Introduction
Cd is frequently used in various industrial activities and is a ubiquitous environmental toxicant, also present in tobacco smoke. An important route of exposure is the circulatory system whereas blood vessels are considered to be main stream organs of Cd toxicity. Cd exposure via the respiratory system has been studied [
1] and it has been reported that Cd causes apoptosis in various cell types
in vitro [
2]. We have shown that CdCl
2 increases blood pressure in both SHR and WKY rats and chronic exposure of VSMCs of rats with CdCl
2 leads to apoptosis (unpublished results). However, the mechanism involved in the elevation of blood pressure and the apoptotic effects of CdCl
2 has not been determined.
The evidence indicates that the mitogen-activated protein kinase (MAPK) cascade is involved in the regulation of cell growth, differentiation, and various cellular stress responses which is mediated through intracellular signal transduction in response to a variety of stimuli [
3,
4]. More recently, MAPK has been studied in response to other stresses [
5–
7] to gain an understanding of the MAPK signaling mechanisms, specifically, in cardiovascular disorders [
8,
9].
Also, the cellular mechanisms involved in triggering and development of atherosclerosis induced by Cd may include MAPKs and other signal transduction systems, such as protein kinase C (PKC). Since the discovery that PKC is the high affinity intracellular receptor of phorbol esters, it is widely accepted that PKC plays a role in the regulation of proliferation and differentiation [
10,
11]. As these processes involve nuclear regulatory control, it is thought that PKC must be involved in mediating nuclear responses to both mitogenic and differentiation factors [
12,
13]. In fact, substantial evidence indicates a direct role for PKC in linking cell membrane receptor binding events to responses at the genome level.
It is our hypothesis that CdCl2 alters the calcium transient mechanism through cadmium-induced stimulation of MAPK (ERK 1 & 2) which is mediated partially through calcium-dependent PKC mechanism. In this study we are providing evidence to support this hypothesis by reporting the results of specific MAPKs and PKC-a/β activation following stimulation with CdCl2 in VSMCs from a hypertensive phenotype. The results indicate that ERK 1 and 2 along with PKC-a/β are reduced or inhibited within 5 min of stimulation in SHR cells which may be the results of Cd competing with intercellular calcium ([Ca2+]i) in activation of the pathways for these intermediates.
Materials and Methods
Materials
Dulbecco’s Modified Eagle’s medium was obtained from Cambrex BioScience (Walkersville, MD), fetal bovine serum, and penicillin/streptomycin, both purchased from Biowhittaker (Walkersville, MD). For experimental procedures, CdCl2 was obtained from Sigma Chemical Company (St. Louis, MO) along with the calcium chelator, BAPTA-AM. The PKC inhibitor, GF109203X, was purchased from Tocris (Ballwin, MO). SDS-Polyacrlymide gels (precast) were ordered from BioRad Laboratories (Hercules, CA) and nitrocellulose membrane from Amersham Biosciences (Piscataway, NJ). Primary antibodies for MAPK and Protein Kinase C-a/β were ordered from Cell Signaling (Beverly, MA) and used for western blotting analysis. To detect and image the primary MAPK and PKC primary antibodies, Anti-rabbit IgG secondary antibodies (horseradish peroxidase linked) was obtained from Amersham Bioscience (Piscataway, NJ) and chemiluminescent (ECL) detection system was obtained from Pierce Biotechnology (Rockford, IL).
Cell Culture and Treatment
Vascular smooth muscle cells were obtained from Dr. Evangeline Motley (Meharry Medical College) and cultured in DMEM containing 10% fetal calf serum, penicillin, and streptomycin. Sub-cultured VSMCs from passages 3–12, used in the experiments, were seeded into conventional plastic tissue culture plates (Falcon) and cultured in DMEM containing 10% fetal calf serum in a humidified atmosphere of 5% CO2 and O2. CdCl2 was prepared in DMEM at 50 mM just before application. After cells were confluent, cells were incubated for the desired time periods with desired concentrations of CdCl2.
