Air Pollution Exposure—A Trigger for Myocardial Infarction?

Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Interview
2.3. Air Pollution and Meteorology
2.4. Statistical Analysis
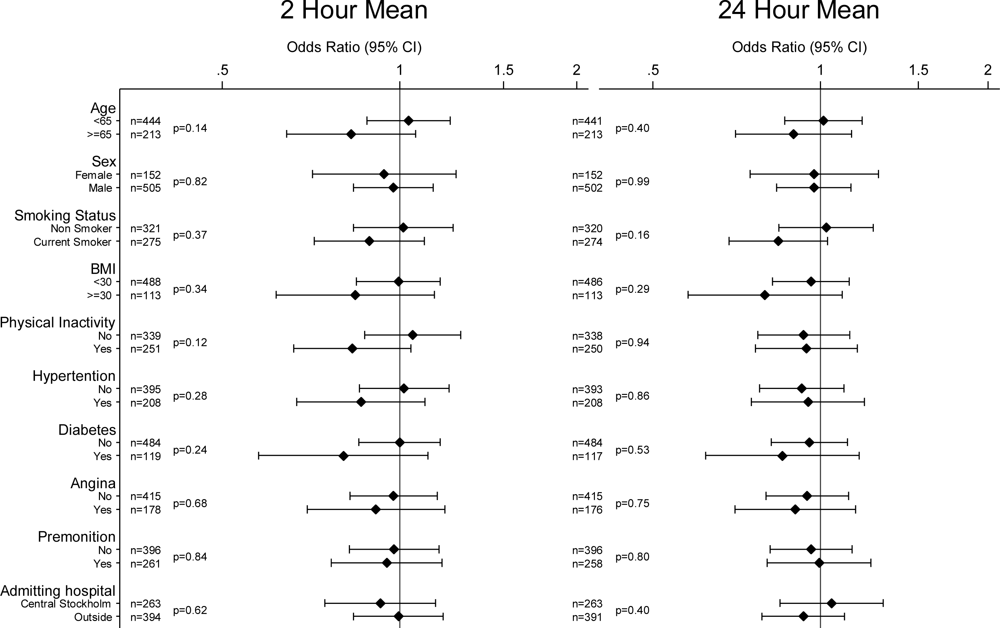
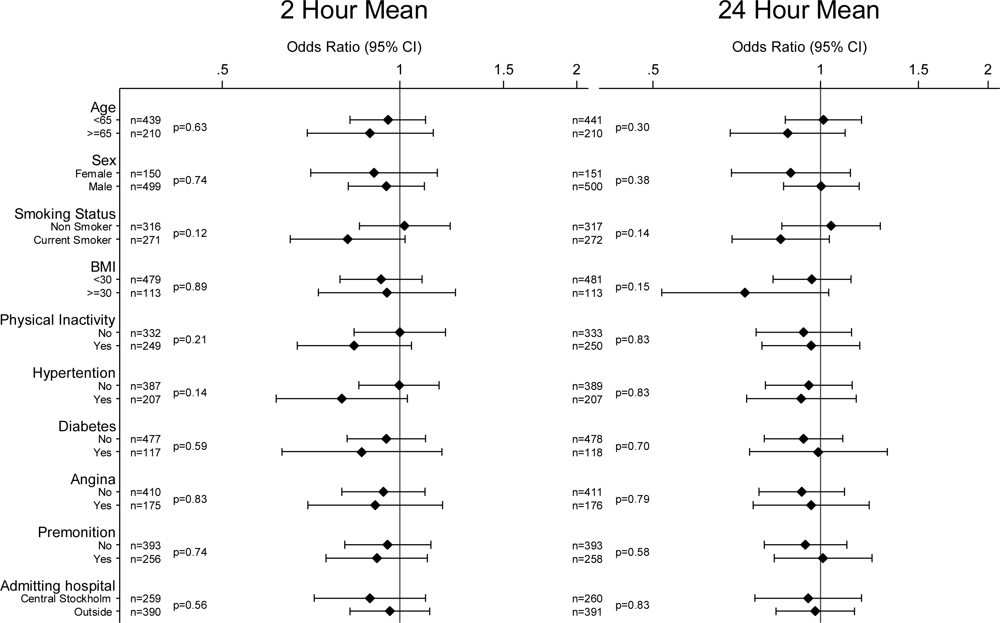
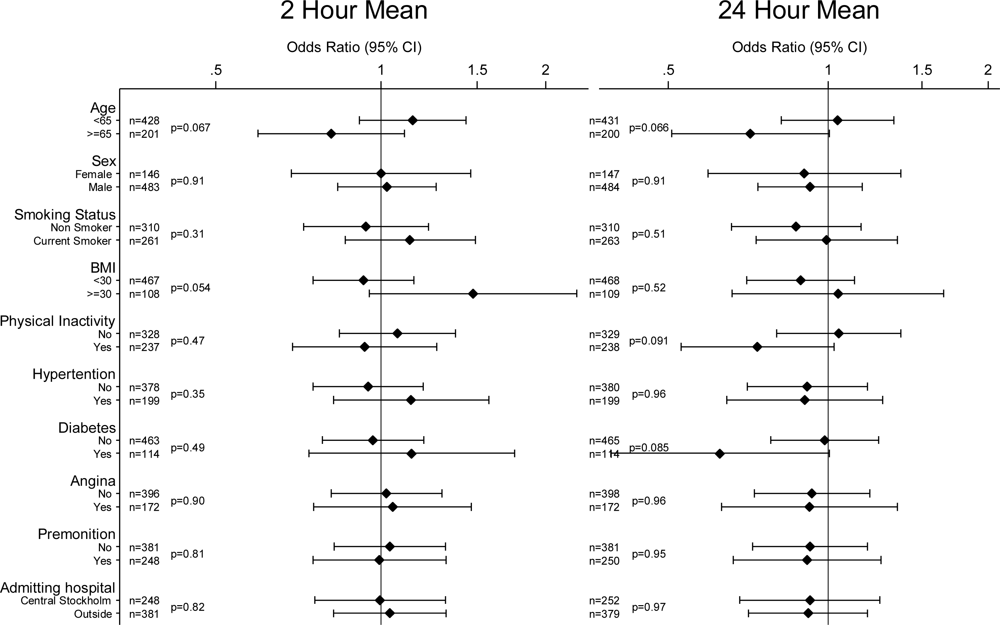
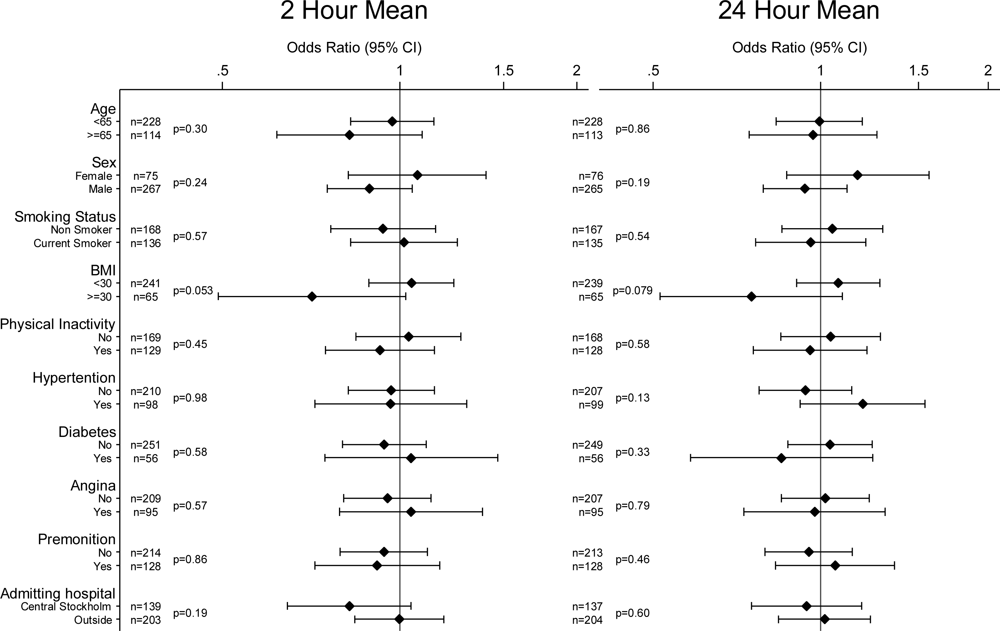
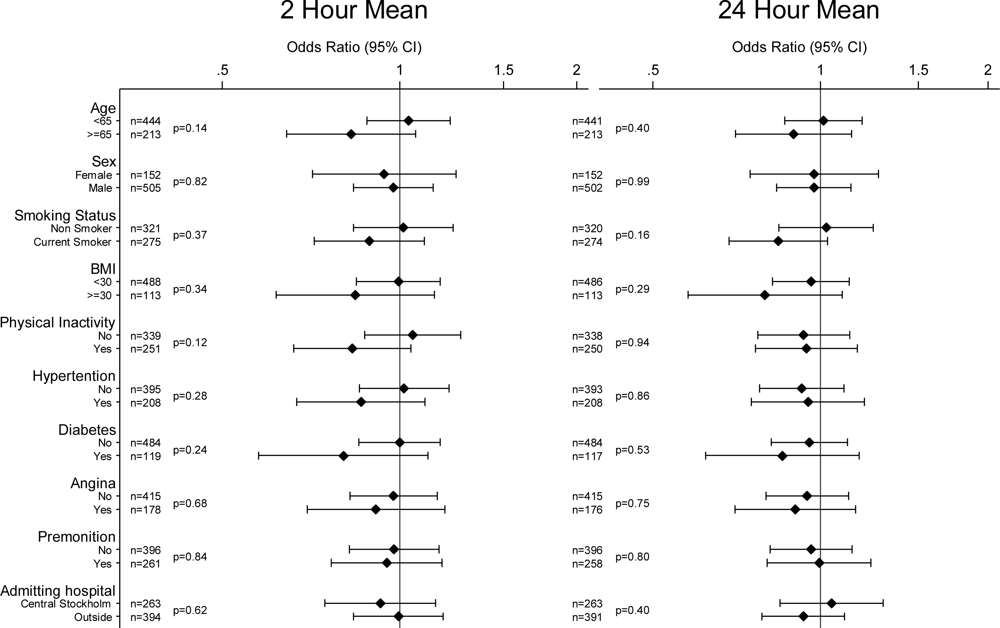
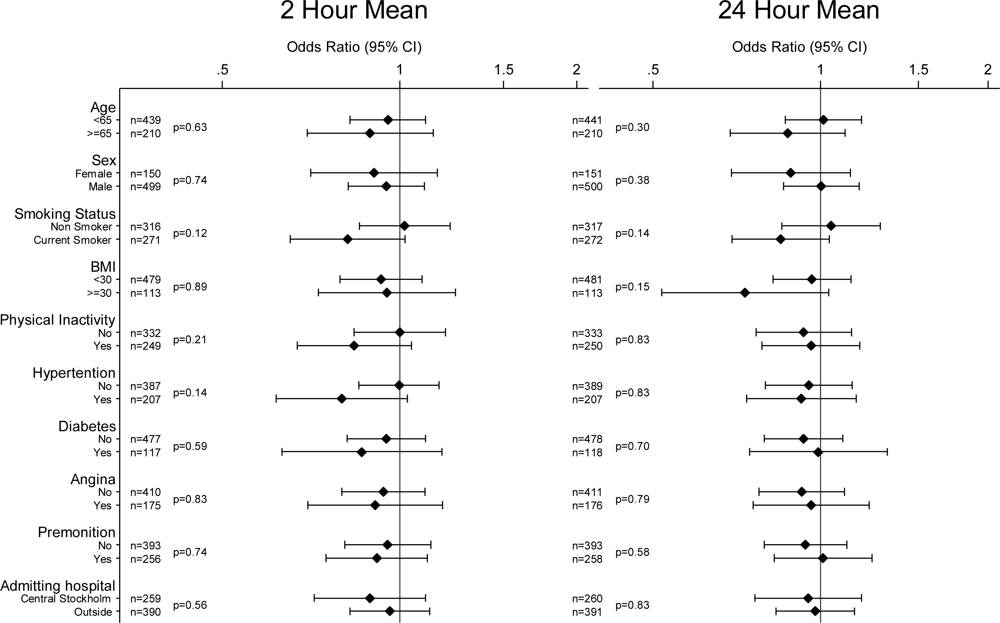
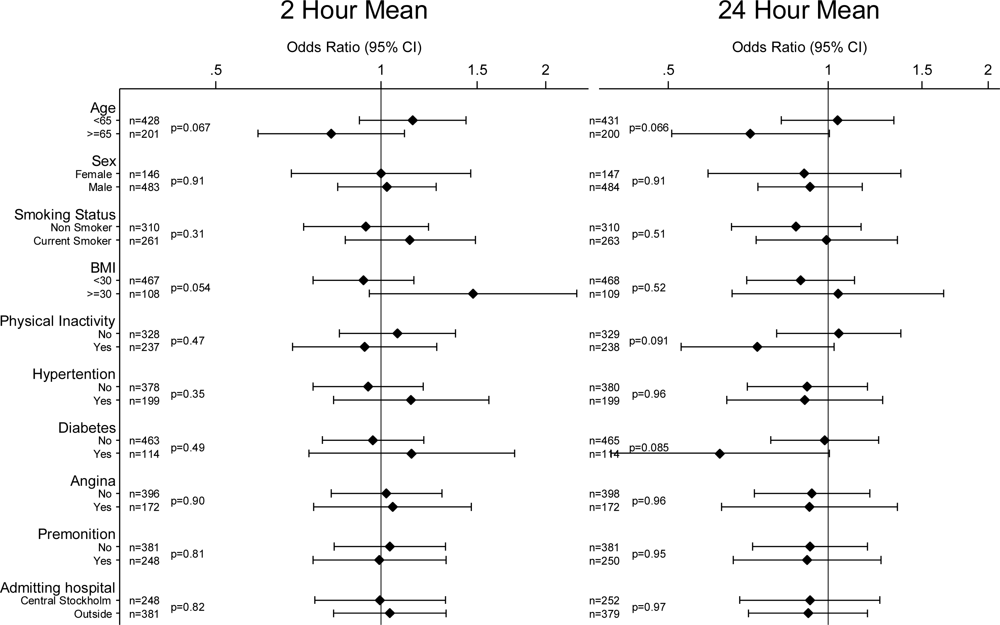
2. Results
4. Discussion
5. Conclusions
Acknowledgments
References and Notes
- Poloniecki, JD; Atkinson, RW; de Leon, AP; Anderson, HR. Daily time series for cardiovascular hospital admissions and previous day’s air pollution in London, UK. Occup. Environ. Med 1997, 54, 535–540. [Google Scholar]
- Bell, ML; Davis, DL. Reassessment of the lethal London fog of 1952: novel indicators of acute and chronic consequences of acute exposure to air pollution. Environ Health Perspect 2001, 109(Suppl. 3), 389–394. [Google Scholar]
- Dominici, F; Peng, RD; Bell, ML; Pham, L; McDermott, A; Zeger, SL; Samet, JM. Fine particulate air pollution and hospital admission for cardiovascular and respiratory diseases. JAMA 2006, 295, 1127–1134. [Google Scholar]
- Host, S; Larrieu, S; Pascal, L; Blanchard, M; Declercq, C; Fabre, P; Jusot, JF; Chardon, B; Le Tertre, A; Wagner, V; Prouvost, H; Lefranc, A. Short-term associations between fine and coarse particles and hospital admissions for cardiorespiratory diseases in six French cities. Occup. Environ. Med 2008, 65, 544–551. [Google Scholar]
- Le Tertre, A; Medina, S; Samoli, E; Forsberg, B; Michelozzi, P; Boumghar, A; Vonk, JM; Bellini, A; Atkinson, R; Ayres, JG; Sunyer, J; Schwartz, J; Katsouyanni, K. Short-term effects of particulate air pollution on cardiovascular diseases in eight European cities. J. Epidemiol. Community Health 2002, 56, 773–779. [Google Scholar]
