
Wastewater Metavirome Diversity: Exploring Replicate Inconsistencies and Bioinformatic Tool Disparities

 , ,
, ,  , , , , , , ,
, , , , , , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
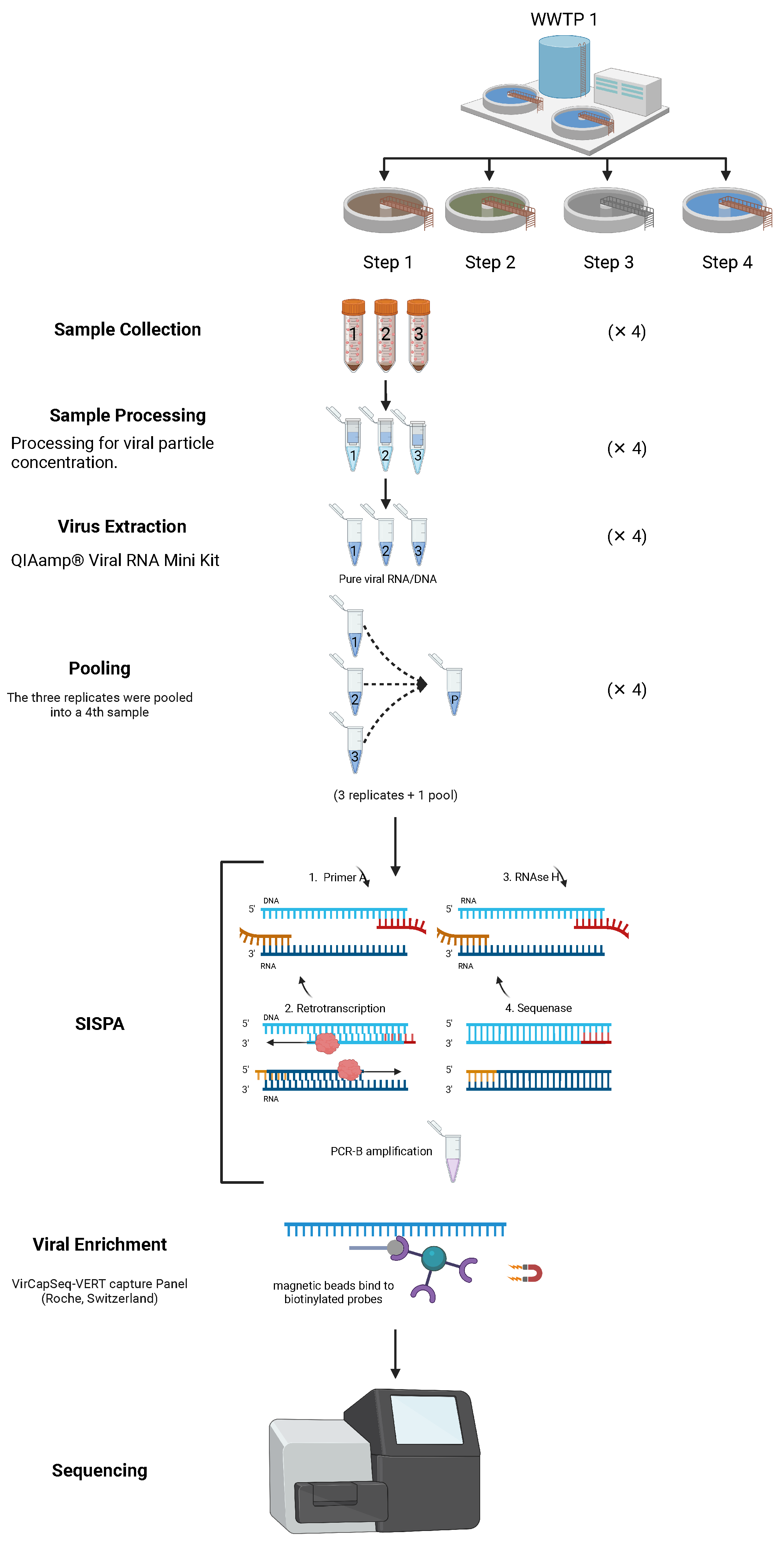
2.1. Wastewater Collection, Processing and Nucleic Acid Extraction
2.2. Sequence-Independent Single-Primer Amplification (SISPA), Viral Enrichment and Sequencing
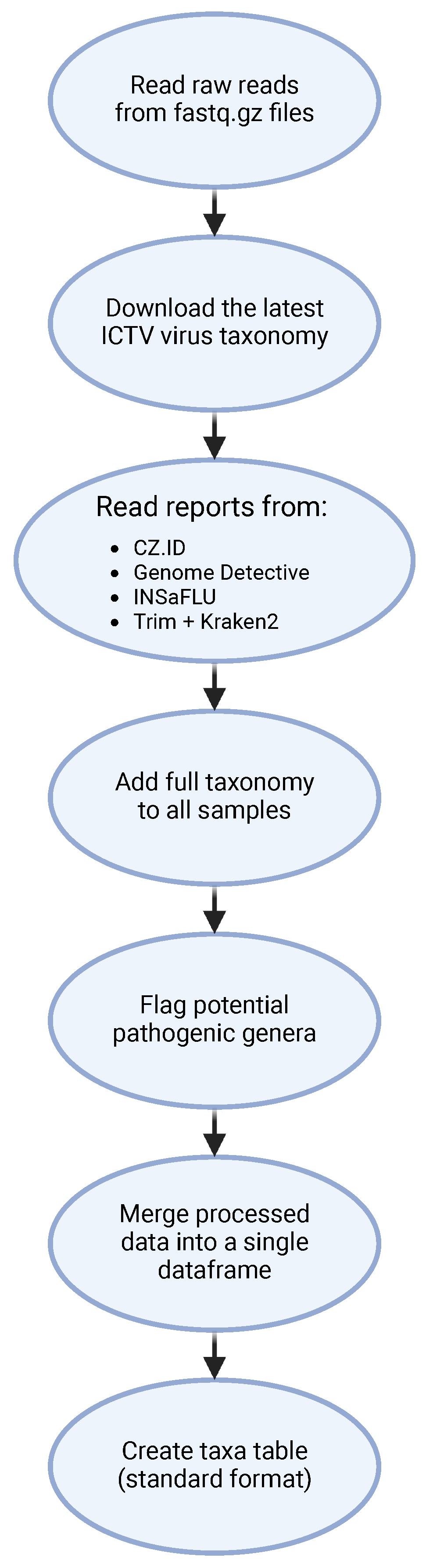
2.3. Bioinformatic Analysis
2.4. Automating the Analysis of Viral Identification
2.5. Statistical Analysis
3. Results
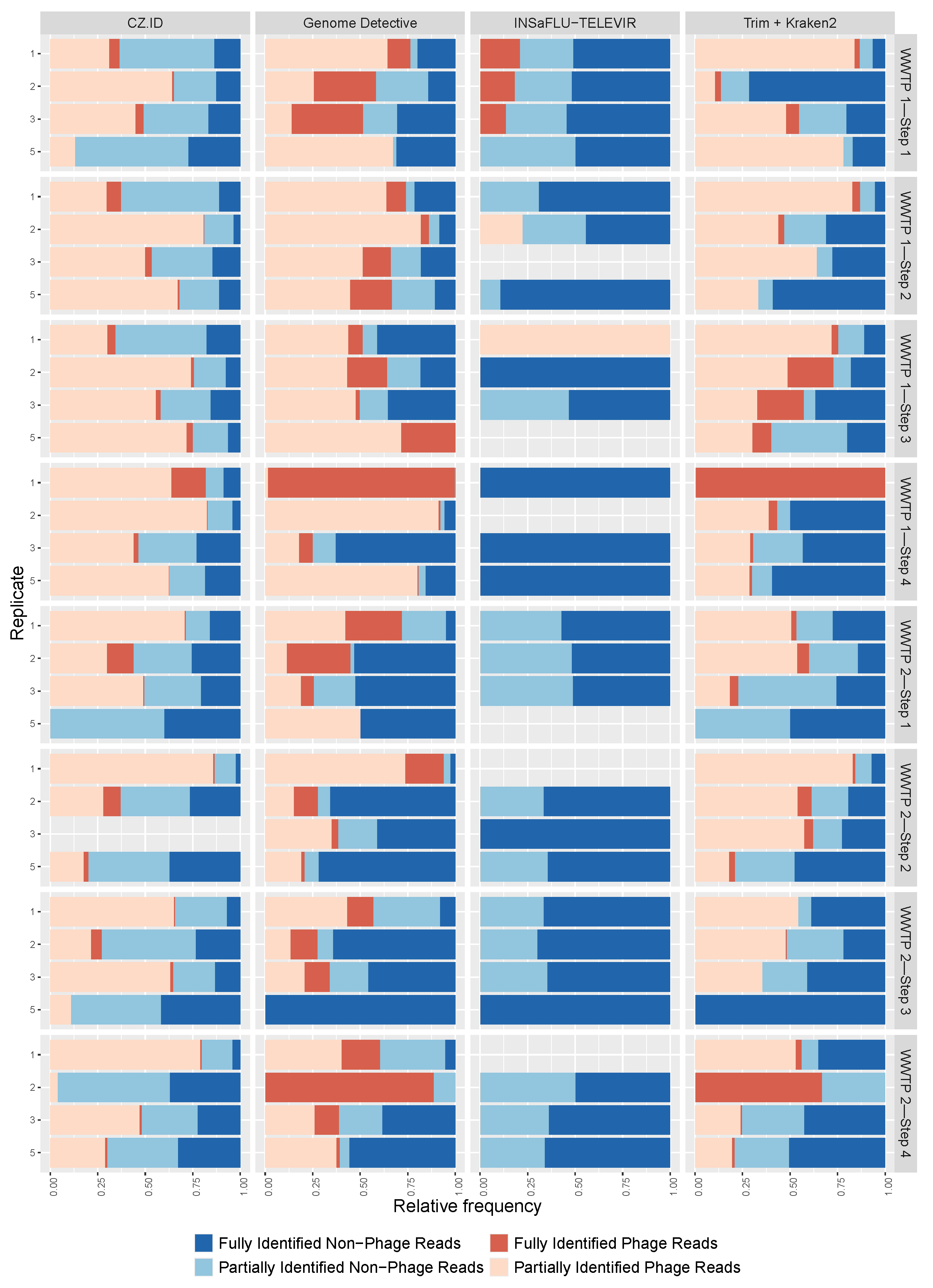
3.1. Sequencing Performance and Viral Identification
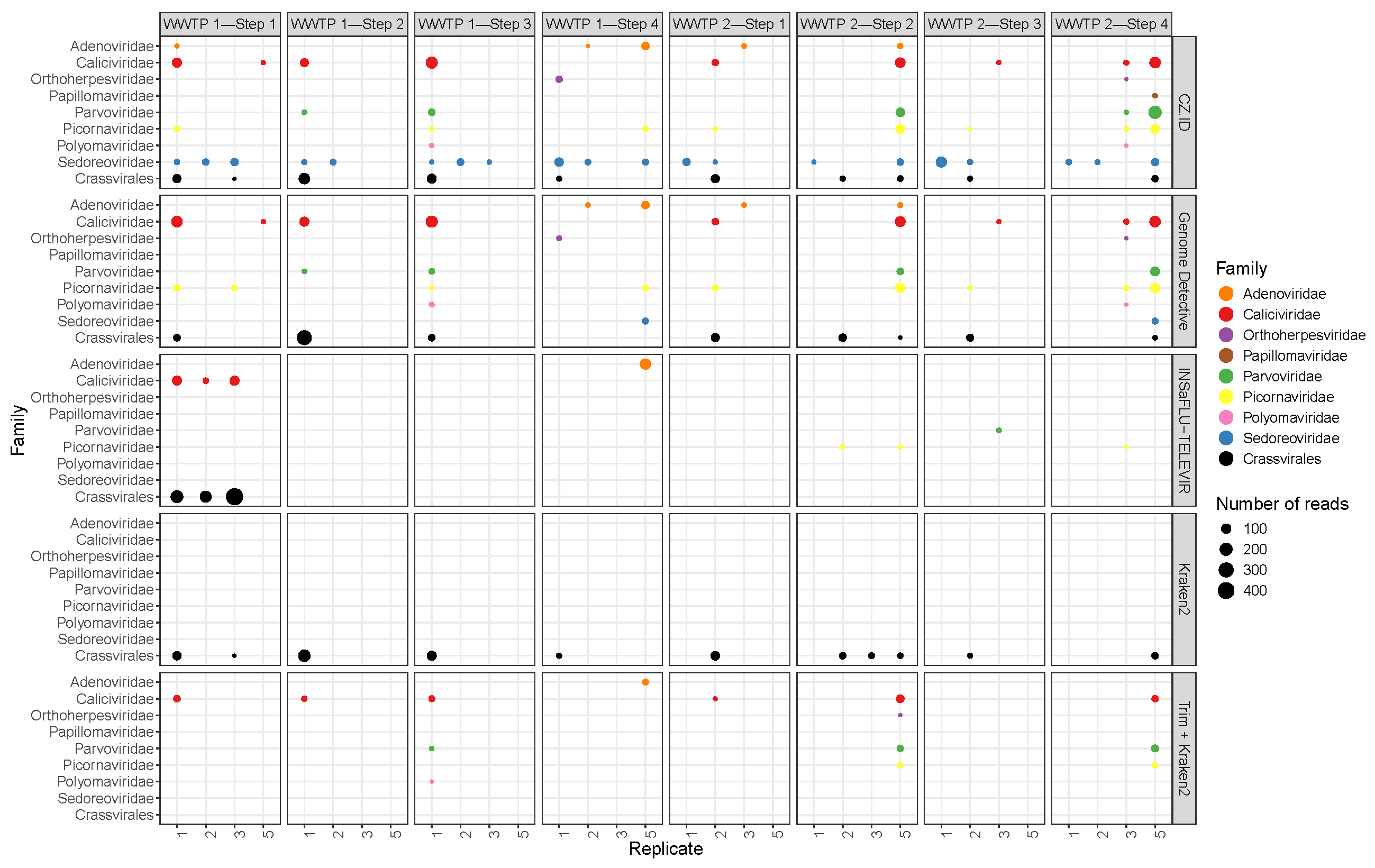
3.2. Phage and Non-Phage Viral Reads
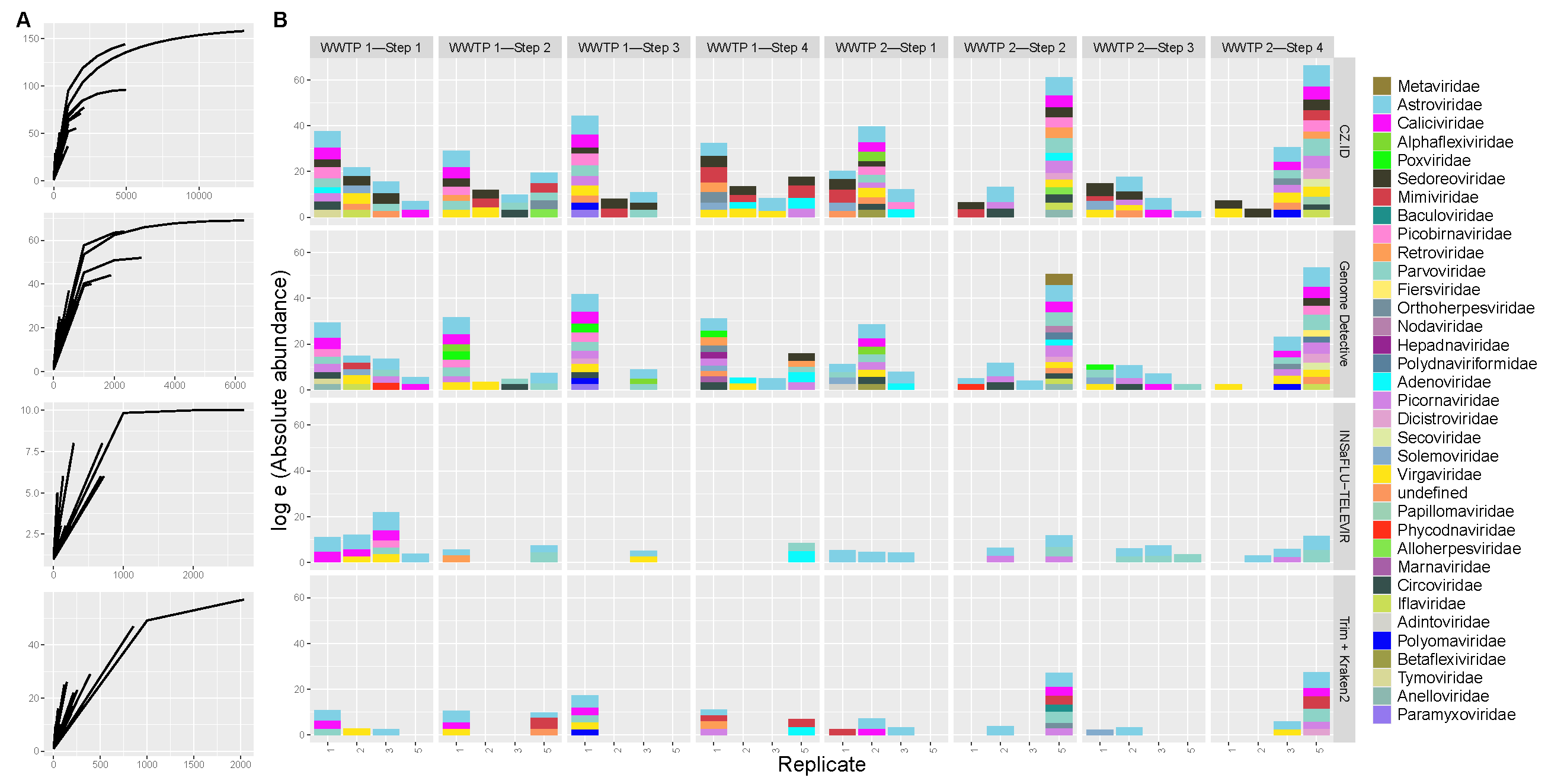
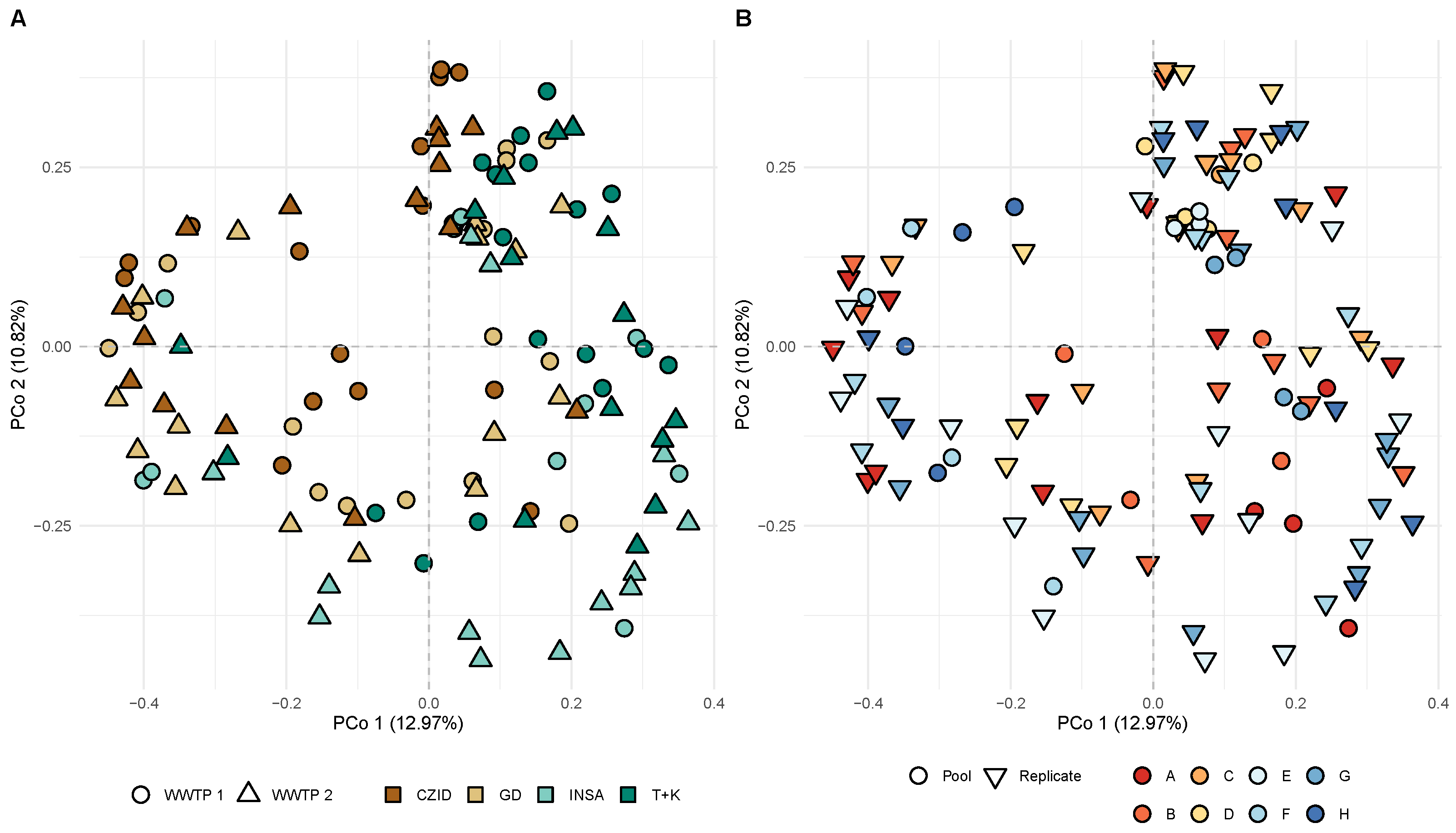
3.3. Viral Abundance and Diversity
3.4. Pathogenic Viruses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hellmér, M.; Paxéus, N.; Magnius, L.; Enache, L.; Arnholm, B.; Johansson, A.; Bergström, T.; Norder, H. Detection of Pathogenic Viruses in Sewage Provided Early Warnings of Hepatitis A Virus and Norovirus Outbreaks. Appl. Environ. Microbiol. 2014, 80, 6771–6781. [Google Scholar] [CrossRef]
- Adriaenssens, E.M.; Farkas, K.; Harrison, C.; Jones, D.L.; Allison, H.E.; McCarthy, A.J. Viromic Analysis of Wastewater Input to a River Catchment Reveals a Diverse Assemblage of RNA Viruses. mSystems 2018, 3, e00025-18. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuijse, D.F.; Oude Munnink, B.B.; Phan, M.V.T.; the Global Sewage Surveillance Project Consortium; Hendriksen, R.S.; Bego, A.; Rees, C.; Neilson, E.H.; Coventry, K.; Collignon, P.; et al. Setting a baseline for global urban virome surveillance in sewage. Sci. Rep. 2020, 10, 13748. [Google Scholar] [CrossRef] [PubMed]
- Tisza, M.; Javornik Cregeen, S.; Avadhanula, V.; Zhang, P.; Ayvaz, T.; Feliz, K.; Hoffman, K.L.; Clark, J.R.; Terwilliger, A.; Ross, M.C.; et al. Wastewater sequencing reveals community and variant dynamics of the collective human virome. Nat. Commun. 2023, 14, 6878. [Google Scholar] [CrossRef]
- Lago, P.M.; Gary, H.E.; Pérez, L.S.; Cáceres, V.; Olivera, J.B.; Puentes, R.P.; Corredor, M.B.; Jímenez, P.; Pallansch, M.A.; Cruz, R.G. Poliovirus detection in wastewater and stools following an immunization campaign in Havana, Cuba. Int. J. Epidemiol. 2003, 32, 772–777. [Google Scholar] [CrossRef]
- Ryerson, A.B.; Lang, D.; Alazawi, M.A.; Neyra, M.; Hill, D.T.; St. George, K.; Fuschino, M.; Lutterloh, E.; Backenson, B.; Rulli, S.; et al. Wastewater Testing and Detection of Poliovirus Type 2 Genetically Linked to Virus Isolated from a Paralytic Polio Case—New York, March 9–11 October 2022. MMWR. Morb. Mortal. Wkly. Rep. 2022, 71, 1418–1424. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, G.; Brandtner, D.; Mancini, P.; Veneri, C.; Bonanno Ferraro, G.; Bonadonna, L.; Lucentini, L.; Suffredini, E. Key SARS-CoV-2 Mutations of Alpha, Gamma, and Eta Variants Detected in Urban Wastewaters in Italy by Long-Read Amplicon Sequencing Based on Nanopore Technology. Water 2021, 13, 2503. [Google Scholar] [CrossRef]