Western Blot Analysis of MAPK and PKC Expression
Cells grown in 6 well plates were stimulated in the presence or absence of the desired concentrations of CdCl2 at 37ºC for specified durations. The reactions were terminated by the replacement of medium with 100 μl of SDS-polyacrylamide gel electrophoresis buffer, pH 6.8, containing 62.5 mM Tris-HCl, 2% SDS, 10% glycerol, 50 mM dithiothreitol, and 0.1% bromophenol blue. Samples were sonicated briefly, followed by a one minute flash spin. Samples were then boiled for three min and centrifuged at 10,000 RPM for 5 min and supernatant (20 μg) was subjected to a 10% SDS-polyacrylamide gel. Proteins in the gel were transferred to a nitrocellulose membrane using the semi-dry transfer method. The membrane was then blocked for non-specific binding for one hr with 5% non-fat dry milk. Primary antibodies specific for ERK 1 & 2 and PKC-a/β were gently rocked at 4ºC over the membrane overnight and washed before the membrane was submerged in the secondary antibody for one hr. After washing, the membrane was incubated with secondary anti-rabbit antibodies and immunoreactive proteins were detected by the ECL.
Cell Viability
VSMCs viability was assessed by using trypan blue exclusion. Briefly, the cells were plated in wells of 6 –well cluster plate and grown to confluence for 48 hr. After treatment with CdCl2, the cells were harvested by trypsin digestion, centrifuged at 1200 × g for 10 min at 4ºC, and