Molecular Basis and Current Treatment for Alcoholic Liver Disease

Abstract
:1. Introduction
2. Assessing the Problem
3. Genetic Predisposition
3.1. Candidate Genes Predisposing Alcoholic Liver Disease
3.2. Candidate Genes Predisposing Alcohol Use Disorders
3.2.1. Endophenotypes
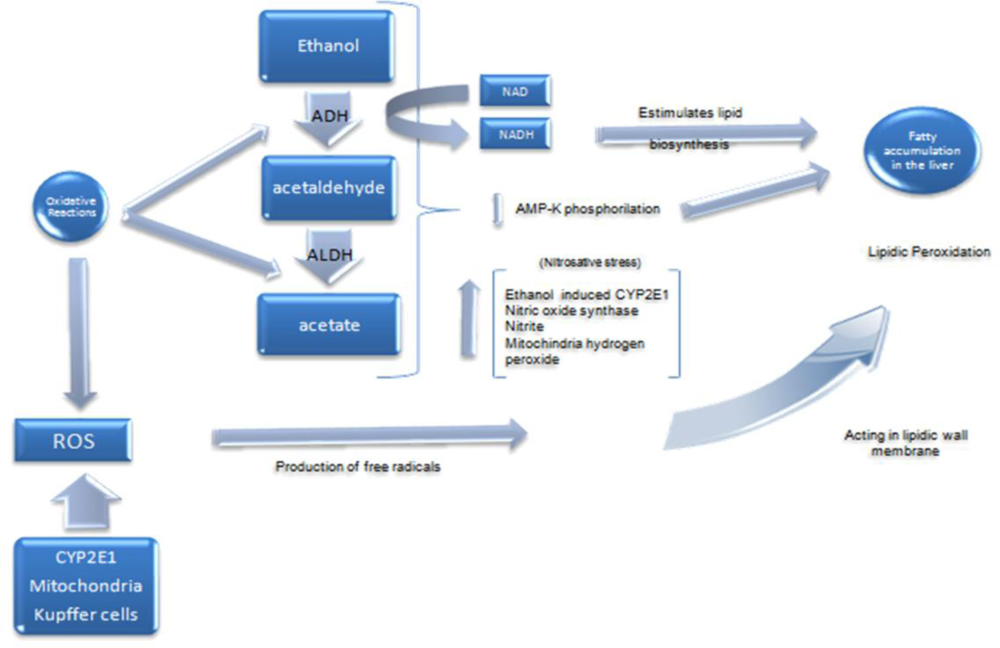
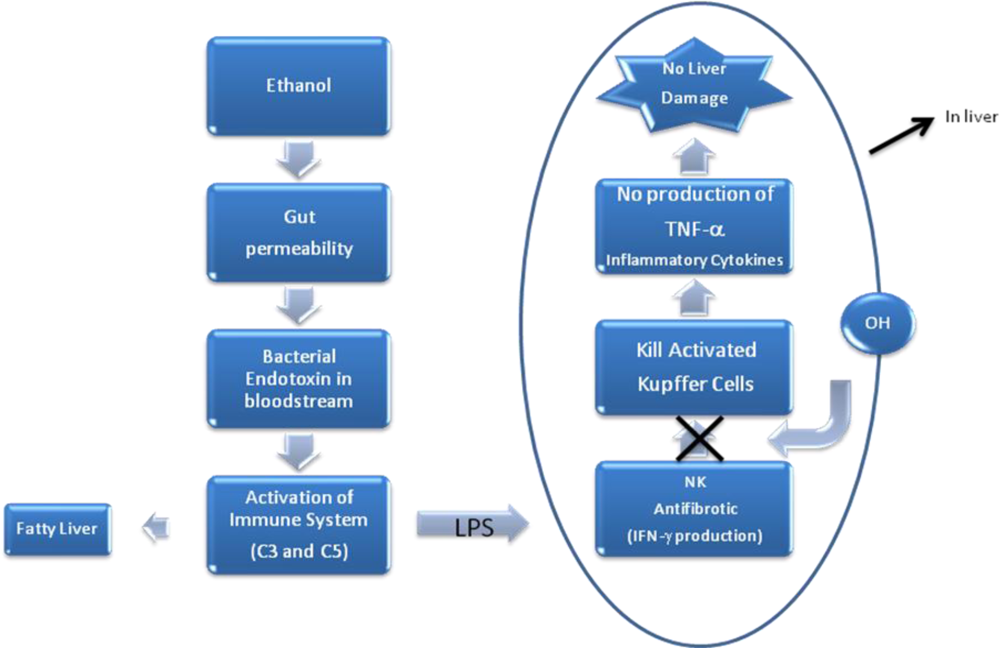
4. Pathophysiology
4.1. Clinical Presentation of Alcoholic Liver Disease
5. Diagnostic Tests
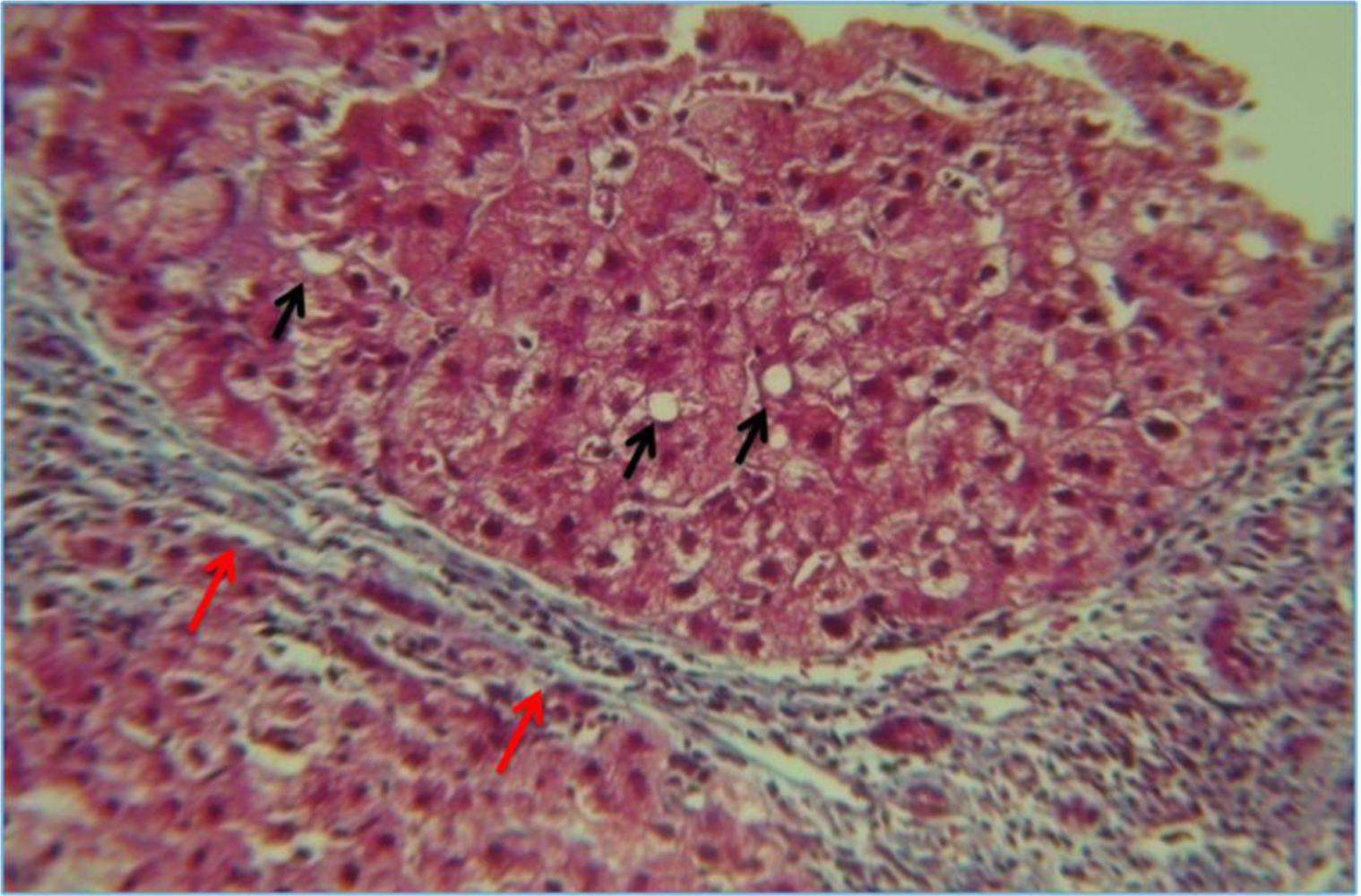
5.1. Steatosis and Steatohepatitis
5.2. Alcoholic Hepatitis
5.3. Fibrosis and Cirrhosis
6. Complications
7. Current Treatment
7.1. Alcoholic Liver Disease
7.1.1. Nutrition
7.1.2. Pharmacotherapy
7.1.3. About transplant
7.2. Alcohol Use Disorders
7.2.1. Pharmacotherapy
7.2.2. Alcoholics anonymous
8. Conclusions
References
- Global Status Report on Alcohol 2004; World Health Organization, Dept. of Mental Health and Substance Abuse: Geneva, Switzerland, 2004; p. 88.
