
Toxicity Assessment of Octachlorostyrene in Human Liver Carcinoma (HepG2) Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Consumables
2.2. Cell Culture
2.3. Cell Viability Assay
2.4. Membrane Integrity Assay
2.5. Measurement of ROS and Several Other Oxidative Stress Parameters
2.6. MMP and ATP Quantification
2.7. Intracellular Ca2+ Measurements
2.8. Apoptosis and AnnexinV-FITC/PI Assay
2.9. Statistical Analyses
3. Results and Discussions
3.1. Cell Viability and Cytotoxicity
3.2. ROS Level and Antioxidant Parameters in HepG2 Cells
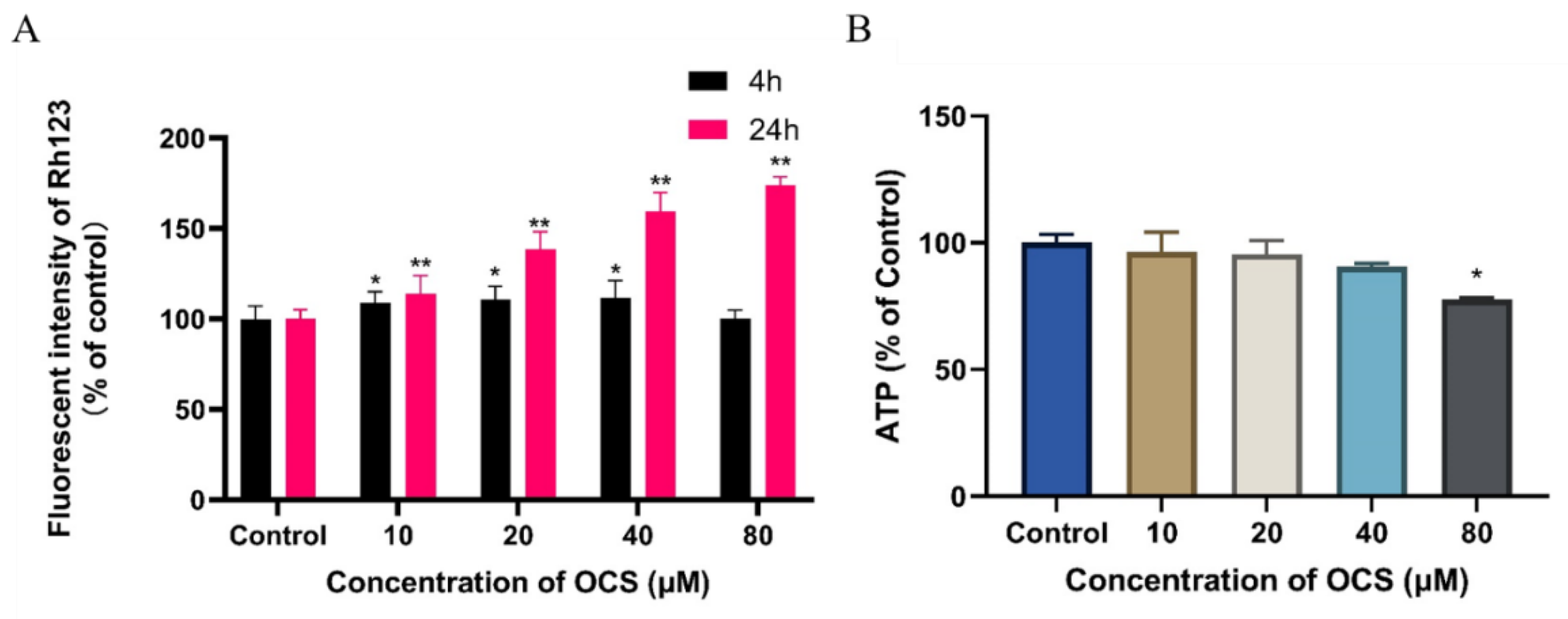
3.3. Effect on MMP and ATP Level
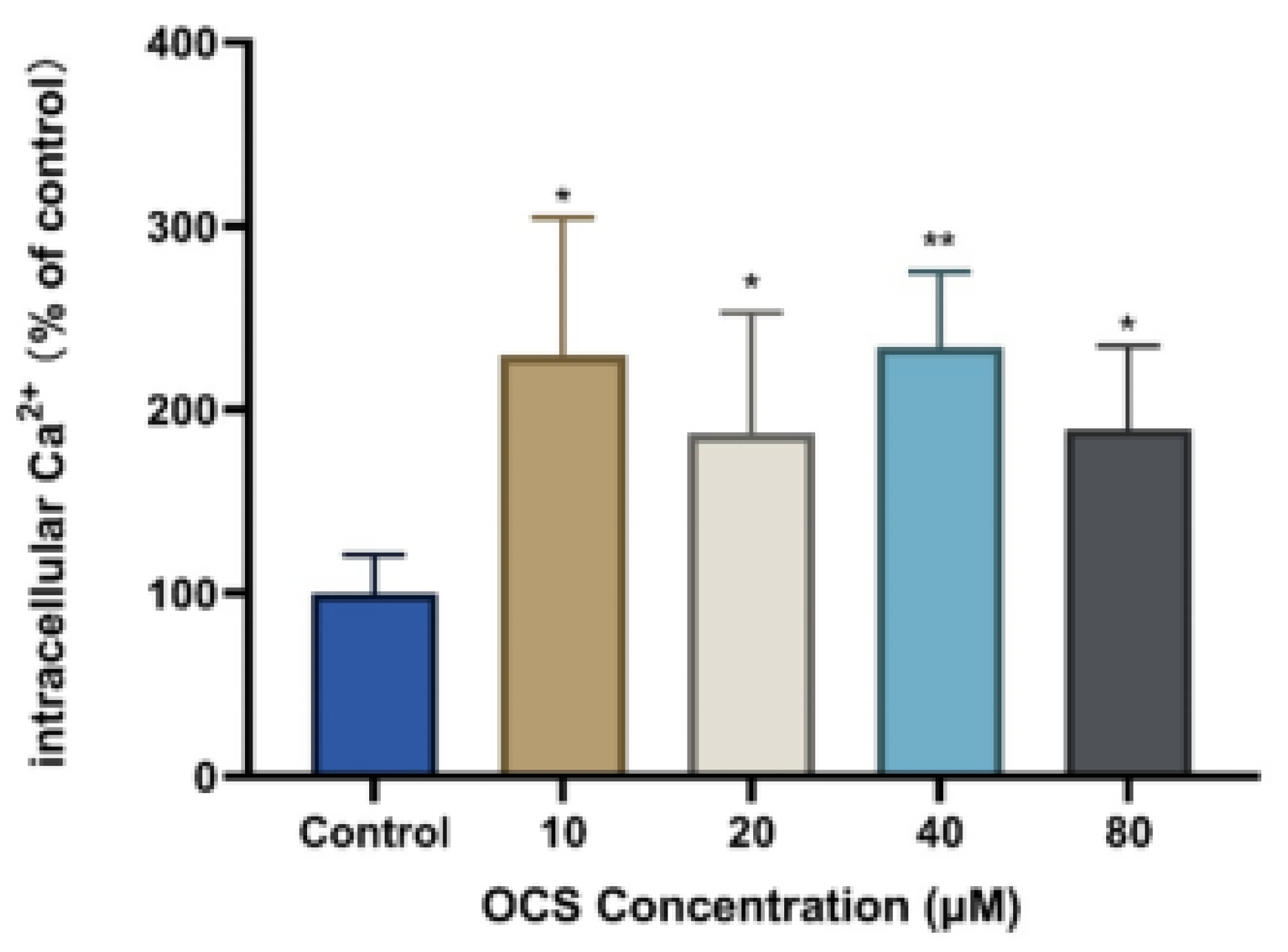
3.4. Intercellular Ca2+ and Apoptosis
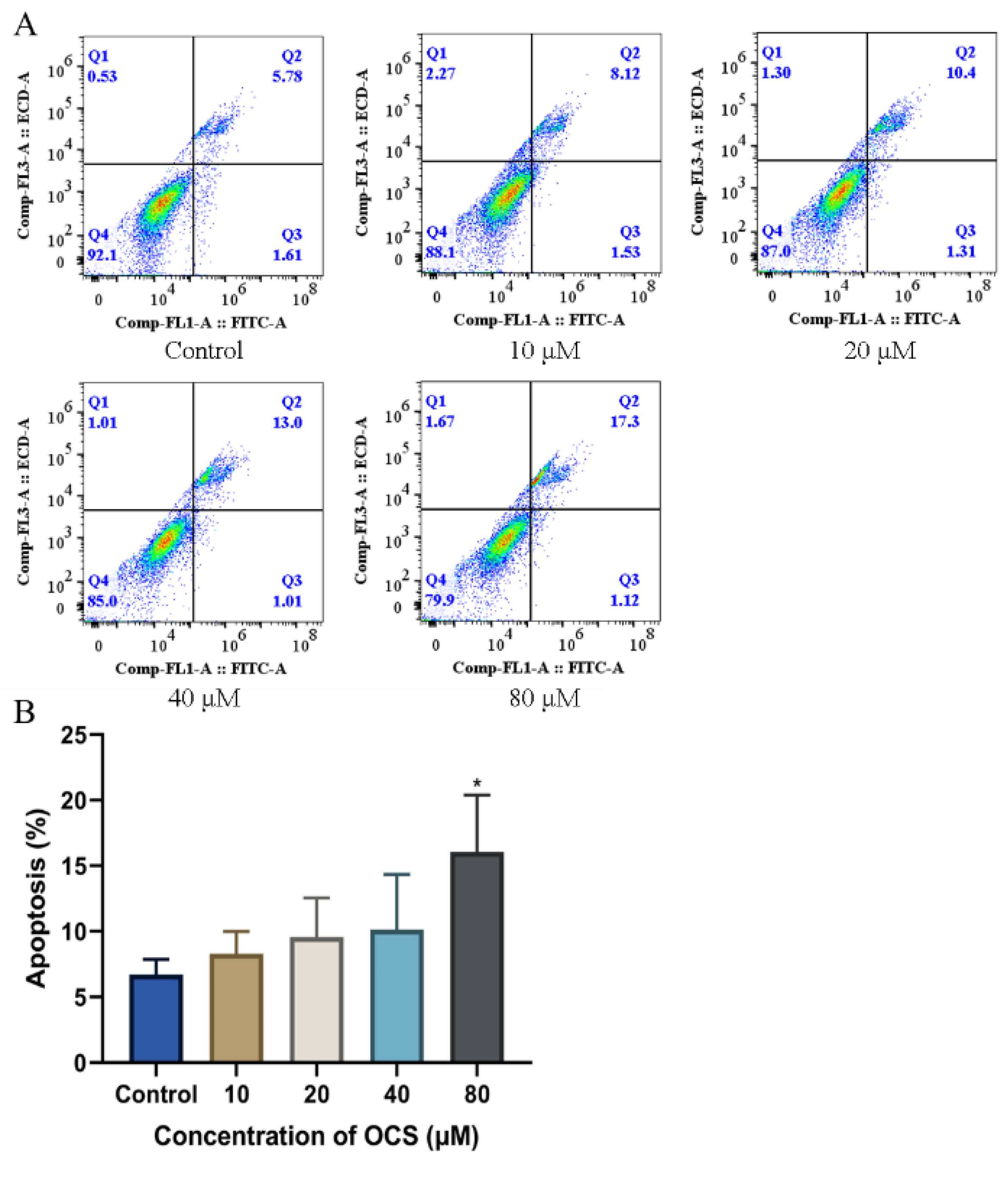
3.5. OCS Induces Apoptosis in HepG2 Cells
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Wilson, L.K.; Harris, M.L.; Trudeau, S.; Ikonomou, M.G.; Elliott, J.E. Properties of blood, porphyrins, and exposure to legacy and emerging persistent organic pollutants in surf scoters (Melanitta perspicillata) overwintering on the south coast of British Columbia, Canada. Arch. Environ. Contam. Toxicol. 2010, 59, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Letcher, R.J.; Gebbink, W.A.; Sonne, C.; Born, E.W.; McKinney, M.A.; Dietz, R. Bioaccumulation and biotransformation of brominated and chlorinated contaminants and their metabolites in ringed seals (Pusa hispida) and polar bears (Ursus maritimus) from East Greenland. Environ. Int. 2009, 35, 1118–1124. [Google Scholar] [CrossRef]
- Braune, B.M.; Gaston, A.J.; Mallory, M.L. Temporal trends of legacy organochlorines in eggs of Canadian Arctic seabirds monitored over four decades. Sci. Total Environ. 2019, 646, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.C.; Ikonomou, M.G.; MacPherson, N.; Sampson, T.; Patterson, D.A.; Dubetz, C. Tissue residue concentrations of organohalogens and trace elements in adult Pacific salmon returning to the Fraser River, British Columbia, Canada. Environ. Toxicol. Chem. 2011, 30, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Carrizo, D.; Grimalt, J.O. Gas chromatographic-mass spectrometric analysis of polychlorostyrene congener mixtures in sediments, human sera and cord sera. J. Chromatogr. A 2009, 1216, 5723–5729. [Google Scholar] [CrossRef]