examined by adding an equivalent volume of a 0.4% trypan blue solution to an aliquot of the re-suspended cells. The re-suspended cells were incubated for 5 min and the stained and unstained cells were counted by means of a hemocytometer.
Statistics
Each experiment was repeated at least 3 times. The effects of cadmium or antagonists on MAPK and PKC expression were plotted using a statistic program, prism (GraphPad Software, San Diego, CA). We performed densitometric analysis of images from Western blotting with a molecular imaging system (BioRad). The results are the mean±SEM and compared by ANOVA of Student-t test where appropriate. A value of p<0.05 was considered significant.
Results
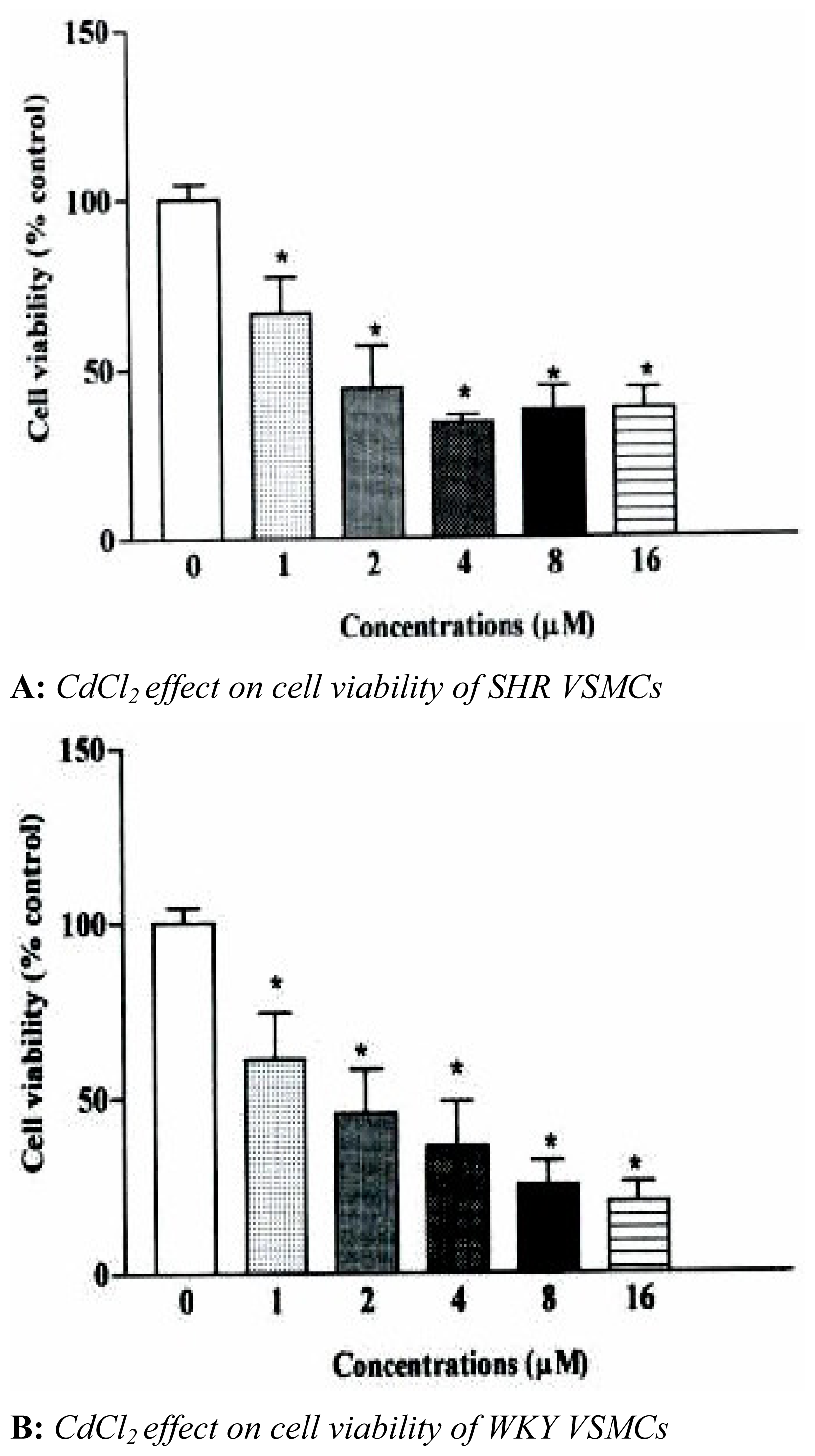
The effects of CdCl
2 on VSMCs viability are shown in
figure 1. CdCl
2 (1–16 μM) exposure of SHR and WKY VSMCs for 24 hr resulted in a statistically significant concentration-related decrease in cell viability for both cell types. VSMCs viability for both cell types decreased 33 and 39%, respectively, when cells were treated with 1 μM of CdCl
2. A significant decrease in cell viability was observed between the two cell types of 6%. Maximum decreases in cell viability observed after cells were treated with concentrations of CdCl
2 (8 and 16 μM) were 66±3.1 and 62±4.5% for SHR cells. This decrease in cell viability was observed to be significantly higher in WKY cell types, 63±1.5 and 75±5.0%, respectively.