- Mann, JK; Tager, IB; Lurmann, F; Segal, M; Quesenberry, CP, Jr; Lugg, MM; Shan, J; Van Den Eeden, SK. Air pollution and hospital admissions for ischemic heart disease in persons with congestive heart failure or arrhythmia. Environ. Health Perspect 2002, 110, 1247–1252. [Google Scholar]
- Ruidavets, JB; Cournot, M; Cassadou, S; Giroux, M; Meybeck, M; Ferrieres, J. Ozone air pollution is associated with acute myocardial infarction. Circulation 2005, 111, 563–569. [Google Scholar]
- von Klot, S; Peters, A; Aalto, P; Bellander, T; Berglind, N; D’Ippoliti, D; Elosua, R; Hormann, A; Kulmala, M; Lanki, T; Lowel, H; Pekkanen, J; Picciotto, S; Sunyer, J; Forastiere, F. Ambient air pollution is associated with increased risk of hospital cardiac readmissions of myocardial infarction survivors in five European cities. Circulation 2005, 112, 3073–3079. [Google Scholar]
- Dockery, DW; Luttmann-Gibson, H; Rich, DQ; Link, MS; Mittleman, MA; Gold, DR; Koutrakis, P; Schwartz, JD; Verrier, RL. Association of air pollution with increased incidence of ventricular tachyarrhythmias recorded by implanted cardioverter defibrillators. Environ. Health Perspect 2005, 113, 670–674. [Google Scholar]
- Rich, DQ; Schwartz, J; Mittleman, MA; Link, M; Luttmann-Gibson, H; Catalano, PJ; Speizer, FE; Dockery, DW. Association of short-term ambient air pollution concentrations and ventricular arrhythmias. Am. J. Epidemiol 2005, 161, 1123–1132. [Google Scholar]
- Ljungman, PL; Berglind, N; Holmgren, C; Gadler, F; Edvardsson, N; Pershagen, G; Rosenqvist, M; Sjogren, B; Bellander, T. Rapid effects of air pollution on ventricular arrhythmias. Eur. Heart J 2008, 29, 2894–2901. [Google Scholar]
- Wellenius, GA; Bateson, TF; Mittleman, MA; Schwartz, J. Particulate air pollution and the rate of hospitalization for congestive heart failure among medicare beneficiaries in Pittsburgh, Pennsylvania. Am. J. Epidemiol 2005, 161, 1030–1036. [Google Scholar]
