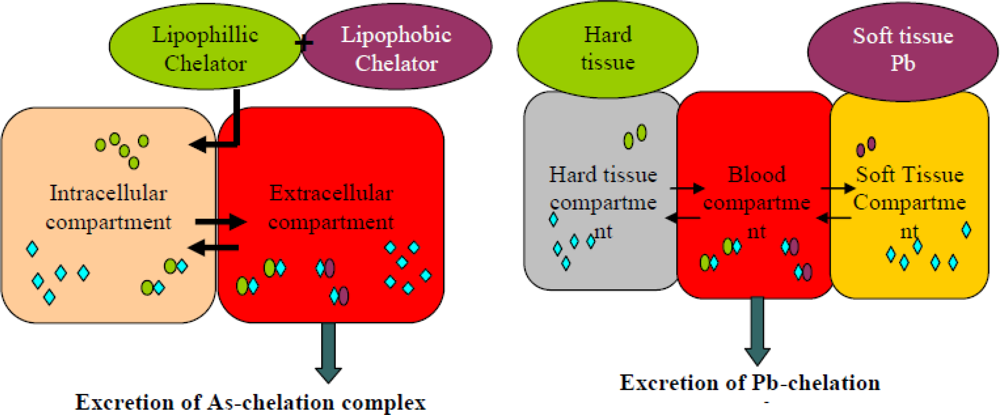
5. Newer Strategies: Combination Therapy
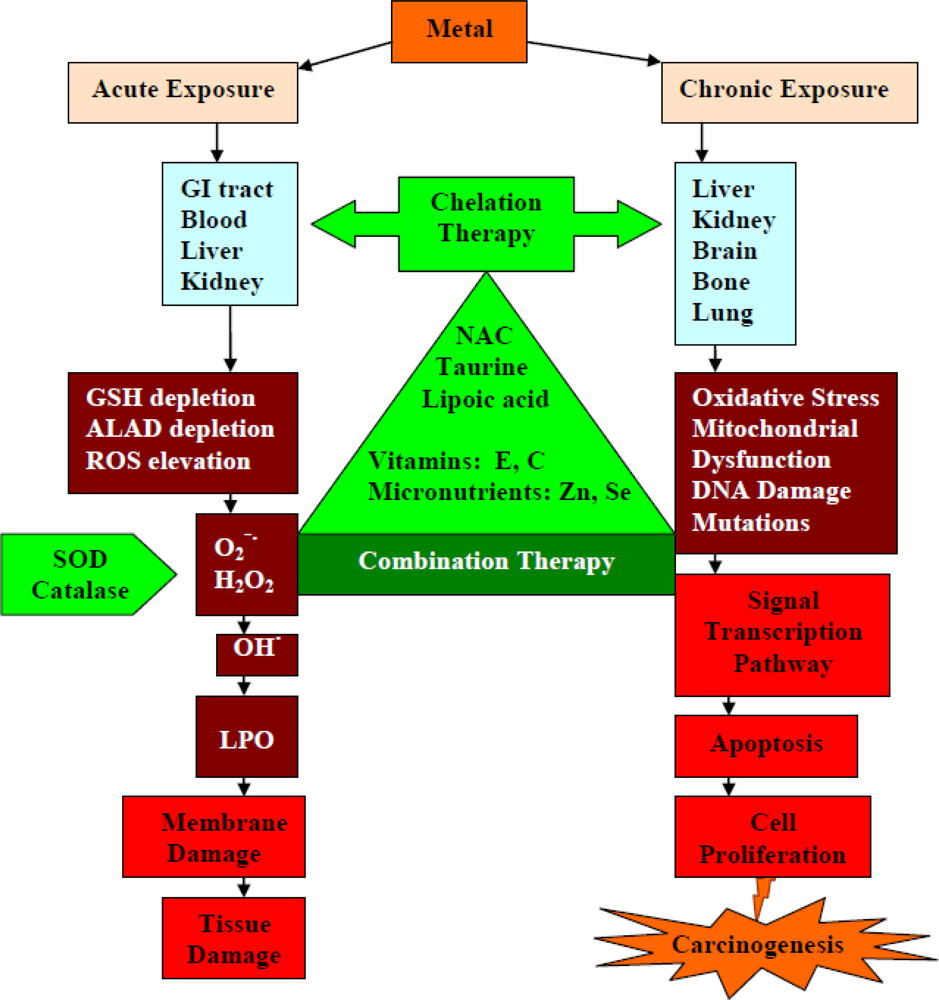
6. Oxidative Stress in Metal Toxicity and the Role of Antioxidants
7. Therapeutic Recommendations for Heavy Metal Poisoning
- Prevention of further metal absorption into the system
- Elimination of metal from the circulation
- Inactivation of metal bioavailable in the system
7.1. Prevention of Further Metal Absorption into the System
7.2. Elimination of Metal from the Circulation
7.3. Inactivation of Metal Bioavailability in the System
7.3.1. Lead
7.3.2. Arsenic
7.3.4. Cadmium
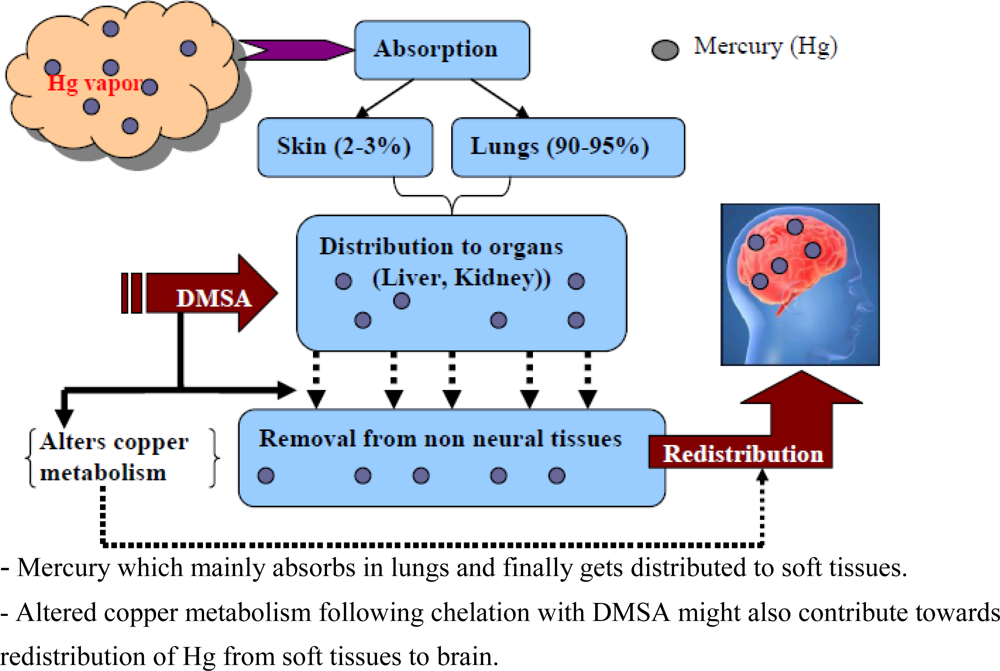
7.3.5. Mercury
7.3.6. Iron
8. Role of Antioxidants during Chelation
9. Conclusions
Acknowledgments
References
- Morgan, T; Gilbert, T; Drew; Harry, DK. CLXII—Researches on residual affinity and co-ordination. Part II Acetyl acetones of selenium and tellurium. J. Chem. Soc 1920, 117, 1456–1465. [Google Scholar]
- Andersen, O. Principles and recent developments in chelation treatment of metal intoxication. Chem. Rev 1999, 99, 2683–2710. [Google Scholar]
- Jones, MM. Design of new chelating agents for removal of intracellular toxic metals. In Coordination Chemistry: A Century of Progress; Kauffman, GB, Ed.; The American Chemical Society: Washington, DC, USA, 1994; pp. 427–438. [Google Scholar]
- Baum, CR. Treatment of mercury intoxication. Curr. Opin. Pediatr 1999, 11, 265–268. [Google Scholar]
- Guldager, B; Jorgensen, PJ; Grandjean, P. Metal excretion and magnesium retention in patients with intermittent claudication treated with intravenous disodium EDTA. Clin. Chem 1996, 42, 1938–1942. [Google Scholar]
- Fournier, L; Thomas, G; Garnier, R; Buisine, A; Houze, P; Pradier, F. 2, 3-Dimercaptosuccinic acid treatment of heavy metal poisoning in humans. Med. Toxicol 1988, 3, 499–504. [Google Scholar]
- Andersen, O. Oral cadmium exposure in mice: toxicokinetics and efficiency of chelating agents. Crit. Rev. Toxicol 1989, 20, 83–112. [Google Scholar]
- Tilbrook, GS; Hider, RC. Iron chelators for clinical use metal ions. Biol. Syst 1998, 35, 691–730. [Google Scholar]
- Singh, S; Khodr, H; Tayler, MI; Hider, RC. Therapeutic iron chelators and their potential side-effects. Biochem. Soc. Symp 1995, 61, 127–137. [Google Scholar]
- Kojima, Y; Binz, PA; Kägi, JH. Metallothionein IV; Klaassen, C, Ed.; Birkhäuser Verlag: Basel, Switzerland, 1999; Volume 66, pp. 3–6. [Google Scholar]
- Cobbett, C; Goldsbrough, P. Phytochelatins and metallothionein: roles in heavy metal detoxification and homeostasis. Annu. Rev. Plant Physiol 2002, 53, 159–182. [Google Scholar]
- Klaassen, CD. Heavy metals and heavy metal antagonists. In The Pharmacological Basis of Therapeutics; Goodman, L, Gilman, A, Eds.; McGraw Hill, Medical Publishing Division: New York, NY, USA, 2006; pp. 1825–1872. [Google Scholar]
- Quan, H; Ghali, WA; Verhoef, MJ; Norris, CN; Galbraith, PD; Knudtson, ML. Use of chelation therapy after coronary angiography. Am. J. Med 2001, 111, 686–691. [Google Scholar]