- Brumfield, K.D.; Leddy, M.; Usmani, M.; Cotruvo, J.A.; Tien, C.; Dorsey, S.; Graubics, K.; Fanelli, B.; Zhou, I.; Registe, N.; et al. Microbiome Analysis for Wastewater Surveillance during COVID-19. mBio 2022, 13, e00591-22. [Google Scholar] [CrossRef]
- Torres, C.; Barrios, M.E.; Cammarata, R.V.; Cisterna, D.M.; Estrada, T.; Martini Novas, S.; Cahn, P.; Blanco Fernández, M.D.; Mbayed, V.A. High diversity of human polyomaviruses in environmental and clinical samples in Argentina: Detection of JC, BK, Merkel-cell, Malawi, and human 6 and 7 polyomaviruses. Sci. Total Environ. 2016, 542, 192–202. [Google Scholar] [CrossRef]
- Hasing, M.E.; Lee, B.E.; Gao, T.; Li, Q.; Qiu, Y.; Ellehoj, E.; Graber, T.E.; Fuzzen, M.; Servos, M.; Landgraff, C.; et al. Wastewater surveillance monitoring of SARS-CoV-2 variants of concern and dynamics of transmission and community burden of COVID-19. Emerg. Microbes Infect. 2023, 12, 2233638. [Google Scholar] [CrossRef]
- Gomes, J.; Frasson, D.; Quinta-Ferreira, R.M.; Matos, A.; Martins, R.C. Removal of Enteric Pathogens from Real Wastewater Using Single and Catalytic Ozonation. Water 2019, 11, 127. [Google Scholar] [CrossRef]
- Prado, T.; De Castro Bruni, A.; Barbosa, M.R.F.; Garcia, S.C.; De Jesus Melo, A.M.; Sato, M.I.Z. Performance of wastewater reclamation systems in enteric virus removal. Sci. Total Environ. 2019, 678, 33–42. [Google Scholar]
- Dlamini, M.; Msolo, L.; Ehi Ebomah, K.; Nontongana, N.; Ifeanyi Okoh, A. A systematic review on the incidence of influenza viruses in wastewater matrices: Implications for public health. PLoS ONE 2024, 19, e0291900. [Google Scholar]
- Adams, C.; Kirby, A.E.; Bias, M.; Riser, A.; Wong, K.K.; Mercante, J.W.; Reese, H. Detecting Mpox Cases Through Wastewater Surveillance—United States, August 2022–May 2023. MMWR. Morb. Mortal. Wkly. Rep. 2024, 73, 37–43. [Google Scholar] [CrossRef]
- Allayeh, A.K.; Al-Daim, S.A.; Ahmed, N.; El-Gayar, M.; Mostafa, A. Isolation and Genotyping of Adenoviruses from Wastewater and Diarrheal Samples in Egypt from 2016 to 2020. Viruses 2022, 14, 2192. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, S.; Pimenta, R.; Nunes, F.; Cunha, M.V.; Santos, R. Detection of dengue virus and chikungunya virus in wastewater in Portugal: An exploratory surveillance study. Lancet Microbe 2024, 5, 100911. [Google Scholar] [CrossRef]
- Wolfe, M.K.; Paulos, A.H.; Zulli, A.; Duong, D.; Shelden, B.; White, B.J.; Boehm, A.B. Wastewater Detection of Emerging Arbovirus Infections: Case Study of Dengue in the United States. Environ. Sci. Technol. Lett. 2024, 11, 9–15. [Google Scholar]
- Haldar, T.; Katarmal, P.; Roy, B.; Koratkar, S. Dengue and chikungunya virus dynamics, identification, and monitoring in wastewater. Environ. Monit. Assess. 2024, 196, 1166. [Google Scholar] [CrossRef]
- Condez, A.C.; Nunes, M.; Filipa-Silva, A.; Leonardo, I.; Parreira, R. Human Polyomaviruses (HPyV) in Wastewater and Environmental Samples from the Lisbon Metropolitan Area: Detection and Genetic Characterization of Viral Structural Protein-Coding Sequences. Pathogens 2021, 10, 1309. [Google Scholar] [CrossRef] [PubMed]