- Peters, A; Dockery, DW; Muller, JE; Mittleman, MA. Increased particulate air pollution and the triggering of myocardial infarction. Circulation 2001, 103, 2810–2815. [Google Scholar]
- D’Ippoliti, D; Forastiere, F; Ancona, C; Agabiti, N; Fusco, D; Michelozzi, P; Perucci, CA. Air pollution and myocardial infarction in Rome: a case-crossover analysis. Epidemiology 2003, 14, 528–535. [Google Scholar]
- Lin, CA; Amador Pereira, LA; de Souza Conceicao, GM; Kishi, HS; Milani, R, Jr; Ferreira Braga, AL; Nascimento Saldiva, PH. Association between air pollution and ischemic cardiovascular emergency room visits. Environ. Res 2003, 92, 57–63. [Google Scholar]
- Peters, A; von Klot, S; Heier, M; Trentinaglia, I; Cyrys, J; Hormann, A; Hauptmann, M; Wichmann, HE; Lowel, H. Particulate air pollution and nonfatal cardiac events. Part I. Air pollution, personal activities, and onset of myocardial infarction in a case-crossover study. Res Rep Health Eff Inst 2005. [Google Scholar]
- Sullivan, J; Sheppard, L; Schreuder, A; Ishikawa, N; Siscovick, D; Kaufman, J. Relation between short-term fine-particulate matter exposure and onset of myocardial infarction. Epidemiology 2005, 16, 41–48. [Google Scholar]
- Zanobetti, A; Schwartz, J. The effect of particulate air pollution on emergency admissions for myocardial infarction: a multicity case-crossover analysis. Environ. Health Perspect 2005, 113, 978–982. [Google Scholar]
- Lanki, T; Pekkanen, J; Aalto, P; Elosua, R; Berglind, N; D’Ippoliti, D; Kulmala, M; Nyberg, F; Peters, A; Picciotto, S; Salomaa, V; Sunyer, J; Tiittanen, P; von Klot, S; Forastiere, F. Associations of traffic related air pollutants with hospitalisation for first acute myocardial infarction: the HEAPSS study. Occup. Environ. Med 2006, 63, 844–851. [Google Scholar]
- Zanobetti, A; Schwartz, J. Air pollution and emergency admissions in Boston, MA. J. Epidemiol. Community Health 2006, 60, 890–895. [Google Scholar]