- Damgaard, I.N.; Skakkebaek, N.E.; Toppari, J.; Virtanen, H.E.; Shen, H.; Schramm, K.W.; Petersen, J.H.; Jensen, T.K.; Main, K.M.; Nordic Cryptorchidism Study Group. Persistent pesticides in human breast milk and cryptorchidism. Environ. Health Perspect. 2006, 114, 1133–1138. [Google Scholar] [CrossRef]
- Soleman, S.R.; Fujitani, T.; Fujii, Y.; Harada, K.H. Levels of Octachlorostyrene in Mothers’ Milk and Potential Exposure Among Infants in Sendai City, Japan 2012. Int. J. Environ. Res. Public Health 2020, 17, 3064. [Google Scholar] [CrossRef]
- Park, E.J.; Park, K. Induction of oxidative stress in human Chang liver cells by octachlorostyrene, the persistent and bioaccumulative toxicant. Toxicol. In Vitro 2008, 22, 367–375. [Google Scholar] [CrossRef]
- Holme, J.A.; Dybing, E. Induction of liver microsomal cytochrome P-450 and associated monooxygenases by octachlorostyrene in the rat. Acta Pharmacologica et Toxicologica 1982, 50, 41–49. [Google Scholar] [CrossRef]
- Yanagiba, Y.; Ito, Y.; Kamijima, M.; Gonzalez, F.J.; Nakajima, T. Octachlorostyrene induces cytochrome P450, UDP-glucuronosyltransferase, and sulfotransferase via the aryl hydrocarbon receptor and constitutive androstane receptor. Toxicol. Sci. 2009, 111, 19–26. [Google Scholar] [CrossRef]
- Kamata, R.; Nakajima, D.; Shiraishi, F. Agonistic effects of diverse xenobiotics on the constitutive androstane receptor as detected in a recombinant yeast-cell assay. Toxicol. In Vitro 2018, 46, 335–349. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Choung, S.Y. Whole genomic expression analysis of octachlorostyrene-induced chronic toxicity in Caenorhabditis elegans. Arch. Pharm. Res. 2009, 32, 1585–1592. [Google Scholar] [CrossRef] [PubMed]