- Alcohol and Injury in Emergency Departments Summary of the Report From the WHO Collaborative on Alcohol and Injuries; World Health Organization, Dept. Of Mental Health and Substance Abuse, Dept. of Injuries and Violence Prevention.: Geneva, Switzerland, 2007; p. 13.
- Mann, RE; Smart, RG; Govoni, R. The epidemiology of alcoholic liver disease. Alcohol. Res. Health 2003, 27, 209–219. [Google Scholar]
- Malhi, H; Gores, GJ. Cellular and molecular mechanisms of liver injury. Gastroenterology 2008, 134, 1641–1654. [Google Scholar]
- Song, BJ; Moon, KH; Olsson, NU; Salem, N, Jr. Prevention of alcoholic fatty liver and mitochondrial dysfunction in the rat by long-chain polyunsaturated fatty acids. J. Hepatol 2008, 49, 262–273. [Google Scholar]
- Lucey, MR; Mathurin, P; Morgan, TR. Alcoholic hepatitis. N. Engl. J. Med 2009, 360, 2758–2769. [Google Scholar]
- Siu, L; Foont, J; Wands, JR. Hepatitis C virus and alcohol. Semin. Liver. Dis 2009, 29, 188–199. [Google Scholar]
- Mayfield, RD; Harris, RA; Schuckit, MA. Genetic factors influencing alcohol dependence. Br. J. Pharmacol 2008, 154, 275–287. [Google Scholar]
- Day, CP; Bassendine, MF. Genetic predisposition to alcoholic liver disease. Gut 1992, 33, 1444–1447. [Google Scholar]
- Ducci, F; Goldman, D. Genetic approaches to addiction: Genes and alcohol. Addiction 2008, 103, 1414–1428. [Google Scholar]
- Michitaka, K; Nishiguchi, S; Aoyagi, Y; Hiasa, Y; Tokumoto, Y; Onji, M. Etiology of liver cirrhosis in Japan: A nationwide survey. J. Gastroenterol 2010, 45, 86–94. [Google Scholar]
- Strat, YL; Ramoz, N; Schumann, G; Gorwood, P. Molecular genetics of alcohol dependence and related endophenotypes. Curr. Genomics 2008, 9, 444–451. [Google Scholar]
- Noble, EP. The D2 dopamine receptor gene: A review of association studies in alcoholism and phenotypes. Alcohol 1998, 16, 33–45. [Google Scholar]
- Polich, J; Bloom, FE. P300, alcoholism heritability, and stimulus modality. Alcohol 1999, 17, 149–156. [Google Scholar]
- Vaughn, MG; Beaver, KM; DeLisi, M; Howard, MO; Perron, BE. Dopamine D4 receptor gene exon III polymorphism associated with binge drinking attitudinal phenotype. Alcohol 2009, 43, 179–184. [Google Scholar]
- Namkoong, K; Cheon, KA; Kim, JW; Jun, JY; Lee, JY. Association study of dopamine D2, D4 receptor gene, GABAA receptor beta subunit gene, serotonin transporter gene polymorphism with children of alcoholics in Korea: a preliminary study. Alcohol 2008, 42, 77–81. [Google Scholar]
- Wu, D; Cederbaum, AI. Alcohol, oxidative stress, and free radical damage. Alcohol. Res. Health 2003, 27, 277–284. [Google Scholar]
- Lu, Y; Cederbaum, AI. CYP2E1 and oxidative liver injury by alcohol. Free Radic. Biol. Med 2008, 44, 723–738. [Google Scholar]
- Bousquet-Dubouch, MP; Nguen, S; Bouyssie, D; Burlet-Schiltz, O; French, SW; Monsarrat, B; Bardag-Gorce, F. Chronic ethanol feeding affects proteasome-interacting proteins. Proteomics 2009, 9, 3609–3622. [Google Scholar]
- Crossley, IR; Neuberger, J; Davis, M; Williams, R; Eddleston, AL. Ethanol metabolism in the generation of new antigenic determinants on liver cells. Gut 1986, 27, 186–189. [Google Scholar]
- Cubero, FJ; Urtasun, R; Nieto, N. Alcohol and liver fibrosis. Semin. Liver Dis 2009, 29, 211–221. [Google Scholar]
- Cahill, A; Cunningham, CC; Adachi, M; Ishii, H; Bailey, SM; Fromenty, B; Davies, A. Effects of alcohol and oxidative stress on liver pathology: the role of the mitochondrion. Alcohol. Clin. Exp. Res 2002, 26, 907–915. [Google Scholar]
- Gyamfi, MA; Damjanov, I; French, S; Wan, YJ. The pathogenesis of ethanol versus methionine and choline deficient diet-induced liver injury. Biochem. Pharmacol 2008, 75, 981–995. [Google Scholar]