- Lokken, RP; Wellenius, GA; Coull, BA; Burger, MR; Schlaug, G; Suh, HH; Mittleman, MA. Air pollution and risk of stroke: underestimation of effect due to misclassification of time of event onset. Epidemiology 2009, 20, 137–142. [Google Scholar]
- Moller, J; Hallqvist, J; Diderichsen, F; Theorell, T; Reuterwall, C; Ahlbom, A. Do episodes of anger trigger myocardial infarction? A case-crossover analysis in the Stockholm Heart Epidemiology Program (SHEEP). Psychosom. Med 1999, 61, 842–849. [Google Scholar]
- Hallqvist, J; Moller, J; Ahlbom, A; Diderichsen, F; Reuterwall, C; de Faire, U. Does heavy physical exertion trigger myocardial infarction? A case-crossover analysis nested in a population-based case-referent study. Am. J. Epidemiol 2000, 151, 459–467. [Google Scholar]
- Moller, J; Ahlbom, A; Hulting, J; Diderichsen, F; de Faire, U; Reuterwall, C; Hallqvist, J. Sexual activity as a trigger of myocardial infarction. A case-crossover analysis in the Stockholm Heart Epidemiology Programme (SHEEP). Heart 2001, 86, 387–390. [Google Scholar]
- Moller, J; Theorell, T; de Faire, U; Ahlbom, A; Hallqvist, J. Work related stressful life events and the risk of myocardial infarction. Case-control and case-crossover analyses within the Stockholm heart epidemiology programme (SHEEP). J. Epidemiol. Community Health 2005, 59, 23–30. [Google Scholar]
- Berglind, N; Bellander, T; Forastiere, F; von Klot, S; Aalto, P; Elosua, R; Kulmala, M; Lanki, T; Lowel, H; Peters, A; Picciotto, S; Salomaa, V; Stafoggia, M; Sunyer, J; Nyberg, F. Ambient air pollution and daily mortality among survivors of myocardial infarction. Epidemiology 2009, 20, 110–118. [Google Scholar]
- Maclure, M. The case-crossover design: a method for studying transient effects on the risk of acute events. Am. J. Epidemiol 1991, 133, 144–153. [Google Scholar]
- Levy, D; Lumley, T; Sheppard, L; Kaufman, J; Checkoway, H. Referent selection in case-crossover analyses of acute health effects of air pollution. Epidemiology 2001, 12, 186–192. [Google Scholar]