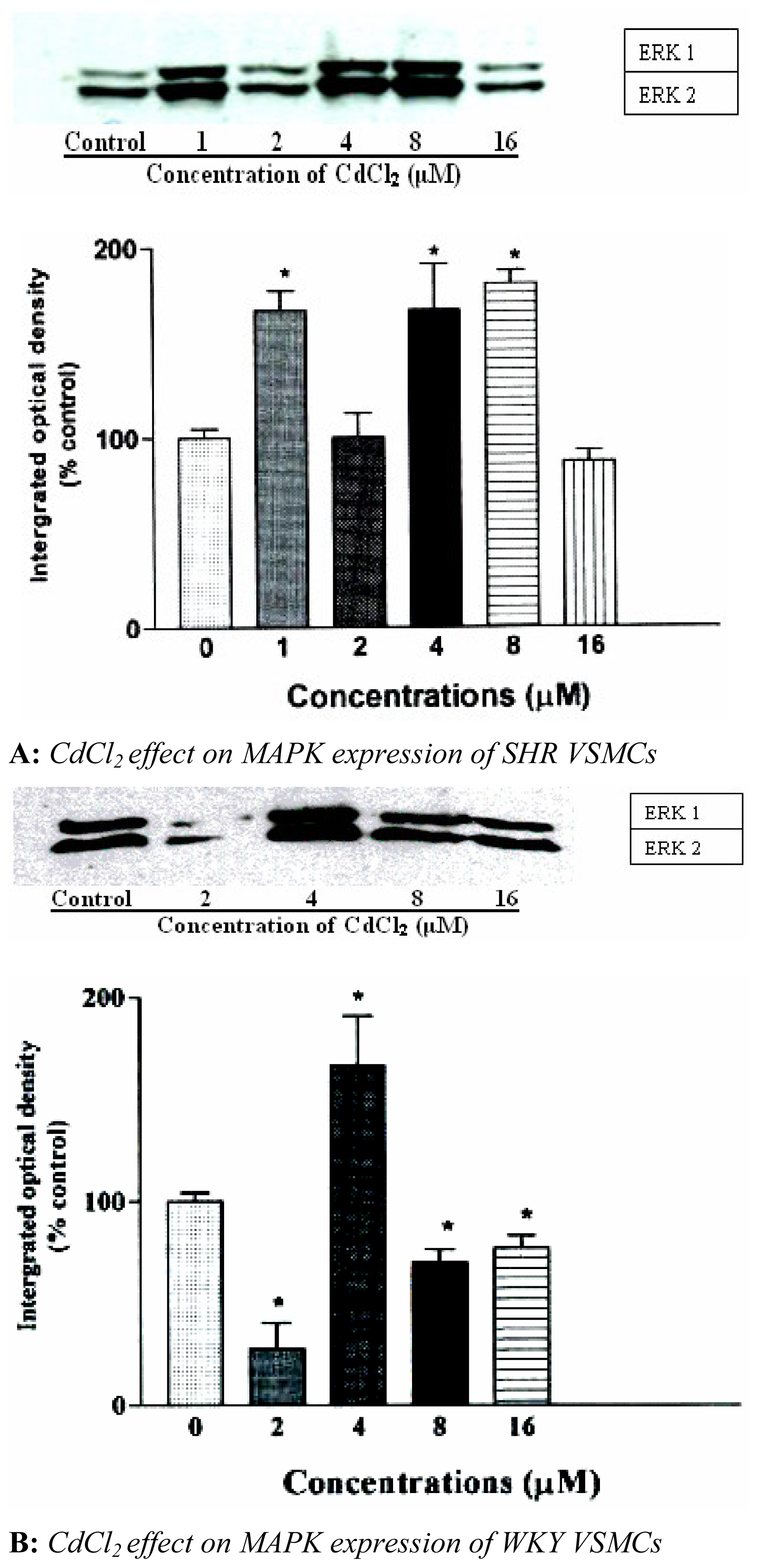
We also investigated the effect of CdCl
2 on expression of ERK 1 and 2. The results indicated that CdCl
2 activated ERK 1 and 2 in a biphasic manner in VSMCs of SHR. Maximum activation of ERK 1 and 2 was observed with 1 and 4 μM of CdCl
2 with 8 μM having similar effects on MAPK expression as that was observed with 4 μM. Expression of ERK 1 and 2 increased 80±3.6 and 85±4.2%, respectively, of control and was suppressed with 2 and 16 μM of CdCl
2 shown in
figure 2A. Treating WKY VSMCs with similar concentrations of CdCl
2, expression of ERK 1 and 2 was initially reduced with 2 μM, whereas, 4 μM increased expression by 77±5.1% of control shown in
figure 2B. It was also observed that 16 μM reduced expression of ERK 1 and 2 in a similar manner as that observed when VSMCs of SHR were treated with similar concentrations of CdCl
2.
