Abstract
Estrogen receptors (ER) are known as nuclear receptors. They exist in the cytoplasm of human cells and serves as a DNA binding transcription factor that regulates gene expression. However the estrogen receptor also has additional functions independent of DNA binding. The human estrogen receptor comes in two forms, alpha and beta. This work focuses on the alpha form of the estrogen receptor. The ERα is found in breast cancer cells, ovarian stroma cells, endometrium, and the hypothalamus. It has been suggested that exposure to DDE, a metabolite of DDT, and other pesticides causes conformational changes in the estrogen receptor. Before examining these factors, this work examines the protein unfolding from the antagonist form found in the 3ERT PDB crystal structure. The 3ERT PDB crystal structure has the estrogen receptor bound to the cancer drug 4-hydroxytamoxifen. The 4-hydroxytamoxifen ligand was extracted before the simulation, resulting in new conformational freedom due to absence of van der Waals contacts between the ligand and the receptor. The conformational changes that result expose the binding clef of the co peptide beside Helix 12 of the receptor forming an apo conformation. Two key conformations in the loops at either end of the H12 are produced resulting in the antagonist to apo conformation transformation. The results were produced over a 42ns Molecular Dynamics simulation using the AMBER FF99SB force field.
Introduction
The structure and chemistry of the estrogen receptor has been of extreme research focus for several years, due to the receptors role as a DNA binding transcription factor, which regulates gene expression. There are two forms of the estrogen receptors: form alpha and form beta. Both forms have been linked to cancer of the breast, and are believed to result in human development issues. [1] In fact, in recent years there have been growing apprehensions about environmental chemicals that disrupt oestrogenic signaling and negatively affect reproduction in humans and in wildlife [1]. Interestingly, both forms of estrogen have been linked to these types of problems. Each form is similar to the other. In general the Estrogen receptor (ER) has three domains, the DNA binding domain (DBD), the ligand binding domain (LBD), and the transactivational domain [2]. The ligand binding domain maintains a particular ligand specificity and functionality based on which ligand is bound. In the case of estradiol the ER takes on the transcriptional active conformation, and in the case of 4-hydroxytamoxifen an antagonist conformation with Helix 12, H12, laying in the co-activator binding pocket preventing initiation of a series of molecular events that culminate in the activation or repression of target genes [4, 5, 6]. We use the 3ERT PDB structure [2, 3] in the antagonist form, extract the ligand, and run MD simulations to watch the ER unfold into a conformation similar to the agonist structure. This conformational change is important due to ER implications in cancer research, birth defects [3], and potential deleterious effects to the ER from exposure to pesticides [4].
Celik et. al. showed a conformational change from a structure derived from a crystal defect (1A52 PDB) termed apo translated into an antagonist conformation. Also, and more importantly the work of Celik et al. support the hypothesis of a zipper mechanism where a hydrogen bond between the alpha hydroxy group of estradiol and a Histidine group at the top of the bonding pocket in the estrogen receptor predicated a Glutamic acid –Asparagines water mediated hydrogen bond at the bottom of the binding pocket; thus the zipper mechanism [7].
In this work we will show the antagonist conformer move towards a different apo conformer [7]. The extraction of the 4-hydroxytamoxifen ligand removes van der Waals interactions between the binding pocket and the ligand. Previous simulations by other groups are not sufficiently long enough to see the migration of key components of the estrogen receptor alpha LBD that is evident in our 42ns simulation. This work has impetus in protein folding, drug discovery, in particular selective estrogen receptor modulators, and in the environmental impact on the oestrogen response system.
Methods
We use molecular dynamic simulations to examine the unfolding of the estrogen receptor alpha beginning from the crystal structure 3ERT from the protein data bank. The 3ERT structure is the antagonist form of the estrogen receptor. After extracting the bound, 4-Hydroxytamoxifen, ligand, we reproduced the Histidine, HIS, transformations to HIE (E-ɛ position) and HID (D-δ position) found in the work of Celik et. al. [7]. These transformations would normally allow for the hydrogen bonding found between a hydroxyl group of the ligand and the histidine. We used the AMBER FF99SB [8, 9] force field that was parameterized for DNA double helices. This is important for protein such as ER alpha because of the large number of helices that it contains. A total of twelve of these helices make up the majority of the structure. Therefore, maintaining the integrity and proper interactions between helices is essential. Most molecular mechanics force fields start with five basic terms and are modified with some adaptations as shown in equation 1.