- Kabakov, A.E.; Gabai, V.L. Cell Death and Survival Assays. Methods Mol. Biol. 2018, 1709, 107–127. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Abramov, A.Y. Role of mitochondrial ros in the brain: From physiology to neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Harris, I.S.; DeNicola, G.M. The complex interplay between antioxidants and ROS in cancer. Trends Cell Biol. 2020, 30, 440–451. [Google Scholar] [CrossRef]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51, 1–3. [Google Scholar] [CrossRef]
- Rao, L.G.; Kang, N.N.; Rao, A.V. Lycopene and Other Antioxidants in the Prevention and Treatment of Osteoporosis in Postmenopausal Women. In Aging; Academic Press: Cambridge, MA, USA, 2014; Chapter 24; pp. 247–258. ISBN 9780124055230. [Google Scholar]
- Srinivas, U.S.; Tan, B.W.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef]
- Wang, S.C.; Gao, Z.Y.; Liu, F.F.; Chen, S.Q.; Liu, G.Z. Effects of polystyrene and triphenyl phosphate on growth, photosynthesis and oxidative stress of Chaetoceros meulleri. Sci, Total Environ. 2021, 797, 149180. [Google Scholar] [CrossRef]
- Ighodaro, O.M.; Akinloye, O.A. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alex. J. Med. 2019, 54, 287–293. [Google Scholar] [CrossRef]
- Ballatori, N.; Krance, S.M.; Notenboom, S.; Shi, S.; Tieu, K.; Hammond, C.L. Glutathione dysregulation and the etiology and progression of human diseases. Biol. Chem. 2009, 390, 191–214. [Google Scholar] [CrossRef]
- Bashir, M.A.; Naveed, M.; Ahmad, Z.; Gao, B.; Mustafa, A.; Nunez-Delgado, A. Combined application of biochar and sulfur regulated growth, physiological, antioxidant responses and Cr removal capacity of maize (Zea mays L.) in tannery polluted soils. J. Environ. Manag. 2020, 259, 110051. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lu, X.; Zhu, R.; Zhang, K.; Li, S.; Chen, Z.; Li, L. Betulinic Acid Induces Apoptosis in Differentiated PC12 Cells Via ROS-Mediated Mitochondrial Pathway. Neurochem. Res. 2017, 42, 1130–1140. [Google Scholar] [CrossRef] [PubMed]
- Aquilano, K.; Baldelli, S.; Ciriolo, M.R. Glutathione: New roles in redox signaling for an old antioxidant. Front. Pharmacol. 2014, 5, 196. [Google Scholar] [CrossRef]
