
Diagnostic and Clinical Manifestation Differences of Glucose Transporter Type 1 Deficiency Syndrome in a Family with SLC2A1 Gene Mutation


Abstract
:1. Introduction
2. Presentation of Cases
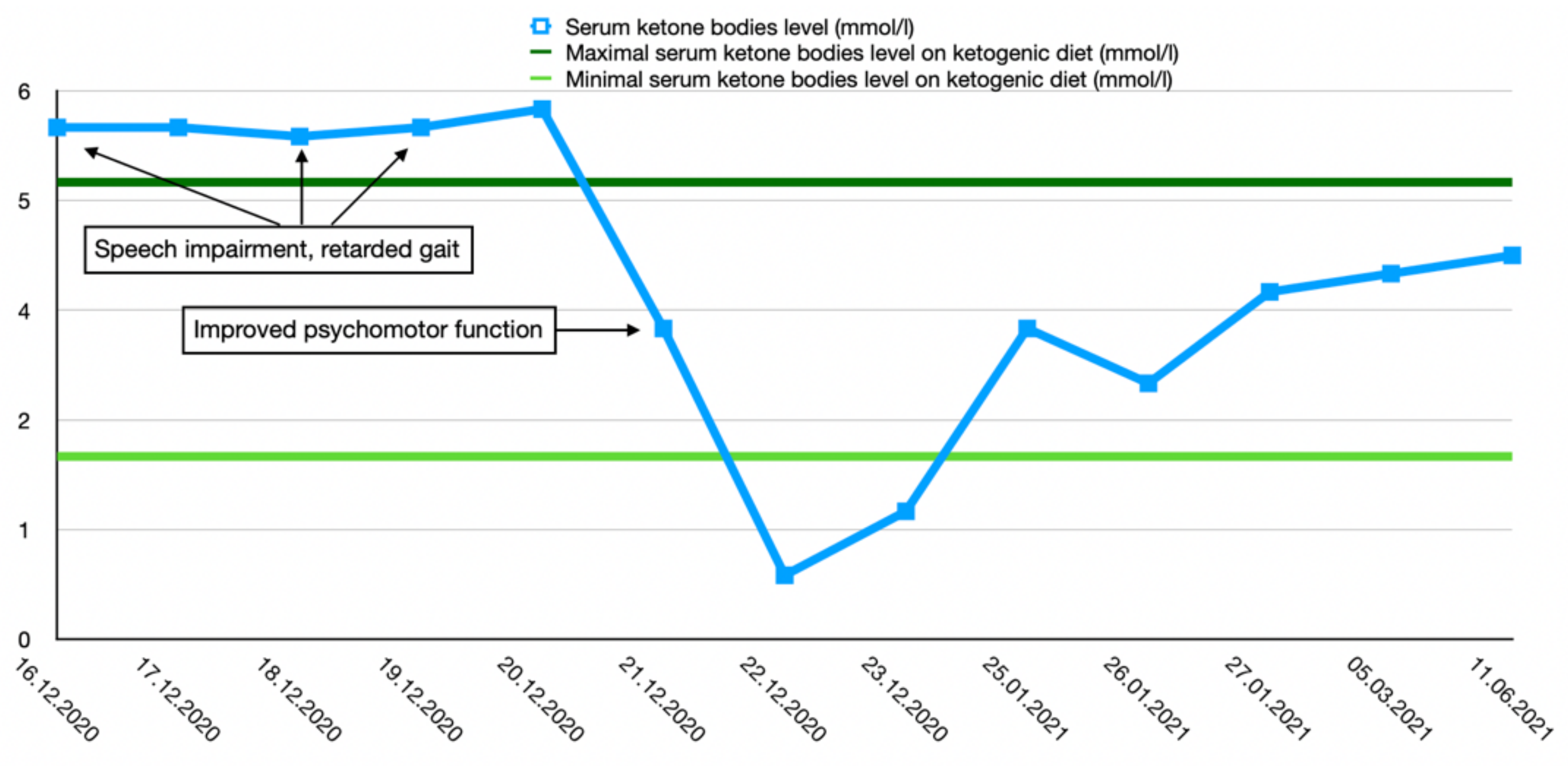
2.1. Case 1
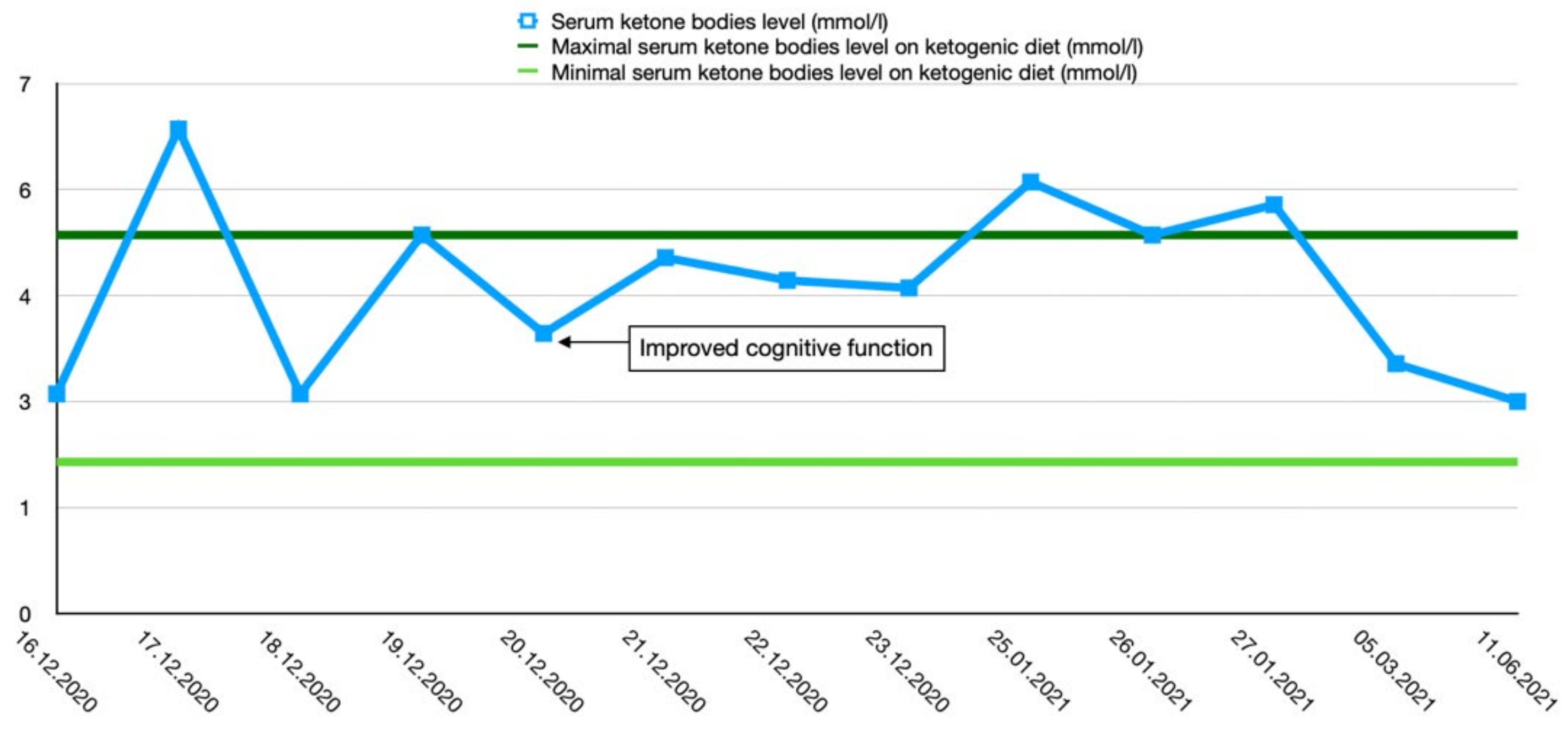
2.2. Case 2
2.3. Treatment
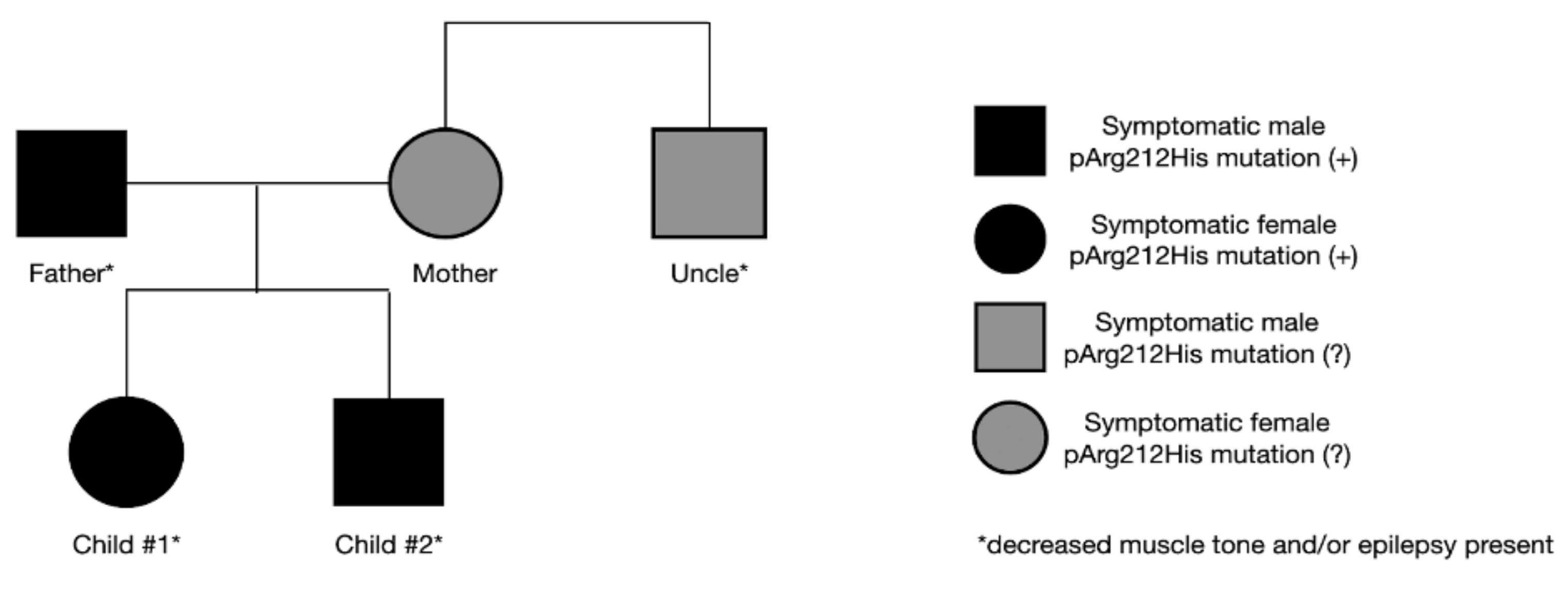
2.4. Patients’ Family History
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Klepper, J.; Scheffer, H. Autosomal recessive inheritance of GLUT1 deficiency syndrome. Neuropediatrics 2009, 40, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Schwantje, M.; Verhagen, L.M. Glucose transporter type 1 deficiency syndrome and the ketogenic diet. J. Inherit. Metab. Dis. 2020, 43, 216–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klepper, J.; Akman, C. Glut1 Deficiency Syndrome (Glut1DS): State of the art in 2020 and recommendations of the international Glut1DS study group. Epilepsia Open 2020, 5, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Koch, H.; Weber, Y.G. The glucose transporter type 1 (Glut1) syndromes. Epilepsy Behav. 2018, 91, 90–93. [Google Scholar] [CrossRef] [PubMed]
- López-Rivera, J.A.; Pérez-Palma, E. A catalogue of new incidence estimates of monogenic neurodevelopmental disorders caused by de novo variants. Brain 2020, 143, 1099–1105. [Google Scholar] [CrossRef] [PubMed]
- Pearson, T.S.; Akman, C. Phenotypic spectrum of glucose transporter type 1 deficiency syndrome (Glut1 DS). Curr. Neurol. Neurosci. Rep. 2013, 13, 342. [Google Scholar] [CrossRef]
- Wang, D.; Pascual, J.M. Glucose Transporter Type 1 Deficiency Syndrome. In GeneReviews; University of Washington: Seattle, DC, USA, 2018. [Google Scholar]