- Thakur, V; Pritchard, MT; McMullen, MR; Wang, Q; Nagy, LE. Chronic ethanol feeding increases activation of NADPH oxidase by lipopolysaccharide in rat Kupffer cells: role of increased reactive oxygen in LPS-stimulated ERK1/2 activation and TNF-alpha production. J. Leukoc. Biol 2006, 79, 1348–1356. [Google Scholar]
- Butura, A; Nilsson, K; Morgan, K; Morgan, TR; French, SW; Johansson, I; Schuppe-Koistinen, I; Ingelman-Sundberg, M. The impact of CYP2E1 on the development of alcoholic liver disease as studied in a transgenic mouse model. J. Hepatol 2009, 50, 572–583. [Google Scholar]
- Qin, L; He, J; Hanes, RN; Pluzarev, O; Hong, JS; Crews, FT. Increased systemic and brain cytokine production and neuroinflammation by endotoxin following ethanol treatment. J. Neuroinflammation 2008, 5, 10. [Google Scholar]
- Mundt, B; Wirth, T; Zender, L; Waltemathe, M; Trautwein, C; Manns, MP; Kuhnel, F; Kubicka, S. Tumour necrosis factor related apoptosis inducing ligand (TRAIL) induces hepatic steatosis in viral hepatitis and after alcohol intake. Gut 2005, 54, 1590–1596. [Google Scholar]
- Pritchard, MT; McMullen, MR; Stavitsky, AB; Cohen, JI; Lin, F; Medof, ME; Nagy, LE. Differential contributions of C3, C5, and decay-accelerating factor to ethanol-induced fatty liver in mice. Gastroenterology 2007, 132, 1117–1126. [Google Scholar]
- Arteel, GE. Silencing a killer among us: ethanol impairs immune surveillance of activated stellate cells by natural killer cells. Gastroenterology 2008, 134, 351–353. [Google Scholar]
- Stewart, S; Prince, M; Bassendine, M; Hudson, M; James, O; Jones, D; Record, C; Day, CP. A randomized trial of antioxidant therapy alone or with corticosteroids in acute alcoholic hepatitis. J. Hepatol 2007, 47, 277–283. [Google Scholar]
- Cotrim, HP; Freitas, LA; Alves, E; Almeida, A; May, DS; Caldwell, S. Effects of light-to-moderate alcohol consumption on steatosis and steatohepatitis in severely obese patients. Eur. J. Gastroenterol. Hepatol 2009, 21, 969–972. [Google Scholar]
- Butterworth, RF. Hepatic encephalopathy—A serious complication of alcoholic liver disease. Alcohol Res. Health 2003, 27, 143–145. [Google Scholar]
- Bergheim, I; McClain, CJ; Arteel, GE. Treatment of alcoholic liver disease. Dig. Dis 2005, 23, 275–284. [Google Scholar]
- Angulo, P. Noninvasive assessment of fibrosis and steatosis in NASH and ASH. Gastroenterol. Clin. Biol 2009, 33, 940–948. [Google Scholar]
- Tiniakos, DG. Liver biopsy in alcoholic and non-alcoholic steatohepatitis patients. Gastroenterol. Clin. Biol 2009, 33, 930–939. [Google Scholar]
- Sebastiani, G. Non-invasive assessment of liver fibrosis in chronic liver diseases: implementation in clinical practice and decisional algorithms. World J. Gastroenterol 2009, 15, 2190–2203. [Google Scholar]
- Liaw, YF; Chu, CM. Hepatitis B virus infection. Lancet 2009, 373, 582–592. [Google Scholar]
- Forrest, EH; Evans, CD; Stewart, S; Phillips, M; Oo, YH; McAvoy, NC; Fisher, NC; Singhal, S; Brind, A; Haydon, G; O'Grady, J; Day, CP; Hayes, PC; Murray, LS; Morris, AJ. Analysis of factors predictive of mortality in alcoholic hepatitis and derivation and validation of the Glasgow alcoholic hepatitis score. Gut 2005, 54, 1174–1179. [Google Scholar]
- Ong, JP; Mullen, KD. Hepatic encephalopathy. Eur J Gastroenterol Hepatol 2001, 13, 325–334. [Google Scholar]
- Jonas, S; Al-Abadi, H; Benckert, C; Thelen, A; Hippler-Benscheid, M; Saribeyoglu, K; Radtke, B; Pratschke, J; Neuhaus, P. Prognostic significance of the DNA-index in liver transplantation for hepatocellular carcinoma in cirrhosis. Ann. Surg 2009, 250, 1008–1013. [Google Scholar]