- Miller, KL; Liebowitz, RS; Newby, LK. Complementary and alternative medicine in cardiovascular diseases: A review of biologically based approaches. Am. Heart J 2004, 147, 401–411. [Google Scholar]
- Ernst, E. Chelation therapy for coronary heart disease: An overview of all clinical investigations. Am. Heart J 2000, 140, 139–141. [Google Scholar]
- Knudtson, ML; Wyse, KG; Galbraith, PD. Chelation therapy for ischemic heart disease. A randomized controlled trail. JAMA 2002, 287, 481–486. [Google Scholar]
- Anderson, TJ; Hubacek, J; Wyse, DG; Knudtson, ML. Effect of chelation therapy on endothelial function in patients with coronary artery disease: PATCH substudy. J. Amer. Coll. Cardiol 2003, 41, 420–425. [Google Scholar]
- Flora, SJS; Bhattacharya, R; Vijayaraghavan, R. Combined therapeutic potential of meso 2,3-dimercaptosuccinic acid and calcium disodium edetate in the mobilization and distribution of lead in experimental lead intoxication in rats. Fund. Appl. Toxicol 1995, 25, 233–240. [Google Scholar]
- Lin-Tan, DT; Lin, JL; Yen, TH; Chen, KH; Huang, YL. Long-term outcome of repeated lead chelation therapy in progressive non-diabetic chronic kidney diseases. Nephrol. Dial. Transplant 2007, 22, 2924–2931. [Google Scholar]
- Lin, JL; Ho, HH; Yu, CC. Chelation therapy for patients with elevated body burden progressive renal insufficiency. A randomized, controlled trail. Ann. Intern. Med 1999, 130, 7–13. [Google Scholar]
- DRUGDEX Drug evaluation, Thomson MICROMEDEX Healthcare Series (Monograph on CD-ROM). 2004; 122.
- Ibim, SE; Trotman, J; Musey, PI; Semafuko, WE. Depletion of essential elements by calcium disodium EDTA treatment in the dog. Toxicology 1992, 73, 229–237. [Google Scholar]
- Flora, SJS; Tandon, SK. Beneficial effects of zinc supplementation during chelation treatment of lead intoxication in rats. Toxicology 1990, 64, 129–139. [Google Scholar]
- Spoor, NL. The use of EDTA and DTPA for accelerating the removal of deposited transuranic elements from human; Harwell: Didcot, UK, 1977. [Google Scholar]
- Llobet, JM; Domingo, JL; Corbella, J. Comparison of the effectiveness of several chelators after single administration on the toxicity, excretion and distribution of cobalt. Arch. Toxicol 1986, 58, 278–281. [Google Scholar]
- Llobet, JM; Domingo, JL; Corbella, J. Antidotes for zinc intoxication in mice. Arch. Toxicol 1988, 61, 321–323. [Google Scholar]
- Gale, GR; Atkins, LM; Walker, EM, Jr; Smith, AB. Comparative effects of diethyldithiocarbamate, dimercaptosuccinate and diethylenetriaminepentaacetate on organ distribution and excretion of cadmium. Ann. Clin. Lab. Sci 1983, 13, 33–44. [Google Scholar]
- Walker, EM, Jr; Gale, GR; Fody, EP; Atkins, LM; Smith, AB; Jones, MM. Comparative antidotal effects of diethyldithicarbamate, dimercaptosuccinate and diethylene triamine pentaacetate against cadmium induced testicular toxicity in mice. Res. Commun. Chem. Pathol. Pharmacol 1986, 51, 231–244. [Google Scholar]
- Radiation emergency assistance center, Training site (REAC/TS). Ca-DTPA (Trisodium calcium diethylenetriaminepentaacetate). Oak Ridge Institute for Science and Education: Oak Ridge, TN, USA, 2002.