- Cavadas, J.; Parreira, R.; Leonardo, I.; Barreto Crespo, M.T.; Nunes, M. Mastadenovirus Molecular Diversity in Waste and Environmental Waters from the Lisbon Metropolitan Area. Microorganisms 2022, 10, 2443. [Google Scholar] [CrossRef]
- Maryam, S.; Ul Haq, I.; Yahya, G.; Ul Haq, M.; Algammal, A.M.; Saber, S.; Cavalu, S. COVID-19 surveillance in wastewater: An epidemiological tool for the monitoring of SARS-CoV-2. Front. Cell. Infect. Microbiol. 2023, 12, 978643. [Google Scholar]
- Carmo Dos Santos, M.; Cerqueira Silva, A.C.; Dos Reis Teixeira, C.; Pinheiro Macedo Prazeres, F.; Fernandes Dos Santos, R.; De Araújo Rolo, C.; De Souza Santos, E.; Santos Da Fonseca, M.; Oliveira Valente, C.; Saraiva Hodel, K.V.; et al. Wastewater surveillance for viral pathogens: A tool for public health. Heliyon 2024, 10, e33873. [Google Scholar]
- Zhao, H.; Yang, M.; Fan, X.; Gui, Q.; Yi, H.; Tong, Y.; Xiao, W. A Metagenomic Investigation of Potential Health Risks and Element Cycling Functions of Bacteria and Viruses in Wastewater Treatment Plants. Viruses 2024, 16, 535. [Google Scholar] [CrossRef]
- Martínez-Puchol, S.; Rusiñol, M.; Fernández-Cassi, X.; Timoneda, N.; Itarte, M.; Andrés, C.; Antón, A.; Abril, J.F.; Girones, R.; Bofill-Mas, S. Characterisation of the sewage virome: Comparison of NGS tools and occurrence of significant pathogens. Sci. Total Environ. 2020, 713, 136604. [Google Scholar] [PubMed]
- Fernandez-Cassi, X.; Timoneda, N.; Martínez-Puchol, S.; Rusiñol, M.; Rodriguez-Manzano, J.; Figuerola, N.; Bofill-Mas, S.; Abril, J.; Girones, R. Metagenomics for the study of viruses in urban sewage as a tool for public health surveillance. Sci. Total Environ. 2018, 618, 870–880. [Google Scholar] [CrossRef]
- Castilletti, C.; Capobianchi, M.R. Polio is back? The risk of poliomyelitis recurrence globally, and the legacy of the severe acute respiratory syndrome coronavirus 2 pandemic. Clin. Microbiol. Infect. 2023, 29, 414–416. [Google Scholar]
- Lapa, D.; Carletti, F.; Mazzotta, V.; Matusali, G.; Pinnetti, C.; Meschi, S.; Gagliardini, R.; Colavita, F.; Mondi, A.; Minosse, C.; et al. Monkeypox virus isolation from a semen sample collected in the early phase of infection in a patient with prolonged seminal viral shedding. Lancet Infect. Dis. 2022, 22, 1267–1269. [Google Scholar] [CrossRef]
- Nooij, S.; Schmitz, D.; Vennema, H.; Kroneman, A.; Koopmans, M.P.G. Overview of Virus Metagenomic Classification Methods and Their Biological Applications. Front. Microbiol. 2018, 9, 749. [Google Scholar] [CrossRef]
- Gulino, K.; Rahman, J.; Badri, M.; Morton, J.; Bonneau, R.; Ghedin, E. Initial Mapping of the New York City Wastewater Virome. mSystems 2020, 5, e00876-19. [Google Scholar] [CrossRef] [PubMed]
- Ballesté, E.; Blanch, A.R.; Muniesa, M.; García-Aljaro, C.; Rodríguez-Rubio, L.; Martín-Díaz, J.; Pascual-Benito, M.; Jofre, J. Bacteriophages in sewage: Abundance, roles, and applications. FEMS Microbes 2022, 3, xtac009. [Google Scholar] [CrossRef]
- Rothman, J.A.; Loveless, T.B.; Griffith, M.L.; Steele, J.A.; Griffith, J.F.; Whiteson, K.L. Metagenomics of Wastewater Influent from Southern California Wastewater Treatment Facilities in the Era of COVID-19. Microbiol. Resour. Announc. 2020, 9, e00907-20. [Google Scholar] [CrossRef] [PubMed]