- Klepper, J. GLUT1 deficiency syndrome in clinical practice. Epilepsy Res. 2012, 100, 272–277. [Google Scholar] [CrossRef]
- Girirajan, S.; Rosenfeld, J.A. Phenotypic heterogeneity of genomic disorders and rare copy-number variants. N. Engl. J. Med. 2012, 367, 321–331. [Google Scholar] [CrossRef] [Green Version]
- Paprocka, J.; Emich-Widera, E. Epilepsy in selected neurometabolic diseases. Neurol. Dziec. 2018, 27, 29–37. [Google Scholar] [CrossRef]
- Verrotti, A.; D’Egidio, C. Glut1 deficiency: When to suspect and how to diagnose? Eur. J. Paediatr. Neurol. 2012, 16, 3–9. [Google Scholar] [CrossRef]
- Akman, C.I.; Yu, J. Diagnosing Glucose Transporter 1 Deficiency at Initial Presentation Facilitates Early Treatment. J. Pediatr. 2016, 6, 171–220. [Google Scholar] [CrossRef] [PubMed]
- Mullen, S.A.; Suls, A. Absence epilepsies with widely variable onset are a key feature of familial GLUT1 deficiency. Neurology 2010, 75, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Pong, A.W.; Geary, B.R. Glucose transporter type I deficiency syndrome: Epilepsy phenotypes and outcomes. Epilepsia 2012, 53, 1503–1510. [Google Scholar] [CrossRef]
- Wilhelmina, G.L. Glucose transporter-1 deficiency syndrome: The expanding clinical and genetic spectrum of a treatable disorder. Brain 2010, 133, 655–670. [Google Scholar]
- Leen, W.G.; Wevers, R.A. Cerebrospinal fluid analysis in the workup of GLUT1 deficiency syndrome: A systematic review. JAMA Neurol. 2013, 70, 1440–1444. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Muzny, D.M. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 2014, 312, 1870–1879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klepper, J.; Willemsen, M. Autosomal dominant transmission of GLUT1 deficiency. Hum. Mol. Genet. 2001, 10, 63–68. [Google Scholar] [CrossRef]