- Roussaeux, CG; MacNabb, LG. Oral administration of D-pencillamine causes neonatal mortality without morphological defects in CD-1 mice. J. Appl. Toxicol 1992, 12, 35–38. [Google Scholar]
- Gupta, B; Srivastava, RK; Saxena, KK; Prasad, DN. A study on the penicillamine induced gastric ulceration in the rat. Ind. J. Pharmacol 1980, 12, 247–252. [Google Scholar]
- Grasedyck, K. D-penicillamine—side effects, pathogenesis and decreasing the risks. Z. Rheumatol 1988, 47, 17–19. [Google Scholar]
- Peters, R; Stocken, L; Thompson, R. British Anti-Lewisite (BAL). Nature 1945, 156, 616–619. [Google Scholar]
- Hoover, TD; Aposhian, HV. BAL increases the arsenic-74 content of rabbit brain. Toxicol. Appl. Pharmacol 1983, 70, 160–162. [Google Scholar]
- Berlin, M; Ullberg, S. Increasing uptake of mercury in mouse brain caused by 2,3-dimercaptopropanol (BAL). Nature 1963, 197, 84–85. [Google Scholar]
- Andersen, O. Chemical and biological considerations in the treatment of metal intoxications by chelating agents. Mini Rev. Med. Chem 2004, 4, 11–21. [Google Scholar]
- Janakiraman, N. Hemolysis during BAL chelation therapy for high blood lead levels in two G6PD deficient children. Clin. Pediatr 1978, 17, 485–487. [Google Scholar]
- Aposhian, HV. DMSA and DMPS—Water soluble antidotes for heavy metal poisoning. Ann. Rev. Pharmacol. Toxicol 1983, 23, 193–215. [Google Scholar]
- Alan, L; Miller, ND. Dimercaptosuccinic Acid (DMSA), a non-toxic, water-soluble treatment for heavy metal toxicity. Altern. Med. Rev 1998, 3, 199–207. [Google Scholar]
- Graziano, JH. Role of 2,3-dimercaptosuccinic acid in the treatment of heavy metal poisoning. Med. Tox 1986, 1, 155–162. [Google Scholar]
- Aposhian, HV; Maiorino, RM; Dart, RC; Perry, DF. Urinary excretion of meso-2,3-dimercaptosuccinic acid in human subjects. Clin. Pharmacol. Ther 1989, 45, 520–526. [Google Scholar]
- Aaseth, J; Alexander, J; Raknerud, N. Treatment of mercuric chloride poisoning with dimercaptosuccinic acid and diuretics: preliminary studies. J. Toxicol. Clin. Toxicol 1982, 19, 173–186. [Google Scholar]
- Grandjean, P; Guldager, B; Larsen, IB. Placebo response in environmental disease. Chelation therapy of patients with symptoms attributed to amalgam fillings. J. Occup. Environ. Med 1997, 39, 707–714. [Google Scholar]
- Ramsey, DT; Casteel, SW; Faggella, AM. Use of orally administered succimer (meso-2, 3-dimercaptosuccinic acid) for treatment of lead poisoning in dogs. J. Am. Vet. Med. Assoc 1996, 208, 371–375. [Google Scholar]
- Aposhian, HV; Morgan, DL; Queen, HL; Maiorino, RM; Aposhian, MM. Vitamin C, glutathione, or lipoic acid did not decrease brain or kidney mercury in rats exposed to mercury vapor. J. Toxicol. Clin. Toxicol 2003, 41, 339–347. [Google Scholar]
- Ewan, KB; Pamphlett, R. Increased inorganic mercury in spinal motor neurons following chelating agents. Neurotoxicology 1996, 17, 343–349. [Google Scholar]
- Gersl, V; Hrdina, R; Vavrova, J; Holeckova, M; Palicka, V; Vogkova, J; Mazurova, Y; Bajgar, J. Effects of repeated administration of dithiol chelating agent- sodium 2,3-dimercapto 1-propanesulphonate (DMPS)- on biochemical and hematological parameters in rabbits. Acta. Medica 1997, 40, 3–8. [Google Scholar]
- Kalia, K; Flora, SJS. Strategies for Safe and Effective Treatment for Chronic Arsenic and Lead Poisoning. J. Occup. Hlth 2005, 47, 1–21. [Google Scholar]
- Aposhian, MM; Maiorino, RM; Xu, Z. Sodium 2,3-dimercapto-1-propanesulfonate (DMPS) treatment does not redistribute lead or mercury to the brain of rats. Toxicology 1996, 109, 49–55. [Google Scholar]
- McNeill Consumer Products Co. Chemet Product Information; McNeill Consumer Products Co: Fort Washington, PA, USA, 1994. [Google Scholar]
- Walker, EM; Stone, A; Milligan, LB; Gale, GR; Atkins, LM; Smith, AB; Jones, MM; Singh, PK; Basinger, MA. Mobilization of lead in mice by administration of monoalkyl esters of meso-2,3-dimercaptosuccinic acid. Toxicology 1992, 76, 79–87. [Google Scholar]
- Flora, SJS; Dubey, R; Kannan, GM; Chauhan, RS; Pant, BP; Jaiswal, DK. Meso-2,3-dimercaptosuccinic acid (DMSA) and monoisoamyl DMSA effect on gallium arsenide induced pathological liver injury in rats. Toxicol. Lett 2002, 132, 9–17. [Google Scholar]
- Flora, SJS; Pande, M; Kannan, GM; Mehta, A. Lead induced oxidative stress and its recovery following co-administration of melatonin or n-acetylcysteine during chelation with succimer in male rats. Cell. Mol. Biol 2004, 50, 543–551. [Google Scholar]
- Jones, MM; Singh, PK; Gale, GR; Smith, AB; Atkins, LM. Cadmium mobilization in vivo by intraperitoneal or oral administration of mono alkyl esters of meso-2,3-dimercaptosuccinic acid. Pharmacol. Toxicol 1992, 70, 336–343. [Google Scholar]
- Flora, SJS; Mehta, A. Monoisoamyl dimercaptosuccinic acid abrogates arsenic-induced developmental toxicity in human embryonic stem cell-derived embryoid bodies: comparison with in vivo studies. Biochem. Pharmacol 2009, 78, 1340–1349. [Google Scholar]
- Mehta, A; Flora, SJS. Possible role of metal redistribution, hepatotoxicity and oxidative stress in chelating agents induced hepatic and renal metallothionein in rats. Food Chem. Toxicol 2001, 39, 1029–1038. [Google Scholar]
- Flora, SJS; Mehta, A; Gautam, P; Jatav, PC; Pathak, U. Essential metal status, prooxidant/antioxidant effects of MiADMSA in male rats: age-related effects. Biol Trace Elem Res 2007, 120, 235–247. [Google Scholar]
- Mehta, A; Kannan, GM; Dube, SN; Pant, BP; Pant, SC; Flora, SJS. Hematological, hepatic and renal alterations after repeated oral or intraperitoneal administration of monoisoamyl DMSA I. Changes in male rats. J. Appl. Toxicol 2002, 22, 359–369. [Google Scholar]
- Flora, SJS; Mehta, A. Haematological, hepatic and renal alterations after repeated oral and intraperitoneal administration of monoisoamyl DMSA. II. Changes in female rats. J. Appl. Toxicol 2003, 23, 97–102. [Google Scholar]
- Blanusa, M; Prester, L; Piasek, M; Kostial, K; Jones, MM; Singh, PK. Monoisoamyl ester of DMSA reduces 203Hg(NO3)2 retention in rats: 1. Chelation therapy during pregnancy. J. Trace Elem. Exp. Med 1997, 10, 173–181. [Google Scholar]
- Mehta, A; Pant, SC; Flora, SJS. Monoisoamyl dimercaptosuccinic acid induced changes in pregnant female rats during late gestation and lactation. Reproduct. Toxicol 2006, 21, 94–103. [Google Scholar]