The first two terms show hook’s law approximations of the bond stretching, and bending from an accepted value of bond length and bond angle, usually determined by experiment or through quantum calculations. The third term expresses the periodic nature of the bond rotation energy or torsional energy. The last two terms are the non-bonding terms. The first is the Leonard Jones potential between two non-bonded atoms. The last of the non-bonded terms models the coulombic interaction of charged atoms separated by some distance r. We used Kollman charges in this work [8, 9]. Using the force calculated from the forcefield we are able to determine the force on the atoms in the system using equation (2):
From there the acceleration, new velocity and then new position of each of the atoms in the system could be determined by integrating Newton’s equation of motion. In this way, the timed evolution of atomic movement molecular dynamic trajectories could be recorded like a movie.10,11 The system studied in this work consisted of the estrogen receptor from the 3ERT crystal structure with the complexed ligand extracted, and changes to Histidine residues were made as mentioned above. Also, the system was solvated with 15212 octahedron waters. The solvation shell was constructed in an octahedron shape in order to maximize computational efficiency. All 79-crystal structure waters were kept for the simulation. Isobaric-Isothermal ensemble molecular dynamics [10–12] were performed on the system using a periodic boundary box with dimensions of 102 Angstroms on each side. The periodic boundary conditions were chosen to insure that the H12 helix would not extend itself beyond the periodic box during simulation while exploring the conformational space. In Figure 1, the solvated 3ERT agonist starting structure of the ERα LBD is depicted absent hydrogen atoms. The ligand has been extracted from the binding pocket; leaving a large space along the loop starting helix 12, H12.
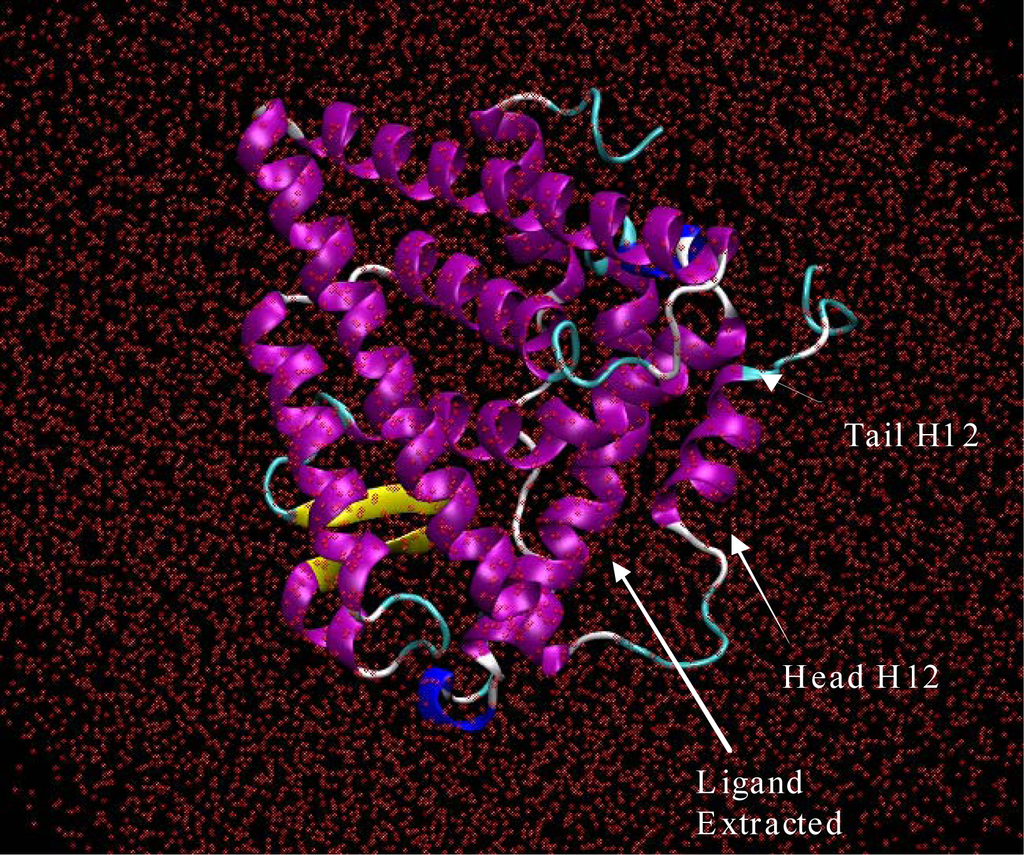
Figure 1.