- Brockmann, K.; Wang, D. Autosomal dominant glut-1 deficiency syndrome and familial epilepsy. Ann. Neurol. 2001, 50, 476–485. [Google Scholar] [CrossRef]
- Winczewska-Wiktor, A.; Hoffman-Zacharska, D. Variety of symptoms of GLUT1 deficiency syndrome in three-generation family. Epilepsy Behav. 2020, 106, 107036. [Google Scholar] [CrossRef]
- Kolic, I.; Radic Nisevic, J. GLUT1 deficiency syndrome-early treatment maintains cognitive development? (Literature Review and Case Report). Genes 2021, 12, 1379. [Google Scholar] [CrossRef]
- Vaudano, A.E.; Olivotto, S. Brain correlates of spike and wave discharges in GLUT1 deficiency syndrome. Neuroimage Clin. 2016, 13, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Leary, L.D.; Wang, D. Seizure characterization and electroencephalographic features in GLUT-1 deficiency syndrome. Epilepsia 2003, 44, 701–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ismayilova, N.; Hacohen, Y. GLUT-1 deficiency presenting with seizures and reversible leukoencephalopathy on MRI imaging. Eur. J. Paediatr. Neurol. 2018, 22, 1161–1164. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C.; Lee, J.W.A. Evaluation of non-coding variation in GLUT1 deficiency. Dev. Med. Child Neurol. 2016, 58, 1295–1302. [Google Scholar] [CrossRef]
- Wang, D.; Pascual, J.M. Glut-1 deficiency syndrome: Clinical, genetic, and therapeutic aspects. Ann. Neurol. 2005, 57, 111–118. [Google Scholar] [CrossRef]
- De Giorgis, V.; Veggiotti, P. GLUT1 deficiency syndrome 2013: Current state of the art. Seizure 2013, 22, 803–811. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Parameter | Reference Range | Changes Typical in GLUT1-DS | Female Patient | Male Patient |