- Taubeneck, MW; Domingo, JL; Llobet, JM; Keen, CL. Meso 2, 3-dimercaptosuccinic acid (DMSA) affects maternal and fetal copper metabolism in Swiss mice. Toxicology 1992, 72, 27–40. [Google Scholar]
- Winship, KA. Toxicity of aluminium: a historical review, Part 2. Adverse Drug React Toxicol. Rev 1993, 12, 177–211. [Google Scholar]
- Hoffbrand, AV; Cohen, A; Hershko, C. Role of deferiprone in chelation therapy for transfusional iron overload. Blood 2003, 102, 17–24. [Google Scholar]
- Kattamis, A; Ladis, V; Berdousi, H; Kelekis, NL; Alexopoulou, E; Papasotiriou, I; Drakaki, K; Kaloumenou, I; Galani, A; Kattamis, C. Iron chelation treatment with combination therapy with deferiprone and deferioxamine: A 12-month trial. Blood Cell. Mol. Dis 2006, 36, 21–25. [Google Scholar]
- Cappellini, MD; Pattoneri, P. Oral iron chelators. Annu. Rev. Med 2009, 60, 25–38. [Google Scholar]
- Kodama, H; Meguro, Y; Tsunakawa, A; Nakazato, Y; Abe, T; Murakita, H. Fate of orally administered triethylenetetramine dihydrochloride: a therapeutic drug for Wilson’s disease. Tohoku J. Exp. Med 1993, 169, 59–66. [Google Scholar]
- Kodama, H; Murata, Y; Iitsuka, T; Abe, T. Metabolism of administered triethylene tetramine dihydrochloride in humans. Life Sci 1997, 61, 899–907. [Google Scholar]
- Bahnemann, R; Leibold, E; Kittel, B; Mellert, W; Jackh, R. Different patterns of kidney toxicity after sub-acute administration of Na-nitrilotriacetic acid and Fe-nitrilotriacetic acid to Wistar rats. Toxicol. Sci 1998, 46, 166–175. [Google Scholar]
- Tandon, SK; Mathur, AK. Chelation in Metal Intoxication. III. Lowering of Nickel Content in Poisoned Rat Organs. Acta Pharmacol. Toxicol 1976, 38, 401–408. [Google Scholar]
- Kaur, G; Hasan, SK; Srivastava, RC. Effect of nitrilotriacetic acid (NTA) on the distribution of manganese-54 in rats. Arch. Toxicol 1980, 45, 203–206. [Google Scholar]
- Anderson, RL. The role of zinc in nitrilotriacetate (NTA)-associated renal tubular cell toxicity. Fd Cosmet. Toxicol 1981, 19, 639–650. [Google Scholar]
- Nixon, GS; Buehler, EV; Niewhuis, RJ. Two year rat feeding study with trisodium nitrilotriacetate and its calcium chelate. Toxicol. Appl. Pharmacol 1972, 21, 244–249. [Google Scholar]
- Alden, CL; Kanerva, RL; Anderson, RL; Adkins, AG. Short-term effects of dietary nitrilotriacetic acid in the male Charles River rat kidney. Vet. Pathol 1981, 18, 549–559. [Google Scholar]
- Hiasa, Y; Kitahori, Y; Konishi, N; Shimoyama, T. Dose-related effect of trisodium nitrilotnacetate monohydrate on renal tumorigenesis initiated with N-ethyl-N-hydroxyethylnitrosamine in rats. Carcinogenesis 1985, 6, 907–910. [Google Scholar]
- Miyashiro, A. Promoting effect of trisodium nitrilotriacetate monohydrate on the development of tumors in kidneys and urinary bladders or rats treated with N-ethyl-N-hydroxyethylnitrosamine or N-butyl-N-(hydroxybutyl) nitrosamine. J. Nara Med Assoc 1984, 35, 550–565. [Google Scholar]
- Dietrich, DR; Swenberg, JA. Preneoplastic lesions in rodent kidney induced spontaneously or by non-genotoxic agents: Predictive nature and comparison to lesion induced by genotoxic carcinogens. Mutat. Res 1991, 248, 239–260. [Google Scholar]
- Hartwig, A; Klyszcz-Nasko, H; Schlepegrell, R; Beyersmann, D. Cellular damage by ferric nitrilitriacetate and ferric citrate in V79 cells: Interrelationship between lipid peroxidation, DNA strand breaks and sister chromatid exchange. Carcinogenesis 1993, 14, 107–112. [Google Scholar]
- Umemura, T; Hasegawa, R; Sai-Kato, K; Nishikawa, A; Furukawa, F; Toyokum, S; Uchida, K; Inouc, T; Kurokawa, Y. Prevention by 2-mercaptoethane sulfonate and N-acetylcysteine of renal oxidative damage in rats treated with ferric nitriltriacetate. Jpn. J. Cancer Res 1996, 87, 882–886. [Google Scholar]
- Angle, CR. Chelation therapies for metal intoxication. In Toxicology of Metals; Chang, LW, Ed.; CRC Press: Boca Raton, FL, USA, 1996; pp. 487–504. [Google Scholar]
- Guha Mazumder, DN; Ghoshal, UC; Saha, J; Santra, A; De, BK; Chatterjee, A; Dutta, S; Angle, CR; Centeno, JA. Randomized placebo-controlled trial of 2,3-dimercapto succinic acid in therapy of chronic arsenicosis due to drinking arsenic contaminated subsoil water. Clin. Toxicol 1998, 36, 683–690. [Google Scholar]
- Kostial, K; Blanusa, M; Plasek, LJ; Samarzila, M; Jones, MM; Singh, PK. Monoisoamyl- and mono-n-hexyl-meso-2,3-dimercaptosuccinate in mobilizing Hg203 retention in relation to age of rats and route of administration. J. Appl. Toxicol 2001, 15, 201–206. [Google Scholar]
- Flora, SJS; Saxena, G; Mehta, A. Reversal of lead-induced neuronal apoptosis by chelation treatment in rats: role of ROS and intracellular Ca2+. J. Pharmacol. Exp. Ther 2007, 322, 108–116. [Google Scholar]
- Cory-Slechta, DA. Mobilisation of lead over the course of DMSA chelation therapy and long term efficacy. J. Pharmacol. Exp. Therap 1988, 246, 84–91. [Google Scholar]
- Flora, SJS; Saxena, G. Lead induced oxidative stress and hematological alterations and their response to combined administration of calcium disodium EDTA with a thiol chelator in rats. J. Biochem. Mol. Toxicol 2004, 18, 221–233. [Google Scholar]
- Mishra, D; Mehta, A; Flora, SJS. Reversal of hepatic apoptosis with combined administration of DMSA and its analogues in guinea pigs: role of glutathione and linked enzymes. Chem. Res. Toxicol 2008, 21, 400–407. [Google Scholar]
- Bhadauria, S; Flora, SJS. Response of arsenic induced oxidative stress, DNA damage and metal imbalance to combined administration of DMSA and monoisoamyl DMSA during chronic arsenic poisoning in rats. Cell. Biol. Toxicol 2007, 23, 91–104. [Google Scholar]
- Flora, SJS; Mehta, A; Rao, PVL; Kannan, GM; Bhaskar, ASB; Dube, SN; Pant, BP. Therapeutic potential of monoisoamyl and monomethyl esters of meso 2, 3-dimercaptosuccinic acid in gallium arsenide intoxicated rats. Toxicology 2004, 195, 127–146. [Google Scholar]
- Flora, SJS; Saxena, G; Gautam, P; Kaur, P; Gill, KD. Lead induced oxidative stress and alterations in biogenic amines in different rat brain regions and their response to combined administration of DMSA and MiADMSA. Chem. Biol. Interac 2007, 170, 209–220. [Google Scholar]
- Flora, SJS; Bhadauria, S; Dhaked, R; Pant, SC. Arsenic induced blood and brain oxidative stress and its response to some thiol chelators in male rats. Life Sci 2005, 77, 2324–2337. [Google Scholar]