Era from the 3ERT PDB crystal structure with the 4-Hydroxytamoxifen extracted from the binding pocket. The system was solvated with 15212 water molecules, shown here without hydrogen atoms. This image was produced using VMD software [13].
This space was solvated by the water molecules using a protocol to relax the crystal waters and the solvation waters into the unoccupied crevasses of 3ERα LBD. The protocol included a series of molecular mechanics energy minimizations and molecular dynamic simulations before the production runs, where trajectories were obtained for dynamic data analysis (not shown here). Initial minimizations were run for 1000 ps to allow water molecules and protons to minimize while the heavy atoms are restrained. Each of these equilibration cycles are continued in succession, reducing the restraints systematically, eventually continued with temperature and pressure coupling to insure the ensemble variable controls are maintained. Final molecular dynamics runs are conducted with only backbone restraints at 2fs time-steps before all the restraints are eliminated with snapshots every 2fs. Thereafter, we produced the production run of 42 ns, which is discussed in this work. In the section that follows we briefly discuss large-scale changes in the antagonist structure to that of the agonist during the 42ns run.
Results
Here we briefly discuss large changes in the structure of our ERα system from the antagonist 3ERT structure at the start of the simulation to the structure, an apo form, resembling the agonist structure at the end of the 42ns simulation. In Figures 2 and three we show the structure of our ERα system color coded with three colors. The blue is the structure at the beginning of the simulation, red at about 20ns into the simulation, and yellow at the end of the 42ns simulation. The largest changes do happen very quickly. However, in previous work, simulations were only about 5ns for any estrogen receptor. This is not long enough to see the extent of dynamics that are present in our simulation.
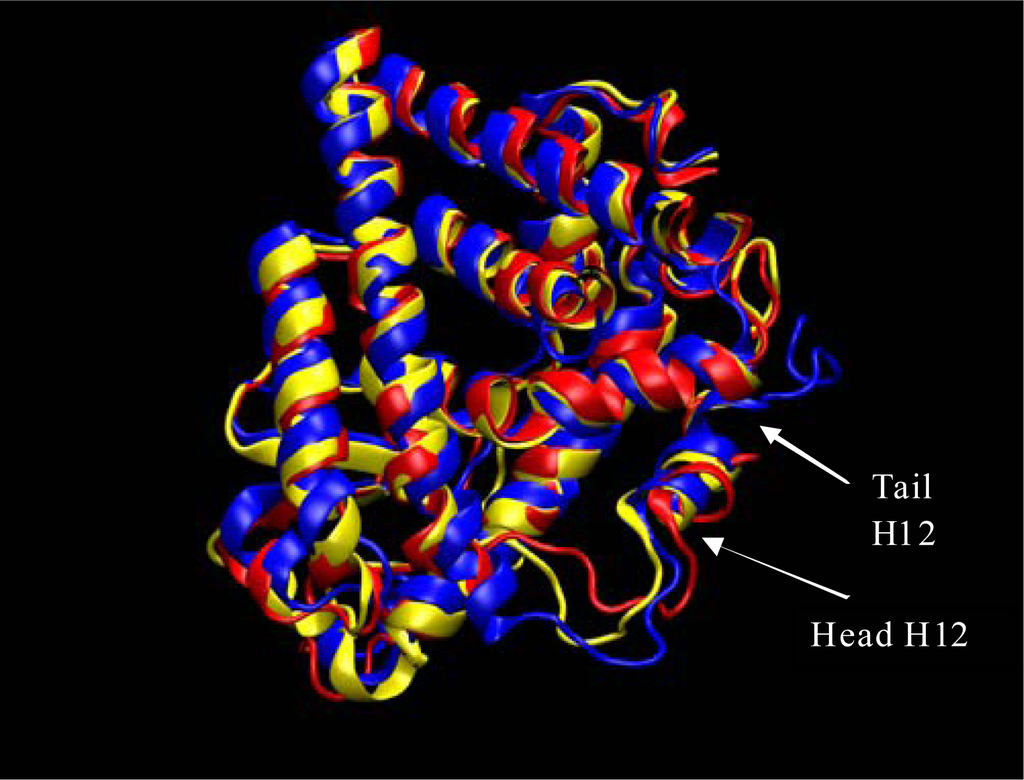
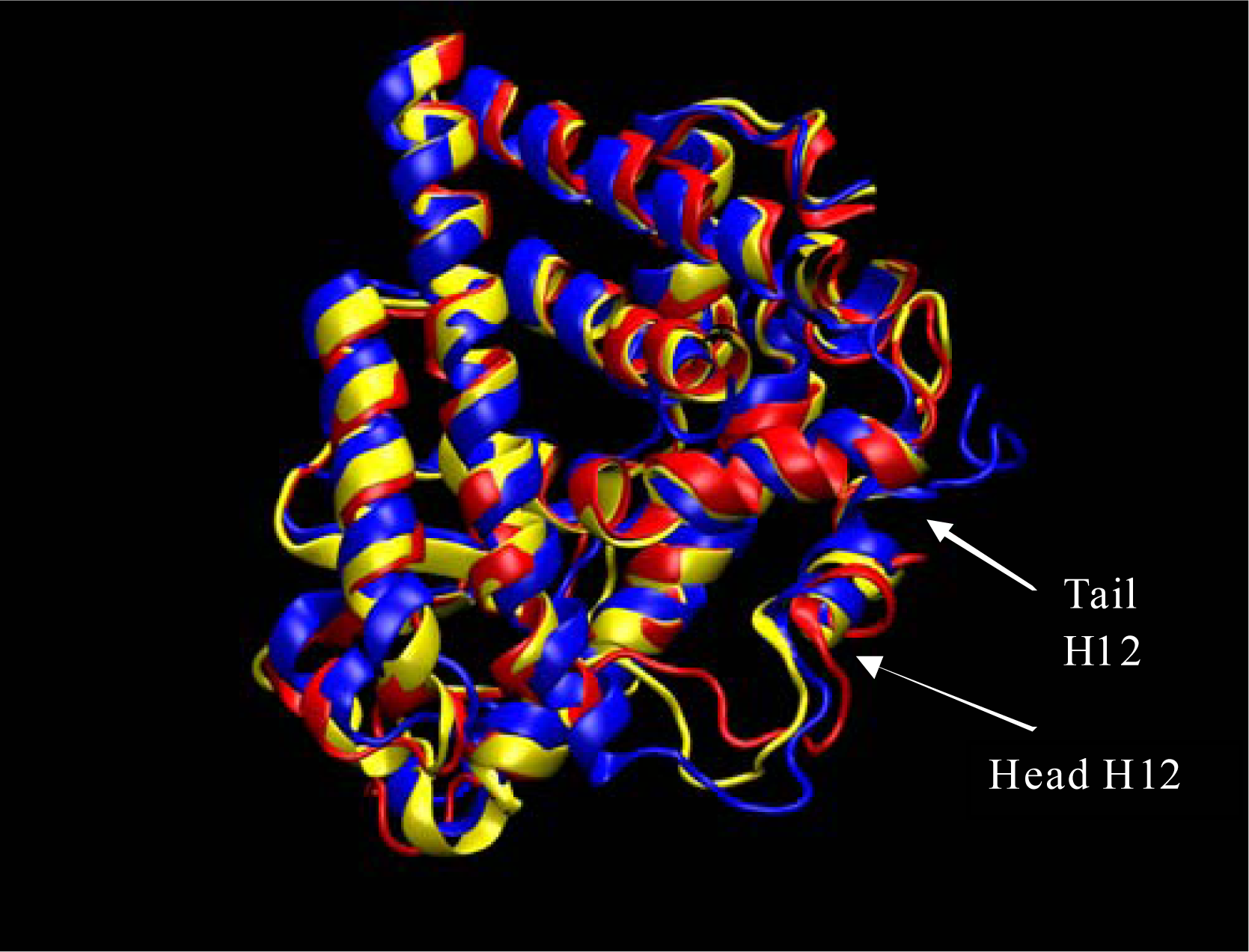
Figure 2.