- Sheth, M; Riggs, M; Patel, T. Utility of the Mayo End-Stage Liver Disease (MELD) score in assessing prognosis of patients with alcoholic hepatitis. BMC Gastroenterol 2002, 2, 2. [Google Scholar] [Green Version]
- Neuberger, J. Developments in liver transplantation. Gut 2004, 53, 759–768. [Google Scholar]
- Johnson, BA. Update on neuropharmacological treatments for alcoholism: scientific basis and clinical findings. Biochem. Pharmacol 2008, 75, 34–56. [Google Scholar]
- Ji, D; Gilpin, NW; Richardson, HN; Rivier, CL; Koob, GF. Effects of naltrexone, duloxetine, and a corticotropin-releasing factor type 1 receptor antagonist on binge-like alcohol drinking in rats. Behav. Pharmacol 2008, 19, 1–12. [Google Scholar]
- Anton, RF; Oroszi, G; O'Malley, S; Couper, D; Swift, R; Pettinati, H; Goldman, D. An evaluation of mu-opioid receptor (OPRM1) as a predictor of naltrexone response in the treatment of alcohol dependence: Results from the Combined Pharmacotherapies and Behavioral Interventions for Alcohol Dependence (COMBINE) study. Arch. Gen. Psychiatry 2008, 65, 135–144. [Google Scholar]
- Anton, RF; Myrick, H; Baros, AM; Latham, PK; Randall, PK; Wright, TM; Stewart, SH; Waid, R; Malcolm, R. Efficacy of a combination of flumazenil and gabapentin in the treatment of alcohol dependence: relationship to alcohol withdrawal symptoms. J. Clin. Psychopharmacol 2009, 29, 334–342. [Google Scholar]
- Newton, BW; Russell, WK; Russell, DH; Ramaiah, SK; Jayaraman, A. Liver proteome analysis in a rodent model of alcoholic steatosis. J. Proteome. Res 2009, 8, 1663–1671. [Google Scholar]
- Anton, RF; O'Malley, SS; Ciraulo, DA; Cisler, RA; Couper, D; Donovan, DM; Gastfriend, DR; Hosking, JD; Johnson, BA; LoCastro, JS; Longabaugh, R; Mason, BJ; Mattson, ME; Miller, WR; Pettinati, HM; Randall, CL; Swift, R; Weiss, RD; Williams, LD; Zweben, A. Combined pharmacotherapies and behavioral interventions for alcohol dependence: the COMBINE study: A randomized controlled trial. JAMA 2006, 295, 2003–2017. [Google Scholar]
- Groh, DR; Jason, LA; Keys, CB. Social network variables in alcoholics anonymous: A literature review. Clin. Psychol. Rev 2008, 28, 430–450. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| 1 | Mouth and oropharynx cancer |
| 2 | Alcohol use disorders |
| 3 | Ischemic heart disease |
| 4 | Liver cirrhosis |
| 5 | Road traffic accidents |
| 6 | Poisonings |
| 7 | Falls |
| 8 | Intentional injuries |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Miranda-Mendez, A.; Lugo-Baruqui, A.; Armendariz-Borunda, J. Molecular Basis and Current Treatment for Alcoholic Liver Disease. Int. J. Environ. Res. Public Health 2010, 7, 1872-1888. https://doi.org/10.3390/ijerph7051872
Miranda-Mendez A, Lugo-Baruqui A, Armendariz-Borunda J. Molecular Basis and Current Treatment for Alcoholic Liver Disease. International Journal of Environmental Research and Public Health. 2010; 7(5):1872-1888. https://doi.org/10.3390/ijerph7051872
Chicago/Turabian StyleMiranda-Mendez, Alejandra, Alejandro Lugo-Baruqui, and Juan Armendariz-Borunda. 2010. "Molecular Basis and Current Treatment for Alcoholic Liver Disease" International Journal of Environmental Research and Public Health 7, no. 5: 1872-1888. https://doi.org/10.3390/ijerph7051872
APA StyleMiranda-Mendez, A., Lugo-Baruqui, A., & Armendariz-Borunda, J. (2010). Molecular Basis and Current Treatment for Alcoholic Liver Disease. International Journal of Environmental Research and Public Health, 7(5), 1872-1888. https://doi.org/10.3390/ijerph7051872