- Shi, H; Shi, X; Liu, KJ. Oxidative mechanism of arsenic toxicity and carcinogenesis. Mol. Cell. Biochem 2004, 255, 67–78. [Google Scholar]
- Kumagai, Y; Sumi, D. Arsenic: Signal transduction, transcription factor, and biotransformation involved in cellular response and toxicity. Ann. Rev. Pharmacol. Toxicol 2007, 47, 243–262. [Google Scholar]
- Hu, Y; Jin, X; Snow, ET. Effect of arsenic on transcription factor AP-1 and NF-kappaB DNA binding activity and related gene expression. Toxicol. Lett 2002, 133, 33–45. [Google Scholar]
- Pi, J; Horiguchi, S; Sun, Y; Nikaido, M; Shimojo, N; Hayashi, T. A potential mechanism for the impairment of nitric oxide formation caused by prolonged oral exposure to arsenate in rabbits. Free Rad. Biol. Med 2003, 35, 102–113. [Google Scholar]
- Rin, K; Kawaguchi, K; Yamanaka, K; Tezuka, M; Oku, N; Okada, S. DNA-strand breaks induced by dimethylarsinic acid, a metabolite of inorganic arsenics, are strongly enhanced by superoxide anion radicals. Biol. Pharm. Bull 1995, 18, 45–58. [Google Scholar]
- Magos, L. Epidemiological and experimental aspects of metal carcinogenesis: physicochemical properties, kinetics, and the active species. Environ. Health Perspect 1991, 95, 157–189. [Google Scholar]
- Angeline, SA; Jefferey, LB; Maria, MM; Eugene, D; Mary, GW; Joshua, WH; Margaret, RK. Arsenic exposure is associated with decreased DNA repair in vitro and in individuals exposed to drinking water arsenic. Environ. Health Perspect 2006, 114, 1193–1198. [Google Scholar]
- Wen-Chien, C; Chunfa, J; Andrew, AK; Richard, JJ; Michael, AT; Chi, VD. Role of NADPH oxidase in arsenic-induced reactive oxygen species formation and cytotoxicity in myeloid leukemia cells. Proc. Natl. Acad. Sci 2004, 101, 4578–4583. [Google Scholar]
- Schiller, CM; Fowler, BA; Woods, JS. Effects of arsenic on pyruvate dehydrogenase activation. Environ. Health Perspect 1977, 19, 205–207. [Google Scholar]
- Nadia, E; Garcia-Medina, ME; Jimenez, C; Marc, C; Luz, MM; Juan, MD; Charles, CH. Conditioned flavor aversion and brain Fos expression following exposure to arsenic. Toxicology 2007, 235, 73–82. [Google Scholar]
- Galan, C; Garcia, BL; Troyano, A; Vilaboa, NE; Fernandez, C; Blas, DE; Aller, P. The role of intracellular oxidation in death induction (apoptosis and necrosis) in human promonocytic cells treated with stress inducers (cadmium, heat, X-rays). Eur. J. Cell. Biol 2001, 80, 312–320. [Google Scholar]
- Watanabe, M; Henmi, K; Ogawa, K; Suzuki, T. Cadmium-dependent generation of reactive oxygen species and mitochondrial DNA breaks in photosynthetic and non-photosynthetic strains of Euglena gracilis. Comp. Biochem. Physiol. C Toxicol. Pharmacol 2003, 134, 227–234. [Google Scholar]
- Casalino, E; Calzaretti, G; Sblano, C; Landriscina, C. Molecular inhibitory mechanisms of antioxidant enzymes in rat liver and kidney by cadmium. Toxicology 2002, 30, 37–50. [Google Scholar]
- Waisberg, M; Joseph, P; Hale, B; Beyersmann, D. Molecular and cellular mechanisms of cadmium carcinogenesis. Toxicology 2003, 192, 95–117. [Google Scholar]
- Watjen, W; Beyersmann, D. Cadmium-induced apoptosis in C6 glioma cells: influence of oxidative stress. Biometals 2004, 17, 65–78. [Google Scholar]
- Ognjanovic, BI; Pavlovic, SZ; Maletic, SD; Zikic, RV; Stajn, AS; Radojicic, RM; Saicic, ZS; Petrovic, VM. Protective influence of vitamin E on antioxidant defense system in the blood of rats treated with cadmium. Physiol. Res 2003, 52, 563–570. [Google Scholar]
- Quig, D. Cysteine metabolism and metal toxicity. Alter. Med. Rev 1998, 3, 262–270. [Google Scholar]
- Flora, SJS. Nutritional components modify metal absorption, toxic response and chelation therapy. J. Nutri. Environ. Med 2002, 12, 51–65. [Google Scholar]
- Pande, M; Flora, SJS. Lead induced oxidative damage and its response to combined administration of α-Lipoic acid and succimers in rats. Toxicology 2002, 177, 187–196. [Google Scholar]
- Gautam, P; Flora, SJS. Oral supplementation of gossypin during lead exposure protects alteration in heme synthesis pathway and brain oxidative stress in rats. Nutrition 2010, 26, 563–570. [Google Scholar]
- Pachauri, V; Saxena, G; Mehta, A; Mishra, D; Flora, SJS. Combinational chelation therapy abrogates lead-induced neurodegeneration in rats. Toxicol. Appl. Pharmacol 2009, 240, 255–264. [Google Scholar]
- Flora, SJS; Pande, M; Bhadauria, S; Kannan, GM. Combined administration of taurine and meso 2, 3-dimercaptosuccinic acid in the treatment of chronic lead intoxication in rats. Hum. Exp. Toxicol 2004, 23, 157–166. [Google Scholar]
- Flora, SJS; Chouhan, S; Kannan, GM; Mittal, M; Swarnakar, H. Combined administration of taurine and monoisoamyl DMSA protects arsenic induced oxidative injury in rats. Oxidat. Med. Cell. Long 2008, 1, 39–45. [Google Scholar]
- Banner, W; Koch, M; Capin, DM; Hopf, SB; Chang, S; Tong, TG. Experimental chelation therapy in chromium, lead, and boron intoxication with N-acetylcysteine and other compounds. Toxicol. Appl. Pharmacol 1986, 83, 142–147. [Google Scholar]
- Flora, SJS. Arsenic induced oxidative stress and its reversibility following combined administration of N-acetylcysteine and meso-2,3-dimercaptosuccinic acid in rats. Clin. Exp. Pharmacol. Physiol 1999, 26, 865–869. [Google Scholar]
- Pande, M; Mehta, A; Pant, BP; Flora, SJS. Combined administration of a chelating agent and an antioxidant in the prevention and treatment of acute lead intoxication in rats. Environ. Toxicol. Pharmacol 2001, 9, 173–184. [Google Scholar]
- Mittal, M; Flora, SJS. Effects of individual and combined exposure to sodium arsenite and sodium fluoride on tissue oxidative stress, arsenic and fluoride levels in male mice. Chem. Biol. Interact 2006, 162, 128–139. [Google Scholar]
- Bhatt, K; Flora, SJS. Oral co-administration of α- lipoic acid, quercetin and captopril prevents gallium arsenide toxicity in rats. Environ. Toxicol. Pharmacol 2009, 240, 236–244. [Google Scholar]
- Saxena, G; Flora, SJS. Changes in brain biogenic amines and heme- biosynthesis and their response to combined administration of succimers and Centella asiatica in lead poisoned rats. J. Pharm. Pharmacol 2006, 58, 547–559. [Google Scholar]
- Flora, SJS; Singh, S; Tandon, SK. Chelation in metal intoxication XVIII: Combined effects of thiamine and calcium disodium versenate on lead toxicity. Life Sci 1986, 38, 67–71. [Google Scholar]