- Forastiere, F; Stafoggia, M; Picciotto, S; Bellander, T; D’Ippoliti, D; Lanki, T; von Klot, S; Nyberg, F; Paatero, P; Peters, A; Pekkanen, J; Sunyer, J; Perucci, CA. A case-crossover analysis of out-of-hospital coronary deaths and air pollution in Rome, Italy. Am. J. Respir. Crit. Care Med 2005, 172, 1549–1555. [Google Scholar]
- Rosenlund, M; Berglind, N; Pershagen, G; Hallqvist, J; Jonson, T; Bellander, T. Long-term exposure to urban air pollution and myocardial infarction. Epidemiology 2006, 17, 383–390. [Google Scholar]
- Rosenlund, M; Picciotto, S; Forastiere, F; Stafoggia, M; Perucci, CA. Traffic-related air pollution in relation to incidence and prognosis of coronary heart disease. Epidemiology 2008, 19, 121–128. [Google Scholar]
- Committee on the Medical Effects of Air Pollutants, Cardiovascular Disease and Air Pollution; Department of Health: London, UK, 2006.
- Chen, LC; Nadziejko, C. Effects of subchronic exposures to concentrated ambient particles (CAPs) in mice. V. CAPs exacerbate aortic plaque development in hyperlipidemic mice. Inhal. Toxicol 2005, 17, 217–224. [Google Scholar]
- Donaldson, K; Tran, CL. Inflammation caused by particles and fibers. Inhal. Toxicol 2002, 14, 5–27. [Google Scholar]
- Sun, Q; Wang, A; Jin, X; Natanzon, A; Duquaine, D; Brook, RD; Aguinaldo, JG; Fayad, ZA; Fuster, V; Lippmann, M; Chen, LC; Rajagopalan, S. Long-term air pollution exposure and acceleration of atherosclerosis and vascular inflammation in an animal model. JAMA 2005, 294, 3003–3010. [Google Scholar]
- Suwa, T; Hogg, JC; Quinlan, KB; Ohgami, A; Vincent, R; van Eeden, SF. Particulate air pollution induces progression of atherosclerosis. J. Am. Coll. Cardiol 2002, 39, 935–942. [Google Scholar]
- Mills, NL; Tornqvist, H; Gonzalez, MC; Vink, E; Robinson, SD; Soderberg, S; Boon, NA; Donaldson, K; Sandstrom, T; Blomberg, A; Newby, DE. Ischemic and thrombotic effects of dilute diesel-exhaust inhalation in men with coronary heart disease. N. Engl. J. Med 2007, 357, 1075–1082. [Google Scholar]