- Ari, C.; Pilla, R.; D’Agostino, D. Nutritional/Metabolic Therapies in Animal Models of Amyotrophic Lateral Sclerosis, Alzheimer’s Disease, and Seizures. In Bioactive Nutraceuticals and Dietary Supplements in Neurological and Brain Disease; Academic Press: Cambridge, MA, USA, 2015; pp. 449–459. [Google Scholar] [CrossRef]
- Taylor, M.E.; Close, J.C.T. Dementia. Handb Clin. Neurol 2018, 159, 303–321. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Ni, Z.; Li, R.; Chang, X.; Li, W.; Yang, M.; Zhou, Z. Flurochloridone induces Sertoli cell apoptosis through ROS-dependent mitochondrial pathway. Ecotoxicol. Environ. Saf. 2021, 216, 112183. [Google Scholar] [CrossRef]
- Rossmann, M.P.; Dubois, S.M.; Agarwal, S.; Zon, L.I. Mitochondrial function in development and disease. Dis. Model. Mech. 2021, 14. [Google Scholar] [CrossRef]
- Cantoni, O.; Zito, E.; Fiorani, M.; Guidarelli, A. Arsenite impinges on endoplasmic reticulum-mitochondria crosstalk to elicit mitochondrial ROS formation and downstream toxicity. Semin. Cancer Biol. 2021, 76, 132–138. [Google Scholar] [CrossRef]
- Sun, S.; Zhao, Z.; Rao, Q.; Li, X.; Ruan, Z.; Yang, J. BDE-47 induces nephrotoxicity through ROS-dependent pathways of mitochondrial dynamics in PK15 cells. Ecotoxicol. Environ. Saf. 2021, 222, 112549. [Google Scholar] [CrossRef]
- McMinn, B.; Duval, A.L.; Sayes, C.M. An Adverse Outcome Pathway Linking Organohalogen Exposure to Mitochondrial Disease. J. Toxicol. 2019, 2019, 9246495. [Google Scholar] [CrossRef]
- Sugano, E.; Endo, Y.; Sugai, A.; Kikuchi, Y.; Tabata, K.; Ozaki, T.; Kurose, T.; Takai, Y.; Mitsuguchi, Y.; Honma, Y.; et al. Geranylgeranyl acetone prevents glutamate-induced cell death in HT-22 cells by increasing mitochondrial membrane potential. Eur. J. Pharmacol. 2020, 883, 173193. [Google Scholar] [CrossRef]
- Ma, Y.Y.; Chen, H.W.; Tzeng, C.R. Low oxygen tension increases mitochondrial membrane potential and enhances expression of antioxidant genes and implantation protein of mouse blastocyst cultured in vitro. J. Ovarian Res. 2017, 10, 47. [Google Scholar] [CrossRef]