- Flora, SJS. Influence of simultaneous supplementation of zinc and copper during chelation of lead in rats. Hum. Exp. Toxicol 1991, 10, 331–336. [Google Scholar]
- Flora, SJS; Bhattacharya, R; Sachan, SRS. Dose dependent effects of zinc supplementation during chelation treatment of lead intoxication in rats. Pharmacol. Toxicol 1994, 74, 330–333. [Google Scholar]
- Flora, SJS; Tandon, SK. Beneficial effects of zinc supplementation during chelation treatment of lead intoxication in rats. Toxicology 1990, 64, 129–139. [Google Scholar]
- Modi, M; Pathak, U; Kalia, K; Flora, SJS. Arsenic antagonism studies with monoisoamyl DMSA and zinc in male mice. Environ. Toxicol. Pharmacol 2005, 19, 131–138. [Google Scholar]
- Paul, PC; Misbahuddin, M; Ahmed, ANN; Dewan, ZF; Mannan, MA. Accumulation of arsenic in tissues of iron-deficient rats. Toxicol. Lett 2000, 135, 193–197. [Google Scholar]
- Flora, SJS; Singh, S; Tandon, SK. Thiamine and zinc in prevention or therapy of lead intoxication. J. Inter. Med. Res 1989, 17, 68–75. [Google Scholar]
- Cerklewski, FL. Post-absorptive effect at increased dietary zinc on toxicity and removal of tissue lead in rats. J. Nutr 1984, 114, 550–554. [Google Scholar]
- Kreppel, H; Liu, J; Liu, Y; Reichl, FX; Klaassen, CD. Zinc-induced arsenite tolerance in mice. Fund. Appl. Toxicol 1994, 23, 32–37. [Google Scholar]
- Flora, SJS; Gubrelay, U; Kannan, GM; Mathur, R. Effect of zinc supplementation during chelating agent administration in cadmium intoxication in rats. J. Appl. Toxicol 1998, 18, 357–362. [Google Scholar]
- Jones, MM; Singh, PK; Gale, GR; Atkins, LM; Smith, AB. Esters of meso dimercaptosuccinic acid as cadmium mobilizing agent. Toxicol. Appl. Pharmacol 1988, 95, 507–514. [Google Scholar]
- Schroeter, H; Boyd, C; Spencer, JPE; Williams, RJ; Cadenas, E; Rice-Evans, C. MAPK signaling in neurodegeneration: influences of flavonoids and of nitric oxide. Neurobiol. Aging 2002, 23, 861–880. [Google Scholar]
- Mishra, D; Flora, SJS. Quercetin administration during chelation therapy protects arsenic induced oxidative stress in mouse. Biol. Trace Elem. Res 2008, 122, 137–147. [Google Scholar]
- Tandon, SK; Singh, S; Prasad, S; Khandekar, K; Dwivedi, VK; Chatterjee, M; Mathur, N. Reversal of cadmium induced oxidative stress by chelating agent, antioxidant or their combination in rat. Toxicol. Lett 2003, 145, 211–217. [Google Scholar]
- Shaikh, ZA; Zaman, K; Tang, W; Thanhtam, ZV. Treatment of chronic cadmium nephrotoxicity by N-acetyl cysteine. Toxicol. Lett 1999, 104, 137–142. [Google Scholar]
- Kadima, W; Rabenstein, DL. Nuclear magnetic resonance studies of the solution chemistry of metal complexes. Mixed ligand complexes of cadmium, nitrilotriacetic acid, glutathione, and related ligands. J. Inorg. Biochem 1990, 38, 277–288. [Google Scholar]
- Nagasaki, H; Nakano, H; Boudjeina, K; Jaeck, D; Alexandre, E; Baek, Y; Kitamura, N; Yamaguchi, M; Kumada, K. Efficacy of preconditioning with N-acetylcysteine against reperfusion injury after prolonged cold ischemia in rats liver in which glutathione had been reduced by buthionine sulphoximine. Eur. J. Surg 1998, 164, 139–146. [Google Scholar]
- Kazantzis, G. Diagnosis and treatment of metal poisoning-General aspect. In Handbook on the Toxicology of Metals; Friberg, G, Nordberg, GF, Vouk, VB, Eds.; Elsevier: Amsterdam, The Netherland, 1990; pp. 302–317. [Google Scholar]
- Finkelstein, Y; Markowitz, M; Rosen, J. Low level lead induced neurotoxicity in children: an update on central nervous system effects. Br. Res. Rev 1998, 27, 168–176. [Google Scholar]
- Preventing Lead Poisoning in Young Children: a Statement by the Centers for Disease Control Atlanta, GA; Centers for Disease Control and Prevention: Atlanta, GA, USA, 1991.
- Jaffe, EK. Porphobilnogen synthase, the first source of heme asymmetry. J. Bioenerg. Biomembra 1995, 27, 169–179. [Google Scholar]
- Wetmur, JG. Influence of the common human δ-aminolevulinate dehydratase polymorphism on lead body burden. Environ. Hlth. Perspect 1994, 102, 215–219. [Google Scholar]
- Gurer, H; Ozgunes, H; Neal, R; Spitz, DR; Ercal, N. Antioxidant effects of N-acetyl cysteine and succimer in red blood cells from lead exposed rats. Toxicology 1998, 128, 181–189. [Google Scholar]
- Sandhir, R; Gill, KD. Effect of lead on lipid peroxidation in liver of rats. Biol. Trace Elem. Res 1995, 48, 91–97. [Google Scholar]
- Flora, SJS. Lead in the environment: prevention and treatment. J. Environ. Biol 2002, 23, 29–44. [Google Scholar]
- Stohs, ST; Bagchi, D. Oxidative mechanism in the toxicity of metal ions. Free Rad. Biol. Med 1995, 18, 321–336. [Google Scholar]
- Ercal, N; Gurer-Orhan, H; Aykin-Burns, N. Toxic metals and oxidative stress part I: mechanisms involved in metal-induced oxidative damage. Curr. Top. Med. Chem 2001, 1, 529–539. [Google Scholar]
- Flora, SJS; Pant, BP; Tripathi, N; Kannan, GM; Jaiswal, DK. Distribution of arsenic by diesters of meso 2, 3-dimercaptosuccinic acid during sub-chronic intoxication in rats. J. Occup. Health 1997, 39, 119–123. [Google Scholar]
- Ercal, N; Treratphan, P; Hammond, TC; Mathews, RH; Grannemann, NH; Spitz, DR. In vivo indices of oxidative stress in lead exposed C57BL/6 mice are reduced by treatment with meso-2,3-dimercaptosuccinic acid or N-acetyl cysteine. Free Rad. Biol. Med 1996, 21, 157–161. [Google Scholar]
- Zhang, J; Wang, XF; Lu, ZB; Liu, NO; Zhao, BL. The effects of meso-2,3-dimercaptosuccinic acid and oligomeric procyanidins on acute lead neurotoxicity in rat hippocampus. Free Rad. Biol. Med 2004, 37, 1037–1050. [Google Scholar]
- ATSDR Toxicological Profile for Arsenic; Agency for Toxic Substances and Disease Registry, ATSDR/PB/2000/108021; US Public Health Service: Atlanta, GA, USA, 2000.
- Duxbury, JM; Mayer, AB; Lauren, JG; Hassan, N. Food chain aspects of arsenic contamination in Bangladesh: effects on quality and productivity of rice. J. Environ. Sci. Hlth. A Tox. Hazard Subst. Environ. Eng 2003, 38, 61–69. [Google Scholar]
- Smedley, PL; Kinniburgh, DG. A review of the source, behavior and distribution of arsenic in natural waters. Appl. Geochem 2001, 17, 517–568. [Google Scholar]
- UN Synthesis Report, Arsenic in Drinking Water; United Nations: Geneva, Switzerland, 2001.