- Lucking, AJ; Lundback, M; Mills, NL; Faratian, D; Barath, SL; Pourazar, J; Cassee, FR; Donaldson, K; Boon, NA; Badimon, JJ; Sandstrom, T; Blomberg, A; Newby, DE. Diesel exhaust inhalation increases thrombus formation in man. Eur. Heart J 2008, 29, 3043–3051. [Google Scholar]
- Chuang, KJ; Coull, BA; Zanobetti, A; Suh, H; Schwartz, J; Stone, PH; Litonjua, A; Speizer, FE; Gold, DR. Particulate air pollution as a risk factor for ST-segment depression in patients with coronary artery disease. Circulation 2008, 118, 1314–1320. [Google Scholar]
- Pope, CA, III; Muhlestein, JB; May, HT; Renlund, DG; Anderson, JL; Horne, BD. Ischemic heart disease events triggered by short-term exposure to fine particulate air pollution. Circulation 2006, 114, 2443–2448. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age in years | Current Smoker | 276 / 598 (46%) | |
| Mean (SD) | 60 (7.2) | Previous Smoker | 174 / 598 (29%) |
| Median (range) | 61 (44, 70) | Physical inactivity | 252 / 592 (43%) |
| Age >= 65 years | 214 / 660 (32%) | Hypertension | 209 / 605 (35%) |
| Male | 507 / 660 (77%) | Diabetes | 120 / 605 (20%) |
| BMI >= 30 | 113 / 603 (19%) | Angina | 178 / 595 (30%) |
| Correlation | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Parameter (unit) | n | mean | SD | median | 25% | 75% | PM10 | NO2 | CO | O3 |
| 1 hour | ||||||||||
| PM10 (ug/m3)* | 7,107* | 18.2 | 12.3 | 15.3 | 10.0 | 22.7 | 1.0 | |||
| NO2 (ug/m3) | 15,323 | 26.0 | 14.5 | 23.4 | 14.9 | 34.0 | 0.29 | 1.0 | ||
| CO (mg/m3) | 15,198 | 0.51 | 0.30 | 0.44 | 0.31 | 0.63 | 0.29 | 0.72 | 1.0 | |
| O3 (ug/m3) | 14,618 | 56.8 | 22.8 | 56.4 | 41.1 | 71.8 | 0.31 | −0.18 | −0.16 | 1.0 |
| Temp (°C) | 15,343 | 8.0 | 7.8 | 8.0 | 2.1 | 13.7 | 0.09 | −0.17 | −0.16 | 0.36 |
| Rel. hum. (%) | 15,343 | 70.4 | 17.9 | 74.0 | 57.8 | 85.2 | −0.17 | 0.06 | 0.18 | −0.61 |
| 24-hours | ||||||||||