- Wyler, E.; Lauber, C.; Manukyan, A.; Deter, A.; Quedenau, C.; Teixeira Alves, L.G.; Wylezich, C.; Borodina, T.; Seitz, S.; Altmüller, J.; et al. Pathogen dynamics and discovery of novel viruses and enzymes by deep nucleic acid sequencing of wastewater. Environ. Int. 2024, 190, 108875. [Google Scholar] [CrossRef]
- Tamaki, H.; Zhang, R.; Angly, F.E.; Nakamura, S.; Hong, P.; Yasunaga, T.; Kamagata, Y.; Liu, W. Metagenomic analysis of DNA viruses in a wastewater treatment plant in tropical climate. Environ. Microbiol. 2012, 14, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Giuffrè, L.; Giosa, D.; Galeano, G.; Aiese Cigliano, R.; Paytuví-Gallart, A.; Sutera, A.M.; Tardiolo, G.; Zumbo, A.; Romeo, O.; D’Alessandro, E. Whole-metagenome shotgun sequencing of pig faecal microbiome. Ital. J. Anim. Sci. 2021, 20, 1147–1155. [Google Scholar] [CrossRef]
- Lui, L.M.; Nielsen, T.N. Decomposing a San Francisco estuary microbiome using long-read metagenomics reveals species- and strain-level dominance from picoeukaryotes to viruses. mSystems 2024, 9, e00242-24. [Google Scholar] [CrossRef]
- McCall, C.; Wu, H.; Miyani, B.; Xagoraraki, I. Identification of multiple potential viral diseases in a large urban center using wastewater surveillance. Water Res. 2020, 184, 116160. [Google Scholar] [CrossRef]
- Mejías-Molina, C.; Pico-Tomàs, A.; Martínez-Puchol, S.; Itarte, M.; Torrell, H.; Canela, N.; Borrego, C.M.; Corominas, L.; Rusiñol, M.; Bofill-Mas, S. Wastewater-based epidemiology applied at the building-level reveals distinct virome profiles based on the age of the contributing individuals. Hum. Genom. 2024, 18, 10. [Google Scholar] [CrossRef]
- Parras-Moltó, M.; Rodríguez-Galet, A.; Suárez-Rodríguez, P.; López-Bueno, A. Evaluation of bias induced by viral enrichment and random amplification protocols in metagenomic surveys of saliva DNA viruses. Microbiome 2018, 6, 119. [Google Scholar] [CrossRef]
- Regnault, B.; Bigot, T.; Ma, L.; Pérot, P.; Temmam, S.; Eloit, M. Deep Impact of Random Amplification and Library Construction Methods on Viral Metagenomics Results. Viruses 2021, 13, 253. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Wang, Y.; Zhang, Y.; Zhuang, J.; Radosevich, M. Viral abundance, community structure and correlation with bacterial community in soils of different cover plants. Appl. Soil Ecol. 2021, 168, 104138. [Google Scholar] [CrossRef]
- Rosseel, T.; Scheuch, M.; Höper, D.; De Regge, N.; Caij, A.B.; Vandenbussche, F.; Van Borm, S. DNase SISPA-Next Generation Sequencing Confirms Schmallenberg Virus in Belgian Field Samples and Identifies Genetic Variation in Europe. PLoS ONE 2012, 7, e41967. [Google Scholar] [CrossRef]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef]
- Li, Y.; Ash, K.T.; Joyner, D.C.; Williams, D.E.; Alamilla, I.; McKay, P.J.; Iler, C.; Hazen, T.C. Evaluating various composite sampling modes for detecting pathogenic SARS-CoV-2 virus in raw sewage. Front. Microbiol. 2023, 14, 1305967. [Google Scholar] [CrossRef]