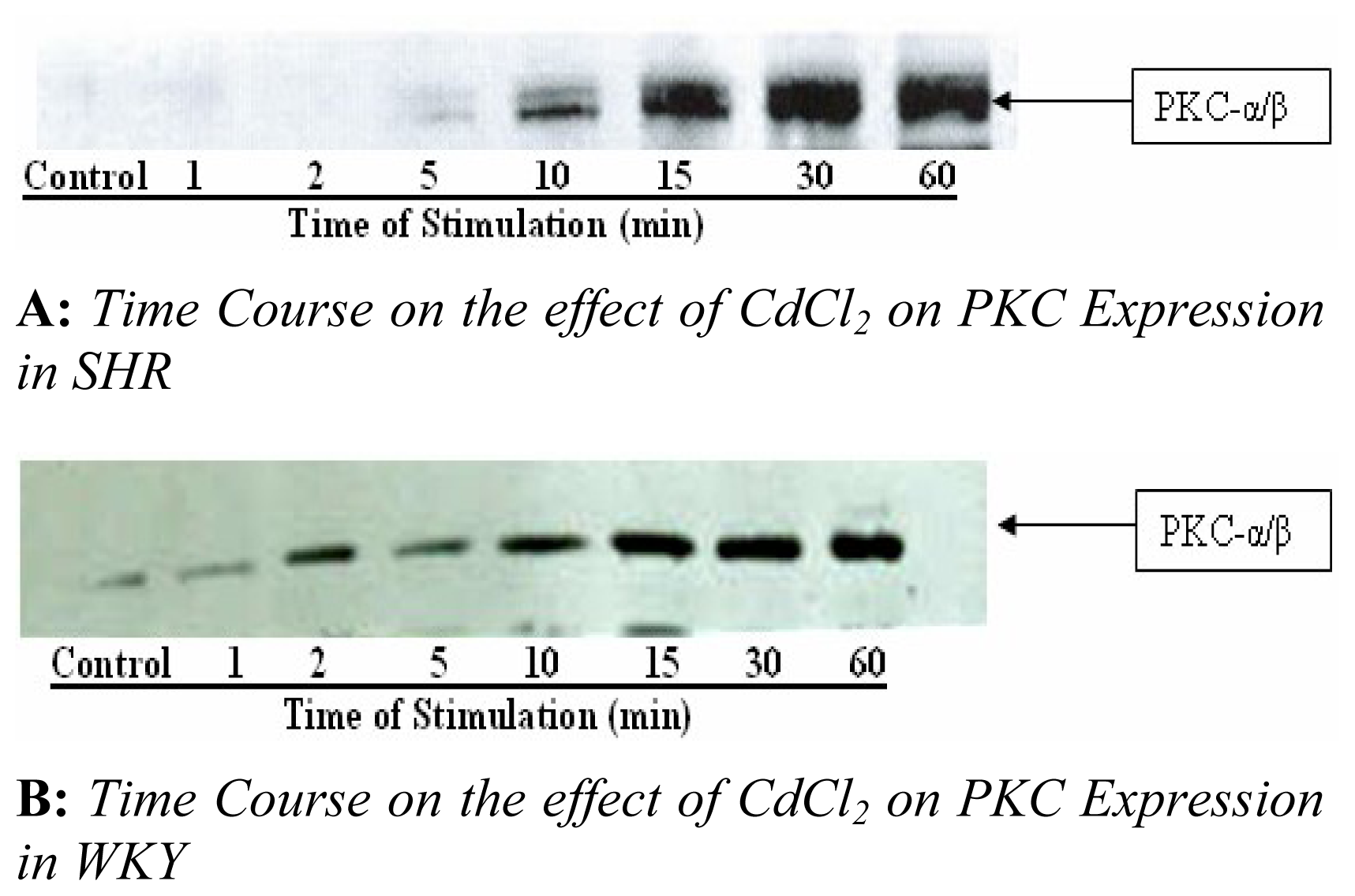
The time of activation of ERK 1 and 2 in VSMCs with 4 μM CdCl
2 is shown in
figure 3A and 3B. The expression of ERK 1 and 2 was suppressed for the first 2 min in SHR VSMCs. Five (5) min of exposure seem to reduce expression which was followed by a time-dependent increase in expression at 15 min in SHR VSMCs shown in
figure 3A. This increase remained present up to 60 min of exposure, whereas, the expression of ERK 1 and 2 increased dramatically within 5 min after exposing WKY VSMCs to CdCl
2. Maximum expression of ERK 1 and 2 in WKY cells was observed within 5 min of exposure followed by a reduction in ERK 2 for 10–60 min shown in
figure 3B.