| PM10 (ug/m3)* | 299* | 18.1 | 9.4 | 15.5 | 11.9 | 21.5 | 1.0 | |||
| NO2 (ug/m3) | 640 | 25.9 | 9.5 | 24.9 | 19.2 | 31.5 | 0.23 | 1.0 | ||
| CO (mg/m3) | 637 | 0.51 | 0.19 | 0.47 | 0.39 | 0.60 | 0.26 | 0.74 | 1.0 | |
| O3 (ug/m3) | 617 | 56.9 | 17.3 | 56.5 | 44.9 | 68.4 | 0.50 | −0.19 | −0.32 | 1.0 |
| Temp (°C) | 640 | 8.0 | 7.4 | 8.3 | 2.2 | 13.6 | 0.10 | −0.26 | −0.32 | 0.33 |
| Rel. hum. (%) | 640 | 70.4 | 14.1 | 71.9 | 59.1 | 82.2 | −0.25 | 0.09 | 0.37 | −0.63 |
| Pollutant | IQR | No. of cases | OR | 95% CI |
|---|---|---|---|---|
| 2-hour average | ||||
| PM10 (ug/m3)* | 12.2 | 342 | 0.93 | (0.81, 1.08) |
| NO2 (ug/m3) | 18.1 | 657 | 0.97 | (0.84, 1.11) |
| CO (mg/m3) | 0.32 | 649 | 0.94 | (0.82, 1.07) |
| O3 (ug/m3) | 29.6 | 629 | 1.02 | (0.85, 1.23) |
| 24-hour average | ||||
| PM10 (ug/m3)* | 9.1 | 341 | 0.99 | (0.85, 1.15) |
| NO2 (ug/m3) | 12.3 | 654 | 0.97 | (0.85, 1.11) |
| CO (mg/m3) | 0.23 | 651 | 0.97 | (0.85, 1.11) |
| O3 (ug/m3) | 24.7 | 631 | 0.92 | (0.75, 1.13) |
| Study center | Study period | N | Mean age | Median age | Male | Prior MI | Prior angina | Diabetes | Hypertension | Current smoker |
|---|---|---|---|---|---|---|---|---|---|---|
| Boston | 1995–1996 | 772 | 62 | - | 63% | 31% | 23% | 19% | 41% | 32% |
| Seattle | 1988–1994 | 5793 | - | 69 | 67% | 31% | 41% | 18% | 49% | 23% |
| Augsburg | 1999–2000 | 691 | 60 | - | 77% | 14% | 24% | 21% | 66% | 36% |
| Stockholm | 1993–1994 | 660 | 60 | 61 | 68% | 0% | 19% | 13% | 36% | 39% |
| Pollutant | City | 1-hour mean | 1-hour IQR | 1-hour 5–95% | Averaging time | Adjusted OR (95% CI)† |
|---|---|---|---|---|---|---|
| PM2.5 | Boston | 12.1 | 27.0 | 2-hour | 1.17 (1.04, 1.32) | |
| Seattle | 12.8 | 10.6 | 2-hour | 1.01 (0.97, 1.05) | ||
| Augsburg | 16.3 | 9.1 | 1-hour | 0.98 (0.87, 1.11) | ||
| PM10 | Boston | 19.4 | 39.2 | 2-hour | 1.11 (1.01, 1.21) | |
| Seattle | 28.3 | 20.5 | ||||
| Augsburg | Same day* | 1.02 (0.97, 1.06) | ||||