- Abdel-Wahab, M.; Bahkali, A.H.; El-Gorban, A.M.; Jones, G. Metagenomics Study of Fungi and Fungi-Like Organisms Associated With the Seagrass Halophila Stipulacea (Forssk.) Asch. From Al-Leith Mangroves, Saudi Arabia. Mycol. Prog. 2021; under review. [Google Scholar]
- Porter, A.F.; Cobbin, J.; Li, C.; Eden, J.; Holmes, E.C. Metagenomic Identification of Viral Sequences in Laboratory Reagents. Viruses 2021, 13, 2122. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.; Luo, R.; Sadakane, K.; Lam, T. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Billington, C.; Kingsbury, J.M.; Rivas, L. Metagenomics Approaches for Improving Food Safety: A Review. J. Food Prot. 2022, 85, 448–464. [Google Scholar] [CrossRef]
- Sharon, B.M.; Hulyalkar, N.V.; Nguyen, V.H.; Zimmern, P.E.; Palmer, K.L.; De Nisco, N.J. Hybrid De Novo Genome Assembly for the Generation of Complete Genomes of Urinary Bacteria using Short- and Long-read Sequencing Technologies. J. Vis. Exp. 2021, 174, 62872. [Google Scholar]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed]
- Ounit, R.; Wanamaker, S.; Close, T.J.; Lonardi, S. CLARK: Fast and accurate classification of metagenomic and genomic sequences using discriminative k-mers. BMC Genom. 2015, 16, 236. [Google Scholar] [CrossRef]
- Kim, D.; Song, L.; Breitwieser, F.P.; Salzberg, S.L. Centrifuge: Rapid and sensitive classification of metagenomic sequences. Genome Res. 2016, 26, 1721–1729. [Google Scholar] [CrossRef]
- Bogaardt, C. Metagenomic Surveillance of Viruses at the Human-Animal Interface. Ph.D. Thesis, The University of Edinburgh, Edinburgh, UK, 2020. [Google Scholar]
- Xu, R.; Rajeev, S.; Salvador, L.C.M. The selection of software and database for metagenomics sequence analysis impacts the outcome of microbial profiling and pathogen detection. PLoS ONE 2023, 18, e0284031. [Google Scholar] [CrossRef]
- Altschul, S. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN analysis of metagenomic data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Mukherjee, A.; D’Ugo, E.; Giuseppetti, R.; Magurano, F.; Cotter, P.D. Metagenomic approaches for understanding microbial communities in contaminated environments: Bioinformatic tools, case studies and future outlook. Metagenomics Bioremediat. 2023, 2023, 103–156. [Google Scholar]
- Arizmendi Cárdenas, Y.O.; Neuenschwander, S.; Malaspinas, A. Benchmarking metagenomics classifiers on ancient viral DNA: A simulation study. PeerJ 2022, 10, e12784. [Google Scholar] [CrossRef]
- Kapoor, V.; Briese, T.; Ranjan, A.; Donovan, W.M.; Mansukhani, M.M.; Chowdhary, R.; Lipkin, W.I. Validation of the VirCapSeq-VERT system for differential diagnosis, detection, and surveillance of viral infections. J. Clin. Microbiol. 2024, 62, e00612-23. [Google Scholar] [CrossRef]
- Vilsker, M.; Moosa, Y.; Nooij, S.; Fonseca, V.; Ghysens, Y.; Dumon, K.; Pauwels, R.; Alcantara, L.C.; Vanden Eynden, E.; Vandamme, A.; et al. Genome Detective: An automated system for virus identification from high-throughput sequencing data. Bioinformatics 2019, 35, 871–873. [Google Scholar] [CrossRef]