ERα simulation initial structure (blue), after about 20ns (red), and 42 ns yellow. The loop region at the head of Helix 12 is fluctuating due to the extraction of the ligand (4-Hydroxytamoxifen) that would have protruded out of the binding pocket. This image was produced from our Amber results using VMD software [13].
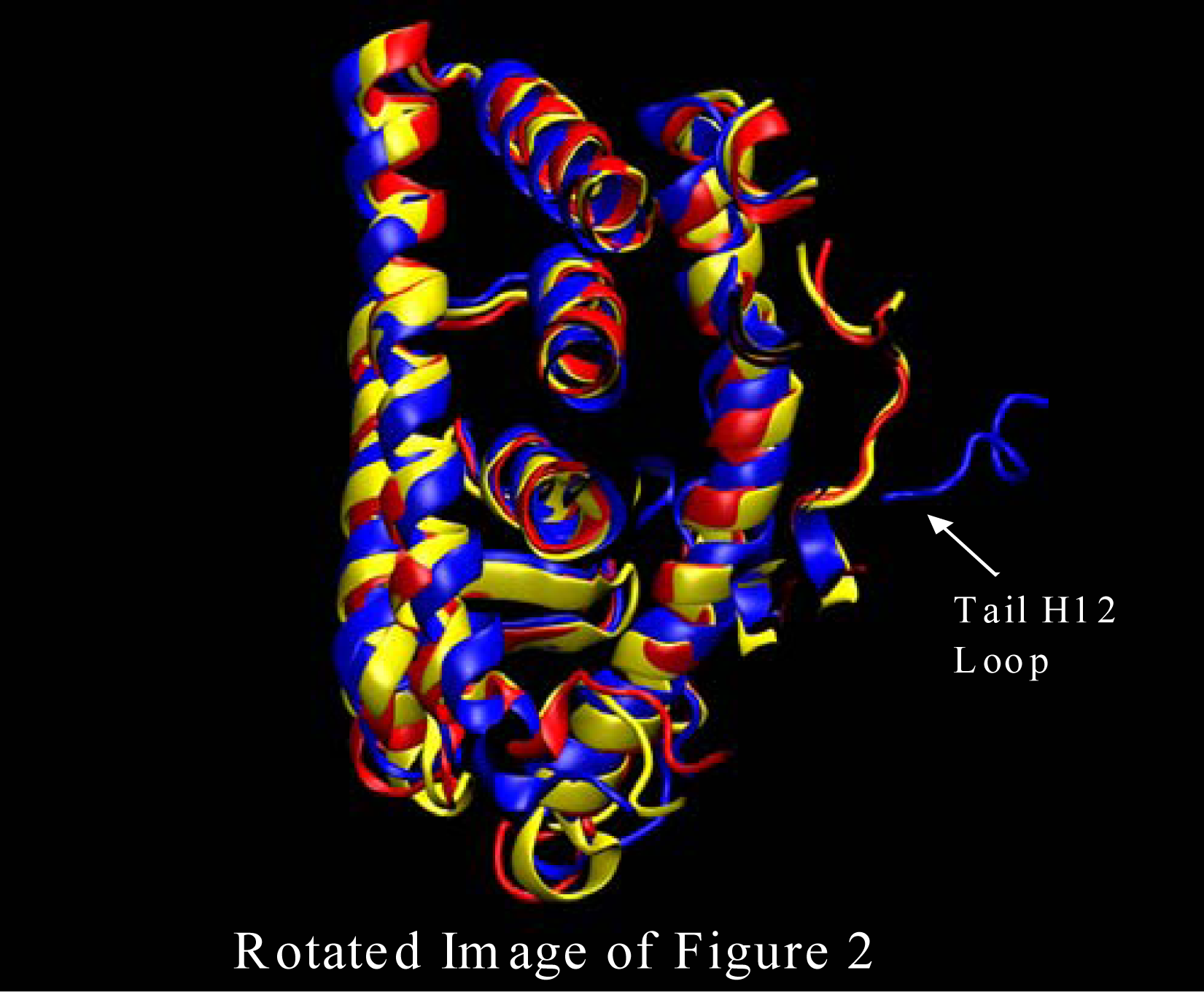
The first major structural change is shown in Figure 2 where the ERα LBD dynamic simulation shows the initial structure in blue, the structure after about 20ns in red, and the 42 ns final structure in yellow. The loop region at the head of Helix 12 is fluctuating due to the extraction of the ligand (4-Hydroxytamoxifen) that would have protruded out of the binding pocket. Without the van der Waals interaction at the opening of the binding pocket the loops at the head of the H12 are free to migrate freely as is shown in Figure 2. Also, shown in Figure 3 the loop at the tail end of helix 12 endures some major changes as well. Once again, the initial structure is in blue, the structure after about 20ns is color coded in red, and the final structure after 42 nanoseconds is in yellow. As can be seen from Figure 3 the tail loop migrates towards helix 9 of the ERα receptor and is maintained there by interactions with the residues in that helix. This all happens within 20ns. This is evident due to the red and yellow structure possessing loops in the tail region of helix 12 in approximately the same region.