| Stockholm | 18.2 | 12.7 | 37.5 | 2-hour | 0.94 (0.84, 1.07) | |
| CO | Boston | 1.3 | 2.1 | 2-hour | 1.02 (0.99, 1.05) | |
| Seattle | 2.3 | 1.6 | 2-hour | 1.00 (1.00, 1.01) | ||
| Augsburg | 0.5 | 0.4 | Same day | 1.00 (0.97, 1.03) | ||
| Stockholm | 0.5 | 0.3 | 0.9 | 2-hour | 0.98 (0.95, 1.02) | |
| NO2 | Boston | 43.2 | 75.2 | 2-hour | 1.01 (0.96, 1.06) | |
| Augsburg | 35.8 | 20.0 | Same day | 1.03 (0.97, 1.10) | ||
| Stockholm | 24.4 | 19.1 | 46.9 | 2-hour | 0.98 (0.91, 1.06) | |
| O3 | Boston | 38.8 | 88.2 | 2-hour | 1.03 (0.98, 1.08) | |
| Augsburg | 43.5 | 48.5 | Same day | 1.00 (0.95, 1.05) | ||
| Stockholm | 56.1 | 30.7 | 74.8 | 2-hour | 1.01 (0.95, 1.07) |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Berglind, N.; Ljungman, P.; Möller, J.; Hallqvist, J.; Nyberg, F.; Rosenqvist, M.; Pershagen, G.; Bellander, T. Air Pollution Exposure—A Trigger for Myocardial Infarction? Int. J. Environ. Res. Public Health 2010, 7, 1486-1499. https://doi.org/10.3390/ijerph7041486
Berglind N, Ljungman P, Möller J, Hallqvist J, Nyberg F, Rosenqvist M, Pershagen G, Bellander T. Air Pollution Exposure—A Trigger for Myocardial Infarction? International Journal of Environmental Research and Public Health. 2010; 7(4):1486-1499. https://doi.org/10.3390/ijerph7041486
Chicago/Turabian StyleBerglind, Niklas, Petter Ljungman, Jette Möller, Johan Hallqvist, Fredrik Nyberg, Mårten Rosenqvist, Göran Pershagen, and Tom Bellander. 2010. "Air Pollution Exposure—A Trigger for Myocardial Infarction?" International Journal of Environmental Research and Public Health 7, no. 4: 1486-1499. https://doi.org/10.3390/ijerph7041486
APA StyleBerglind, N., Ljungman, P., Möller, J., Hallqvist, J., Nyberg, F., Rosenqvist, M., Pershagen, G., & Bellander, T. (2010). Air Pollution Exposure—A Trigger for Myocardial Infarction? International Journal of Environmental Research and Public Health, 7(4), 1486-1499. https://doi.org/10.3390/ijerph7041486