- Kalantar, K.L.; Carvalho, T.; De Bourcy, C.F.A.; Dimitrov, B.; Dingle, G.; Egger, R.; Han, J.; Holmes, O.B.; Juan, Y.; King, R.; et al. IDseq—An open source cloud-based pipeline and analysis service for metagenomic pathogen detection and monitoring. GigaScience 2020, 9, giaa111. [Google Scholar] [CrossRef]
- Borges, V.; Pinheiro, M.; Pechirra, P.; Guiomar, R.; Gomes, J.P. INSaFLU: An automated open web-based bioinformatics suite “from-reads” for influenza whole-genome-sequencing-based surveillance. Genome Med. 2018, 10, 46. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Ruby, J.G.; Bellare, P.; DeRisi, J.L. PRICE: Software for the Targeted Assembly of Components of (Meta) Genomic Sequence Data. G3 Genes Genomes Genet. 2013, 3, 865–880. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Wu, T.D.; Nacu, S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 2010, 26, 873–881. [Google Scholar] [CrossRef]
- Zhao, Y.; Tang, H.; Ye, Y. RAPSearch2: A fast and memory-efficient protein similarity search tool for next-generation sequencing data. Bioinformatics 2012, 28, 125–126. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data [Software]. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 14 February 2024).
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Cantu, V.A.; Sadural, J.; Edwards, R. PRINSEQ++, a multi-threaded tool for fast and efficient quality control and preprocessing of sequencing datasets. PeerJ Preprint 2019, 7, e27553v1. [Google Scholar] [CrossRef]
- Breitwieser, F.P.; Baker, D.N.; Salzberg, S.L. KrakenUniq: Confident and fast metagenomics classification using unique k-mer counts. Genome Biol. 2018, 19, 198. [Google Scholar] [CrossRef] [PubMed]
- The Galaxy Community, J.P.; Abueg, L.A.L.; Afgan, E.; Allart, O.; Awan, A.H.; Bacon, W.A.; Baker, D.; Bassetti, M.; Batut, B.; Bernt, M.; et al. The Galaxy platform for accessible, reproducible, and collaborative data analyses: 2024 update. Nucleic Acids Res. 2024, 52, W83–W94. [Google Scholar] [CrossRef]
- Baldovin, T.; Amoruso, I.; Fonzo, M.; Buja, A.; Baldo, V.; Cocchio, S.; Bertoncello, C. SARS-CoV-2 RNA detection and persistence in wastewater samples: An experimental network for COVID-19 environmental surveillance in Padua, Veneto Region (NE Italy). Sci. Total Environ. 2021, 760, 143329. [Google Scholar]
- Schaeffer, J.; Desdouits, M.; Besnard, A.; Le Guyader, F.S. Looking into sewage: How far can metagenomics help to detect human enteric viruses? Front. Microbiol. 2023, 14, 1161674. [Google Scholar] [CrossRef]
- Hendriksen, R.S.; Munk, P.; Njage, P.; Van Bunnik, B.; McNally, L.; Lukjancenko, O.; Röder, T.; Nieuwenhuijse, D.; Pedersen, S.K.; Kjeldgaard, J.; et al. Global monitoring of antimicrobial resistance based on metagenomics analyses of urban sewage. Nat. Commun. 2019, 10, 1124. [Google Scholar]
- Karlsson, O.E.; Belák, S.; Granberg, F. The Effect of Preprocessing by Sequence-Independent, Single-Primer Amplification (SISPA) on Metagenomic Detection of Viruses. Biosecur. Bioterror. Biodef. Strateg. Pract. Sci. 2013, 11, S227–S234. [Google Scholar] [CrossRef]
- Zhang, J.; Tang, A.; Jin, T.; Sun, D.; Guo, F.; Lei, H.; Lin, L.; Shu, W.; Yu, P.; Li, X.; et al. A panoramic view of the virosphere in three wastewater treatment plants by integrating viral-like particle-concentrated and traditional non-concentrated metagenomic approaches. iMeta 2024, 3, e188. [Google Scholar] [CrossRef]