- Lee, D.G.; Choi, B.K.; Kim, Y.H.; Oh, H.S.; Park, S.H.; Bae, Y.S.; Kwon, B.S. The repopulating cancer cells in melanoma are characterized by increased mitochondrial membrane potential. Cancer Lett. 2016, 382, 186–194. [Google Scholar] [CrossRef]
- Thiel, G.; Schmidt, T.; Rossler, O.G. Ca(2+) Microdomains, Calcineurin and the Regulation of Gene Transcription. Cells 2021, 10, 875. [Google Scholar] [CrossRef] [PubMed]
- La Rovere, R.M.; Roest, G.; Bultynck, G.; Parys, J.B. Intracellular Ca(2+) signaling and Ca(2+) microdomains in the control of cell survival, apoptosis and autophagy. Cell Calcium 2016, 60, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Barak, P.; Parekh, A.B. Signaling through Ca(2+) Microdomains from Store-Operated CRAC Channels. Cold Spring Harb. Perspect. Biol. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Wolf, I.M.A.; Guse, A.H. Ca(2+) Microdomains in T-Lymphocytes. Front. Oncol. 2017, 7, 73. [Google Scholar] [CrossRef]
- Holme, J.A.; Brinchmann, B.C.; Le Ferrec, E.; Lagadic-Gossmann, D.; Ovrevik, J. Combustion Particle-Induced Changes in Calcium Homeostasis: A Contributing Factor to Vascular Disease? Cardiovasc. Toxicol. 2019, 19, 198–209. [Google Scholar] [CrossRef]
- Brinchmann, B.C.; Le Ferrec, E.; Podechard, N.; Lagadic-Gossmann, D.; Shoji, K.F.; Penna, A.; Kukowski, K.; Kubatova, A.; Holme, J.A.; Ovrevik, J. Lipophilic Chemicals from Diesel Exhaust Particles Trigger Calcium Response in Human Endothelial Cells via Aryl Hydrocarbon Receptor Non-Genomic Signalling. Int. J. Mol. Sci. 2018, 19, 1429. [Google Scholar] [CrossRef]
- Zheng, J.; McKinnie, S.M.K.; El Gamal, A.; Feng, W.; Dong, Y.; Agarwal, V.; Fenical, W.; Kumar, A.; Cao, Z.; Moore, B.S.; et al. Organohalogens Naturally Biosynthesized in Marine Environments and Produced as Disinfection Byproducts Alter Sarco/Endoplasmic Reticulum Ca(2+) Dynamics. Environ. Sci. Technol. 2018, 52, 5469–5478. [Google Scholar] [CrossRef]