Next, we examined the effect of CdCl
2 on PKC expression in both SHR and WKY VSMCs shown in
figure 4A and 4B. CdCl
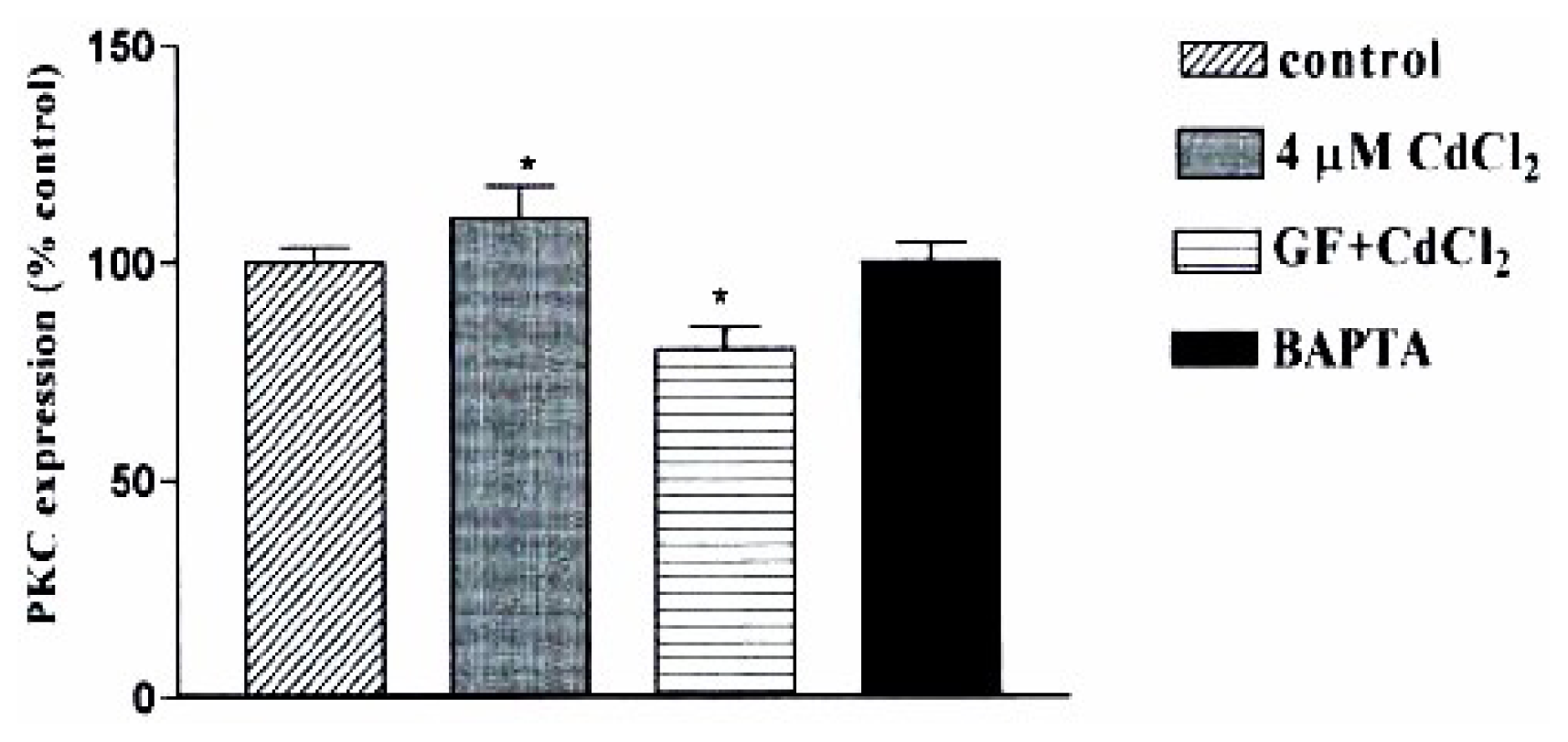
2 increased PKC–a/β expression in a concentration and time dependent manner with maximum expression observed within 15 min of exposing SHR cells to 4 μM, whereas, maximum expression of PKC- a/β was obtained in WKY cells when cells were exposed 30 min or longer. We also investigated whether the [Ca
2+]
i and PKC pathway are link to the CdCl
2 effect observed in SHR cells. GF109203X, the PKC inhibitor, reduced the CdCl
2 induced-effect by 32±2.5% whereas BAPTA suppressed the CdCl
2 effect which is shown in
figure 5.