- Wielinga, P.R.; De Heer, L.; De Groot, A.; Hamidjaja, R.A.; Bruggeman, G.; Jordan, K.; Van Rotterdam, B.J. Evaluation of DNA extraction methods for Bacillus anthracis spores spiked to food and feed matrices at biosafety level 3 conditions. Int. J. Food Microbiol. 2011, 150, 122–127. [Google Scholar]
- Colosi, L.M.; Barry, K.E.; Kotay, S.M.; Porter, M.D.; Poulter, M.D.; Ratliff, C.; Simmons, W.; Steinberg, L.I.; Wilson, D.D.; Morse, R.; et al. Development of Wastewater Pooled Surveillance of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) from Congregate Living Settings. Appl. Environ. Microbiol. 2021, 87, e00433-21. [Google Scholar] [PubMed]
- Ray, K.J.; Cotter, S.Y.; Arzika, A.M.; Kim, J.; Boubacar, N.; Zhou, Z.; Zhong, L.; Porco, T.C.; Keenan, J.D.; Lietman, T.M.; et al. High-throughput sequencing of pooled samples to determine community-level microbiome diversity. Ann. Epidemiol. 2019, 39, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Ko, B.; Van Raamsdonk, J.M. RNA Sequencing of Pooled Samples Effectively Identifies Differentially Expressed Genes. Biology 2023, 12, 812. [Google Scholar] [CrossRef] [PubMed]
- Ibarbalz, F.M.; Orellana, E.; Figuerola, E.L.M.; Erijman, L. Shotgun Metagenomic Profiles Have a High Capacity To Discriminate Samples of Activated Sludge According to Wastewater Type. Appl. Environ. Microbiol. 2016, 82, 5186–5196. [Google Scholar] [CrossRef]
- Adriaenssens, E.M.; Farkas, K.; McDonald, J.E.; Jones, D.L.; Allison, H.E.; McCarthy, A.J. Tracing the fate of wastewater viruses reveals catchment-scale virome diversity and connectivity. Water Res. 2021, 203, 117568. [Google Scholar]
- Itarte, M.; Calvo, M.; Martínez-Frago, L.; Mejías-Molina, C.; Martínez-Puchol, S.; Girones, R.; Medema, G.; Bofill-Mas, S.; Rusiñol, M. Assessing environmental exposure to viruses in wastewater treatment plant and swine farm scenarios with next-generation sequencing and occupational risk approaches. Int. J. Hyg. Environ. Health 2024, 259, 114360. [Google Scholar] [CrossRef]
- Corpuz, M.V.A.; Buonerba, A.; Vigliotta, G.; Zarra, T.; Ballesteros, F.; Campiglia, P.; Belgiorno, V.; Korshin, G.; Naddeo, V. Viruses in wastewater: Occurrence, abundance and detection methods. Sci. Total Environ. 2020, 745, 140910. [Google Scholar] [CrossRef]
- Parashar, U.D.; Gibson, C.J.; Bresee, J.S.; Glass, R.I. Rotavirus and Severe Childhood Diarrhea. Emerg. Infect. Dis. 2006, 12, 304–306. [Google Scholar]
- Bisseux, M.; Colombet, J.; Mirand, A.; Roque-Afonso, A.; Abravanel, F.; Izopet, J.; Archimbaud, C.; Peigue-Lafeuille, H.; Debroas, D.; Bailly, J.; et al. Monitoring human enteric viruses in wastewater and relevance to infections encountered in the clinical setting: A one-year experiment in central France, 2014 to 2015. Eurosurveillance 2018, 23, 17-00237. [Google Scholar]
- Dutilh, B.E.; Cassman, N.; McNair, K.; Sanchez, S.E.; Silva, G.G.Z.; Boling, L.; Barr, J.J.; Speth, D.R.; Seguritan, V.; Aziz, R.K.; et al. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat. Commun. 2014, 5, 4498. [Google Scholar] [CrossRef]