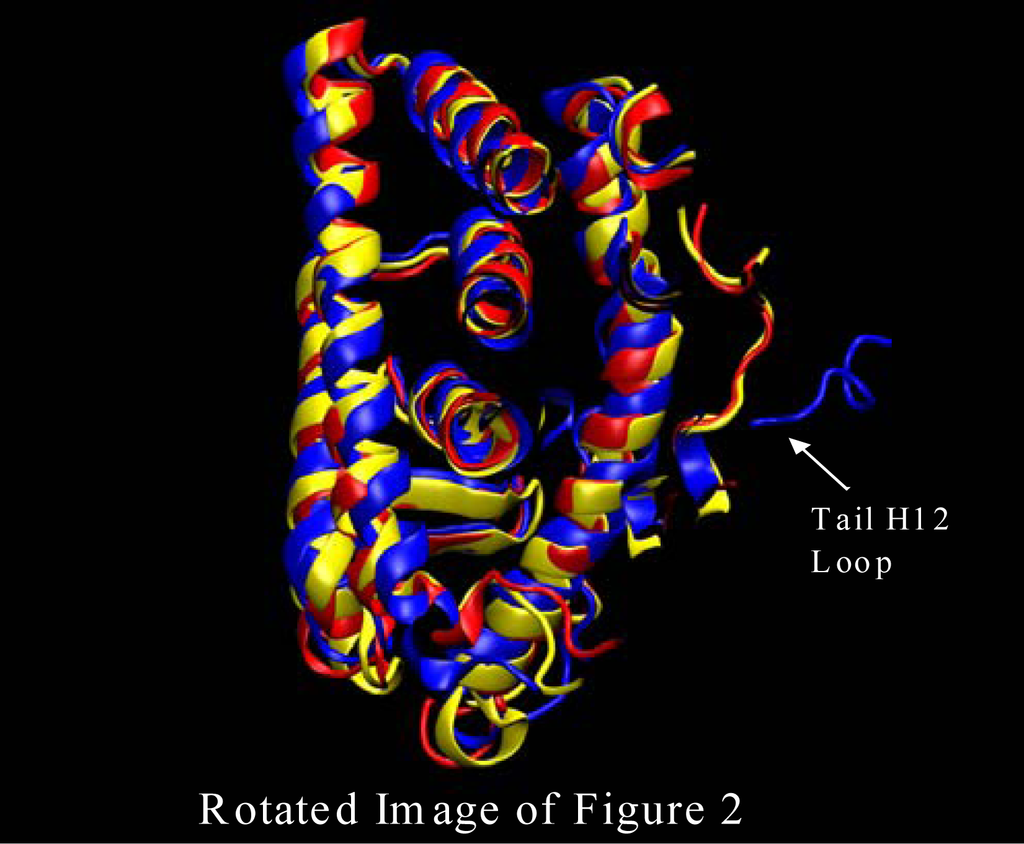
Figure 3.
ERα simulation initial structure (blue), after about 20ns (red), and 42 ns yellow. The loop region at the head of Helix 12 is fluctuating due to the extraction of the ligand (4-Hydroxytamoxifen) that would have protruded out of the binding pocket. This image was produced from our Amber results using VMD software [13].