Discussion
It has also been shown that Cd exposure rapidly increases inositol 1, 4, 5,-triphophate and then triggers Ca
2+ mobilization in various cell types [
14,
15]. Also, it has been shown that pre-treatment with an intracellular Ca
2+ chelator, BAPTA/AM, suppresses CdCl
2 induced MAPKs in renal epithelial and CCRF-CEM cells [
16]. In the present study, we examined the effect of CdCl
2 on MAPK and PKC expression in VSMCs of SHR. The results indicate that CdCl
2 decreased cell viability of both SHR and WKY cells in a concentration dependent manner. Furthermore, it appears that CdCl
2 activates ERK 1 & 2 in a biphasic manner in SHR VSMCs and that PKC a/β expression is more sensitive to CdCl
2 effects than what was observed in WKY VSMCs. Also, CdCl
2 induced effect is suppressed with the intracellular Ca
2+ chelator, BAPTA and is reduced significantly with the PKC inhibitor, GF109203X which may indicate that the mediated effect of Cd is link to the mobilization of calcium through the MAPK and PKC pathways which share a role in the proprogating of the signal generated by Cd.
It is also known that MAPKs mediate intracellular signal transduction in response to a variety of stimuli [
3,
4]. The MAPK family members are themselves activated by reverse dual phosphorylation on the Thr and Tyr residues in the catalytic domain. Three major MAPKs have been identified, the extra cellular signal-regulated kinases (ERK1 and 2), the stress-activated protein kinases c-Jun NH2-terminal kinases (JNK) and p38 mitogen activated protein kinases. It seems that Cd exposure rapidly increases inositol 1, 4, 5,-triphosphate and then triggers Ca
2+ mobilization in various cell types [
14,
15] and that pre-treatment with an intracellular Ca
2+ chelator, BAPTA, suppresses CdCl
2-induced JNK. The results from these studies showed that intracellular Ca
2+ plays a role in CdCl
2-induced activation of ERK, JNK, and 38 MAPK in CCRF-CEM cells [
17].
We previously observed that in hypertensive rat, Cd increased blood pressure and heart rate. Also Cd exposure has been implicated in Hypertension and atherosclerotic lesion [
18,
19]. The frequencies of ten diseases in a Cd-contaminated region in the Netherlands [
20] were investigated and it was found that a significantly higher frequency for atherosclerosis occurred which indicated a relationship between Cd exposure and cardiovascular disease epidemiologically [
21] and in Cd exposed pigeons, not only hypertension but also atherosclerotic lesion was observed [
18].
In conclusion, it was found that Cd can decrease cell viability and can activate MAPKs and PKC expression in SHR VSMCs. An intracellular calcium-dependent pathway is suggested to be involved in the Cd mediated effect which may lead to its contribution to hypertension through the dysfunction of VSMCs.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