- Yutin, N.; Benler, S.; Shmakov, S.A.; Wolf, Y.I.; Tolstoy, I.; Rayko, M.; Antipov, D.; Pevzner, P.A.; Koonin, E.V. Analysis of metagenome-assembled viral genomes from the human gut reveals diverse putative CrAss-like phages with unique genomic features. Nat. Commun. 2021, 12, 1044. [Google Scholar] [CrossRef]
- Wu, Z.; Greaves, J.; Arp, L.; Stone, D.; Bibby, K. Comparative fate of CrAssphage with culturable and molecular fecal pollution indicators during activated sludge wastewater treatment. Environ. Int. 2020, 136, 105452. [Google Scholar] [CrossRef]
- McKee, A.M.; Cruz, M.A. Microbial and Viral Indicators of Pathogens and Human Health Risks from Recreational Exposure to Waters Impaired by Fecal Contamination. J. Sustain. Water Built Environ. 2021, 7, 03121001. [Google Scholar] [CrossRef]
- Sabar, M.A.; Honda, R.; Haramoto, E. CrAssphage as an indicator of human-fecal contamination in water environment and virus reduction in wastewater treatment. Water Res. 2022, 221, 118827. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Taylor, B.; Lin, V.; Altman, T.; Barbera, P.; Meleshko, D.; Lohr, D.; Novakovsky, G.; Buchfink, B.; Al-Shayeb, B.; et al. Petabase-scale sequence alignment catalyses viral discovery. Nature 2022, 602, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Bartoszewicz, J.M.; Seidel, A.; Rentzsch, R.; Renard, B.Y. DeePaC: Predicting pathogenic potential of novel DNA with reverse-complement neural networks. Bioinformatics 2020, 36, 81–89. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos, A.F.B.; Nunes, M.; Filipa-Silva, A.; Pimentel, V.; Pingarilho, M.; Abrantes, P.; Miranda, M.N.S.; Crespo, M.T.B.; Abecasis, A.B.; Parreira, R.; et al. Wastewater Metavirome Diversity: Exploring Replicate Inconsistencies and Bioinformatic Tool Disparities. Int. J. Environ. Res. Public Health 2025, 22, 707. https://doi.org/10.3390/ijerph22050707
Santos AFB, Nunes M, Filipa-Silva A, Pimentel V, Pingarilho M, Abrantes P, Miranda MNS, Crespo MTB, Abecasis AB, Parreira R, et al. Wastewater Metavirome Diversity: Exploring Replicate Inconsistencies and Bioinformatic Tool Disparities. International Journal of Environmental Research and Public Health. 2025; 22(5):707. https://doi.org/10.3390/ijerph22050707
Chicago/Turabian StyleSantos, André F. B., Mónica Nunes, Andreia Filipa-Silva, Victor Pimentel, Marta Pingarilho, Patrícia Abrantes, Mafalda N. S. Miranda, Maria Teresa Barreto Crespo, Ana B. Abecasis, Ricardo Parreira, and et al. 2025. "Wastewater Metavirome Diversity: Exploring Replicate Inconsistencies and Bioinformatic Tool Disparities" International Journal of Environmental Research and Public Health 22, no. 5: 707. https://doi.org/10.3390/ijerph22050707
APA StyleSantos, A. F. B., Nunes, M., Filipa-Silva, A., Pimentel, V., Pingarilho, M., Abrantes, P., Miranda, M. N. S., Crespo, M. T. B., Abecasis, A. B., Parreira, R., & Seabra, S. G. (2025). Wastewater Metavirome Diversity: Exploring Replicate Inconsistencies and Bioinformatic Tool Disparities. International Journal of Environmental Research and Public Health, 22(5), 707. https://doi.org/10.3390/ijerph22050707