Conclusions
The molecular dynamics results here show that van der Waals interactions with the complexed ligand are necessary to hold the ERα LBD in the antagonist position. Once the ligand is removed, helix 12 begins to fluctuate in the loop region at the head of the helix. Directly after the fluctuations in the loop region at the head of the helix, the loop region at the tail of the helix begins to moves into a fairly stable position in close proximity to H9. This movement exposes the binding cleft for the co peptide found to bind to the agonist structure of the ERα receptor. Our preliminary work show that longer simulation times (longer than 5ns) are necessary to see the migration of the H12 and the loops associated with the helix. This work shows a potentially stable apo structure not yet shown in previous works. We will continue these simulations to ensure the stability of this apo structure over time and elucidate the reason for the stability.
Acknowledgments
This work was conducted with the support from the National Oceanic and Atmospheric Administration (NOAA) Climate and Global Change program, CFDA Number: 11.431, the National Institutes of Health RCMI program grant number G12 RR 03020, a National Science Foundation C.R.E.S.T. grant, (number 0630270), and support from the SEAGAP program at the University of Florida. We also appreciate the support provided by the staff of the Quantum Theory Project at the University of Florida.
References
- Sumbayev, VV; Bonefeld-Jørgensen, EC; Wind, T; Andreasen, PA. A novel pesticide-induced conformational state of the oestrogen receptor ligand-binding domain, detected by conformation-specific peptide binding. FEBS Lett 2005, 579, 541–548. [Google Scholar]
- Shiau, AK; Barstad, D; Loria, PM; Cheng, L; Kushner, PJ; Agard, DA; Greene, GL. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 1998, 95, 927–937. [Google Scholar]
- Tanenbaum, DM; Wang, Y; Williams, SP; Sigler, PB. Crystallographic comparison of the estrogen and pogesterone receptor’s ligand binding domain. Proc Nat Acad Sci USA 1998, 95, 5998–6003. [Google Scholar]
- Laak, AM; Jongejan, A; Vermeulen, NP; Wamelink, M; Geerke, D; Meerman, JH. Prediction of ligand binding affinity and orientation of xenoestrogens to the estrogen receptor by molecular dynamics simulations and the linear interaction energy method. J Med Chem 2004, 47(4), 1018–30. [Google Scholar]
- Nilsson, S; Makela, S; Treuter, E; Tujague, M; Thomesen, J; Andersson, G; Enmark, E; Pettersson, K; Warner, M; Gustafsson, JA. Mechanisms of Estrogne action. Physiol Rev 2001, 81, 1535–1565. [Google Scholar]
- McKenna, NM; O’Malley, BW. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell 2002, 108, 465–474. [Google Scholar]
- Celik, L; Lund, JDD; Schiøtt, B. Conformational dynamics of the Estrogen Receptor a: Moleulcar Dynamics Simulations of the Influence of Binding Site Structure on Protein Dynamics. Biochemistry 2007, 46, 1743–1758. [Google Scholar]
- Case, DA; Cheatham, TE, III; Darden, T; Gohlke, H; Luo, R; Merz, KM, Jr; Onufriev, A; Simmerling, C; Wang, B; et al. The Amber biomolecular simulation programs. J Comput Chem 2005, 26, 1668–1688. [Google Scholar]
- Pearlman, DA; Case, DA; Caldwell, JW; Ross, WR; Cheatham, TE, III; DeBolt, S; Ferguson, D; Seibel, G; Kollman, P. AMBER, a computer program for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to elucidate the structures and energies of molecules. Comp Phys Commun 1995, 91, 1–41. [Google Scholar]
- Tuckerman, ME; Matyna, GJ. Understanding modern molecular dynamics: Techniques and applications. J Phys Chem B 2000, 104, 159–178. [Google Scholar]
- Martyna, GJ; Tobias, DJ; Klein, ML. Constant-pressure molecular dynamic algorithms. J Chem Phys 1994, 101, 4177–4189. [Google Scholar]
- Berendsen, HJC; Postma, JPM; van Gunsteren, WF; Dinola, A; Haak, JR. Molecular dynamics with coupling to an external bath. J Chem Phys 1984, 81, 3684–3690. [Google Scholar]
- Humphrey, W; Dalke, A; Schulten, K. VMD - visual molecular dynamics. J Molec Graphics 1996, 14, 33–38. [Google Scholar]
© MDPI. All rights reserved