- Smith, AH; Arroyo, AP; Mazumdar, DN. Arsenic-induced skin lesions among Atacameno people in northern Chile despite good nutrition and centuries of exposure. Environ. Hea. Perspect 2000, 108, 617–620. [Google Scholar]
- Tchounwou, PB; Patlolla, AK; Centeno, JA. Carcinogenic and systemic health effects associated with arsenic exposure-a critical review. Toxicol. Pathol 2003, 31, 575–588. [Google Scholar]
- Chowdhury, UK; Biswas, BK; Chowdhury, TR; Samanta, G; Mandal, BK; Basu, GC; Chanda, CR; Lodh, D; Saha, KC; Mukherjee, SK; Roy, S; Kabir, S; Quamruzzaman, Q; Chakraborti, D. Groundwater arsenic contamination in Bangladesh and West Bengal, India. Environ. Hlth. Perspect 2000, 108, 393–397. [Google Scholar]
- Bode, AM; Dong, Z. The paradox of arsenic: molecular mechanisms of cell transformation and chemotherapeutic effects. Crit. Rev. Oncol. Hematol 2002, 42, 5–24. [Google Scholar]
- Wang, YP; Zhu, HG; Zhang, ZY. Preliminary study on arsenic trioxide induced Tca8113 cell apoptosis. Shanghai Kou Qiang Yi Xue 2002, 11, 343–345. [Google Scholar]
- Gubrelay, U; Mathur, R; Flora, SJS. Treatment of arsenic poisoning: an update. Ind. J. Pharmacol 1998, 30, 209–217. [Google Scholar]
- Guha Mazumder, DN; Ghoshal, UC; Saha, J; Santra, A; De, BK; Chatterjee, A; Dutta, S; Angle, CR; Centeno, JA. Randomized placebo-controlled trial of 2,3-dimercaptosuccinic acid in therapy of chronic arsenicosis due to drinking arsenic-contaminated subsoil water. J. Toxicol. Clin. Toxicol 1998, 36, 683–690. [Google Scholar]
- Suwazono, Y; Kido, T; Nakagawa, H; Nishijo, M; Honda, R; Kobayashi, E; Dochi, M; Nogawa, K. Biological half-life of cadmium in the urine of inhabitants after cessation of cadmium exposure. Biomarkers 2009, 14, 77–81. [Google Scholar]
- Mandel, JS; McLaughlin, JK; Schlehofer, B; Mellemgaard, A; Helmuert, U; Linbald, R; McCredie, M; Adami, U. International renal-cell cancer study. IV. Occupation. Int. J. Cancer 1995, 61, 601–605. [Google Scholar]
- Waalkes, MP; Misra, RR. Cadmium carcinogenicity and genotoxicity. In Toxicology of Metals; Chang, L, Ed.; CRC Press: Boca Raton, FL, USA, 1996; pp. 231–244. [Google Scholar]
- IARC, International Agency for Research on Cancer, Beryllium, cadmium, mercury, and exposures in the glass manufacturing industry. In International Agency for Research on Cancer Monographs on the Evaluation of Carcinogenic Risks to Humans; IARC Scientific Publications: Lyon, France, 1993; Volume 58, pp. 119–237.
- Goyer, RA; Cherian, MG. Renal effects of metals. In Metal Toxicology; Goyer, RA, Klaassen, CD, Waalkes, MP, Eds.; Academic Press: San Diego, CA, USA, 1995; pp. 389–412. [Google Scholar]
- Sheweita, SA. Heavy metal-induced changes in the glutathione levels and glutathione reductase/glutathione S-transferase in the liver of male mice. Int. J. Toxicol 1998, 17, 383–392. [Google Scholar]
- Kelley, C; Sargent, DE; Uno, JK. Cadmium therapeutic agents. Curr. Pharm. Des 1999, 5, 229–240. [Google Scholar]
- Liu, F; Jan, KY. DNA damage in arsenite- and cadmium-treated bovine aortic endothelial cells. Free Radic. Biol. Med 2000, 28, 55–63. [Google Scholar]
- Bagchi, D; Vuchetich, PJ; Bagchi, M; Hassoun, EA; Tran, MX; Tang, L; Stohs, SJ. Induction of oxidative stress by chronic administration of sodium dichromate and cadmium chloride to rats. Free Radic. Biol. Med 1997, 22, 471–478. [Google Scholar]
- Shaikh, ZA; Vu, TT; Zaman, K. Oxidative stress as a mechanism of chronic cadmium-induced hepatotoxicity and renal toxicity and protection by antioxidants. Toxicol. Appl. Pharmacol 1999, 154, 256–263. [Google Scholar]
- Ramirez, DC; Gimenez, MS. Induction of redox changes, inducible nitric oxide synthase and cyclooxygenase-2 by chronic cadmium exposure in mouse peritoneal macrophages. Toxicol. Lett 2003, 145, 121–132. [Google Scholar]
- Waisberg, M; Joseph, P; Hale, B; Beyersmann, D. Molecular and cellular mechanisms of cadmium carcinogenesis. Toxicology 2003, 192, 95–117. [Google Scholar]
- Yamano, T; DeCicco, LA; Rikans, LE. Attenuation of cadmium-induced liver injury in senescent male Fischer 344 rats: role of Kupffer cells and inflammatory cytokines. Toxicol. Appl. Pharmacol 2000, 162, 68–75. [Google Scholar]
- Belyaeva, EA; Dymkowska, D; Wieckowski, MR; Wojtczak, L. Mitochondria as an important target in heavy metal toxicity in rat hepatoma AS-30D cells. Toxicol. Appl. Pharmacol 2008, 231, 34–42. [Google Scholar]
- Dorta, DJ; Leite, S; DeMarco, KC; Prado, IM; Rodrigues, T; Mingatto, FE; Uyemura, SA; Santos, AC; Curti, C. A proposed sequence of events for cadmium induced mitochondrial impairment. J. Inorg. Biochem 2003, 97, 251–257. [Google Scholar]
- Ali, MM; Lal, B; Mathur, N; Chandra, SV. Behavioral toxicity of cadmium in rats in relation to the level of protein nutrition. Nutr. Res 1991, 11, 325. [Google Scholar]
- Klaassen, CD; Liu, J. Role of metallothionein in cadmium-induced hepatotoxicity and nephrotoxicity. Drug Metab. Rev 1997, 29, 79–102. [Google Scholar]
- Amler, S. Liquid mercury: a poisonous plaything. Contemp. Pediatr 2002, 19, 37–56. [Google Scholar]
- Risher, JF; Nickle, RA; Amler, SN. Elemental mercury poisoning in occupational and residential settings: two case studies. Int. J. Hyg. Environ. Hlth 2003, 206, 371–379. [Google Scholar]
- Patterson, JE; Weissberg, BG; Dennison, PJ. Mercury in human breath from dental amalgams. Bull Environ. Contam. Toxicol 1985, 34, 459–468. [Google Scholar]
- Bjorkman, L; Lundekvam, BF; Laegreid, T; Bertelsen, BI; Morild, I; Lilleng, P. Mercury in human brain, blood, muscle and toenails in relation to exposure: an autopsy study. Environ. Health 2007, 6, 30. [Google Scholar]
- Goyer, RA; Clarkson, TW. Toxic effects of metals. In Casarett & Doull’s Toxicology: the Basic of Poisons, 6th ed; Klaassen, CD, Ed.; McGraw-Hill: New York, NY, USA, 2001; pp. 822–826. [Google Scholar]
- Geier, DA; Sykes, LK; Geier, MR. A review of thimerosal (merthiolate) and its ethyl mercury breakdown product: specific historical considerations regarding safety and effectiveness. J. Toxicol. Environ. Health B Crit. Rev 2007, 10, 575–596. [Google Scholar]
- Clarkson, TW. The pharmacology of mercury compounds. Ann. Rev. Pharmacol 1972, 12, 375–406. [Google Scholar]
- Lorscheider, FL; Vimy, MJ; Summers, AO. Mercury exposure from “silver” tooth fillings: emerging evidence questions a traditional dental paradigm. FASEB J 1995, 9, 504–508. [Google Scholar]
- Hultberg, B; Anderson, A; Isaksson, A. Interaction of metals and thiols in cell damage and glutathione distribution: potentiation of mercury toxicity by dithiothreitol. Toxicology 2001, 156, 93–100. [Google Scholar]
- McGoldrick, TA; Lock, EA; Rodilla, V; Hawksworth, GM. Renal cysteine conjugate C-S lyase mediated toxicity of halogenated alkenes in primary cultures of human and rat proximal tubular cells. Arch. Toxicol 2003, 77, 365–370. [Google Scholar]