|---|---|---|---|---|
| CSF glucose | 48–85 mg/dL | <40 mg/dL | No Data | 42.8 mg/dL |
| CSF lactates | 10–29 mg/dL | 5.4–13.5 mg/dL | No Data | 9.1 mg/dL |
| CSF/serum glucose ratio | 0.55–0.75 | 0.19–0.59 | 0.41 | 0.59 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pawlik, W.; Okulewicz, P.; Pawlik, J.; Krzywińska-Zdeb, E. Diagnostic and Clinical Manifestation Differences of Glucose Transporter Type 1 Deficiency Syndrome in a Family with SLC2A1 Gene Mutation. Int. J. Environ. Res. Public Health 2022, 19, 3279. https://doi.org/10.3390/ijerph19063279
Pawlik W, Okulewicz P, Pawlik J, Krzywińska-Zdeb E. Diagnostic and Clinical Manifestation Differences of Glucose Transporter Type 1 Deficiency Syndrome in a Family with SLC2A1 Gene Mutation. International Journal of Environmental Research and Public Health. 2022; 19(6):3279. https://doi.org/10.3390/ijerph19063279
Chicago/Turabian StylePawlik, Weronika, Patrycja Okulewicz, Jakub Pawlik, and Elżbieta Krzywińska-Zdeb. 2022. "Diagnostic and Clinical Manifestation Differences of Glucose Transporter Type 1 Deficiency Syndrome in a Family with SLC2A1 Gene Mutation" International Journal of Environmental Research and Public Health 19, no. 6: 3279. https://doi.org/10.3390/ijerph19063279
APA StylePawlik, W., Okulewicz, P., Pawlik, J., & Krzywińska-Zdeb, E. (2022). Diagnostic and Clinical Manifestation Differences of Glucose Transporter Type 1 Deficiency Syndrome in a Family with SLC2A1 Gene Mutation. International Journal of Environmental Research and Public Health, 19(6), 3279. https://doi.org/10.3390/ijerph19063279