- Favaloro, B.; Allocati, N.; Graziano, V.; Di Ilio, C.; Laurenzi, V.D. Role of Apoptosis in disease. Aging 2012, 4, 330–349. [Google Scholar] [CrossRef]
- An, K.; Zhang, Y.; Liu, Y.; Yan, S.; Hou, Z.; Cao, M.; Liu, G.; Dong, C.; Gao, J.; Liu, G. Neferine induces apoptosis by modulating the ROSmediated JNK pathway in esophageal squamous cell carcinoma. Oncol. Rep. 2020, 44, 1116–1126. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Li, R.; Dai, P.; Li, Z.; Li, Y.; Li, C. Deoxynivalenol induced apoptosis and inflammation of IPEC-J2 cells by promoting ROS production. Environ. Pollut. 2019, 251, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Prima, D.O.; Vorontsova, E.V.; Makarov, A.G.; Makarov, A.Y.; Bagryanskaya, I.Y.; Mikhailovskaya, T.F.; Slizhov, Y.G.; Zibarev, A.V. Halogenated (F, Cl) 1,3-benzodiazoles, 1,2,3-benzotriazoles, 2,1,3-benzothia(selena)diazoles and 1,4-benzodiazines inducing Hep2 cell apoptosis. Mendeleev. Commun. 2017, 27, 439–442. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, G.; Wu, K.; An, J.; Shang, Y.; Zheng, K.; Yu, Z. Toxicity Assessment of Octachlorostyrene in Human Liver Carcinoma (HepG2) Cells. Int. J. Environ. Res. Public Health 2022, 19, 14272. https://doi.org/10.3390/ijerph192114272
Ren G, Wu K, An J, Shang Y, Zheng K, Yu Z. Toxicity Assessment of Octachlorostyrene in Human Liver Carcinoma (HepG2) Cells. International Journal of Environmental Research and Public Health. 2022; 19(21):14272. https://doi.org/10.3390/ijerph192114272
Chicago/Turabian StyleRen, Guofa, Kangming Wu, Jing An, Yu Shang, Kewen Zheng, and Zhiqiang Yu. 2022. "Toxicity Assessment of Octachlorostyrene in Human Liver Carcinoma (HepG2) Cells" International Journal of Environmental Research and Public Health 19, no. 21: 14272. https://doi.org/10.3390/ijerph192114272
APA StyleRen, G., Wu, K., An, J., Shang, Y., Zheng, K., & Yu, Z. (2022). Toxicity Assessment of Octachlorostyrene in Human Liver Carcinoma (HepG2) Cells. International Journal of Environmental Research and Public Health, 19(21), 14272. https://doi.org/10.3390/ijerph192114272