- Gonzalez, P; Dominique, Y; Massabuau, JC; Boudou, A; Bourdineaud, JP. Comparative effects of dietary methyl mercury on gene expression in liver, skeletal muscle, and brain of the zebra fish (Danio rerio). Environ. Sci. Technol 2005, 39, 3972–3980. [Google Scholar]
- Sarafian, TA; Bredesen, DE; Verity, MA. Cellular resistance to methylmercury. Neurotoxicology 1996, 17, 27–36. [Google Scholar]
- Blanusa, M; Varnai, VM; Piasek, M; Kostial, K. Chelators as antidotes of metal toxicity: Therapeutic and Experimental Aspects. Curr. Med. Chem 2005, 12, 2771–2794. [Google Scholar]
- Gonzalez-Ramirez, D; Zuniga-Charles, M; Narro-Juarez, A; Molina-Recio, Y; Hurlbut, KM; Dart, RC; Aposhian, HV. DMPS (2,3-dimercaptopropane-1-sulfonate, dimaval) decreases the body burden of mercury in humans exposed to mercurous chloride. J. Pharmacol. Exp. Ther 1998, 287, 8–12. [Google Scholar]
- Lund, ME; Banner, W, Jr; Clarkson, TW; Berlin, M. Treatment of acute methylmercury ingestion by hemodialysis with N-acetylcysteine (Mucomyst) infusion and 2,3-dimercaptopropane sulfonate. J. Toxicol. Clin. Toxicol 1984, 22, 31–49. [Google Scholar]
- Rassmusen, M; Folsom, AR; Catellier, DJ; Tsai, M; Garg, U; Eckfeldt, JH. A prospective study of coronary heart disease and the hemochromatosis gene (HFE) C282Y mutation: the atherosclerosis risk in communities (ARIC) study. Atherosclerosis 2001, 154, 739–746. [Google Scholar]
- Parikkila, S; Niemela, O; Savolainen, ER; Koistinen, P. HFE mutation do not account for trnasfusional iron overload in patients with acute myeloid leukemia. Transfusion 2001, 41, 828–831. [Google Scholar]
- Berg, D; Gerlach, M; Youdim, MB; Double, KL; Zecca, L; Riederer, P; Becker, G. Brain iron pathways and their relevance to Parkinson’s disease. J. Neurochem 2001, 79, 225–236. [Google Scholar]
- Li, J; Zhu, Y; Singhal, DP. HFE gene mutation in patients with rheumatoid arthritis. J. Rhematol 2000, 27, 2074–2077. [Google Scholar]
- Tenenbein, M. Hepatotoxicity in acute iron poisoning. J. Toxicol. Clin. Toxicol 2001, 39, 721–726. [Google Scholar]
- Salahudeen, AK; Oliver, B; Bower, JD; Roberts, LJ. Increase in plasma esterified F2-isoprostanes following intravenous iron infusion in patients on hemodialysis. Kidney Int 2001, 11, 539–549. [Google Scholar]
- Ponka, P; Tenenbein, M; Eaton, JW. Iron. In Handbook on the Toxicology of Metals, 3rd ed; Nordberg, GF, Fowler, BA, Nordberg, M, Friberg, L, Eds.; Academic Press: San Diego, CA, USA, 2007; pp. 577–598. [Google Scholar]
- Kontoghiorghes, GJ. Comparative efficacy and toxicity of desferioxamine, deferiprone and other iron and aluminium chelating drugs. Toxicol. Lett 1995, 80, 1–18. [Google Scholar]
- Kontoghiorghes, GJ; Kolnagou, A. Effective new treatment of iron overload in the lassaemia using the ICOC combination therapy protocol of deferiprone (L1) and deferoxamine and of new chelating drugs. Haematologica 2006, 91, 34–35. [Google Scholar]
- Stumpf, JL. Deferasirox. Am. J. Health Syst. Pharm 2007, 64, 606–616. [Google Scholar]
- Flora, SJS; Mittal, M; Mehta, A. Heavy metal induced oxidative stress & its possible reversal by chelation therapy. Ind. J. Med. Res 2008, 128, 501–523. [Google Scholar]
- Pachauri, P; Saxena, G; Mehta, A; Mishra, D; Flora, SJS. Combinational chelation therapy abrogates lead induced neurodegeneration in rats. Toxicol. Appl. Pharmacol 2009, 240, 255–265. [Google Scholar]
- Flora, SJS; Pande, M; Mehta, A. Beneficial effect of combined administration of some naturally occurring antioxidants (vitamins) and thiol chelators in the treatment of chronic lead intoxication. Chem. Biol. Inter 2003, 145, 267–280. [Google Scholar]
- Pande, M; Flora, SJS. Lead induced oxidative damage and its response to combined administration of α-lipoic acid and succimers in rats. Toxicology 2002, 177, 187–196. [Google Scholar]
- Kannan, GM; Flora, SJS. Combined administration of n-acetyl cysteine and monoisoamyl DMSA on tissue oxidative stress during arsenic chelation therapy. Biol. Trace Elem. Res 2006, 110, 43–59. [Google Scholar]
- Modi, M; Flora, SJS. Combined administration of iron and monoisoamyl DMSA in the treatment of chronic arsenic intoxication in mice. Cell. Biol. Toxicol 2007, 23, 429–443. [Google Scholar]
- Saxena, G; Flora, SJS. Changes in brain biogenic amines and heme- biosynthesis and their response to combined administration of succimer and Centella asiatica in lead poisoned rats. J. Pharm. Pharmacol 2006, 58, 547–559. [Google Scholar]
- Mishra, D; Gupta, R; Pant, SC; Kushwah, P; Satish, HT; Flora, SJS. Therapeutic potential of combined administration of MiADMSA and Moringa oleifera seed powder on arsenic induced oxidative stress and metal distribution in mouse. Toxicol. Mechanism Meth 2008, 19, 169–182. [Google Scholar]
- Flora, SJS; Mehta, A; Gupta, R. Prevention of arsenic-induced hepatic apoptosis by concomitant administration of garlic extracts in mice. Chem. Biol. Inter 2009, 177, 227–233. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metal | Na | Li | Ba | Sr | Mg | Ca | Mn | Fe | Co | Zn | Cd | Pb | Ni |
| K (log) | 1.7 | 2.8 | 7.8 | 8.6 | 8.7 | 10.6 | 13.4 | 14.4 | 16.1 | 16.1 | 16.4 | 18.3 | 18.4 |
| Therapeutic Strategy | Examples | Refs. | Benefits |
|---|---|---|---|
| Development of newer chelating, agents | MiADMSA MmDMSA MchDMSA | [86] |
|
| Combination therapy with two chelating agents | DMSA+ MiADMSA MiADMSA+CaNa2EDTA | [52,87,89] [85] |
|
| Chelating agent + Antioxidants | DMSA/MiADMSA + NAC DMSA/MiADMSA + LA MiADMSA+ Quercetin DMSA/MiADMSA + Taurine DMSA/MiADMSA+ Vitamin | [115,207] [109] [132] [112,113] [205,108,120] |
|
| Chelating agent + Micronutrients | DMSA+Zn CaNa2EDTA+Zn Fe DMSA+Cu | [16,122,121] [111] [208] [121] |
|
| Chelating agent + Herbal extract. | Centella asiatica Moringa Oleifera Garlic | [209] [210] [211] |
|
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Flora, S.J.S.; Pachauri, V. Chelation in Metal Intoxication. Int. J. Environ. Res. Public Health 2010, 7, 2745-2788. https://doi.org/10.3390/ijerph7072745
Flora SJS, Pachauri V. Chelation in Metal Intoxication. International Journal of Environmental Research and Public Health. 2010; 7(7):2745-2788. https://doi.org/10.3390/ijerph7072745
Chicago/Turabian StyleFlora, Swaran J.S., and Vidhu Pachauri. 2010. "Chelation in Metal Intoxication" International Journal of Environmental Research and Public Health 7, no. 7: 2745-2788. https://doi.org/10.3390/ijerph7072745
APA StyleFlora, S. J. S., & Pachauri, V. (2010). Chelation in Metal Intoxication. International Journal of Environmental Research and Public Health, 7(7), 2745-2788. https://doi.org/10.3390/ijerph7072745