
mRNA-Based Vaccine Designing against Epstein-Barr Virus to Induce an Immune Response Using Immunoinformatic and Molecular Modelling Approaches

 , , , , and
, , , , and 
Abstract
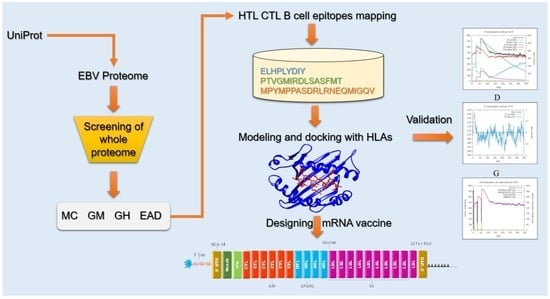
1. Introduction
2. Methodology
2.1. Collection of Proteins
2.2. MHC-I and MHC-II Binding Epitopes Prediction
2.3. Prioritization of T Cell Epitopes
2.4. Linear B Cell Epitope Prediction and Assessment
2.5. Molecular Docking of T Cell Epitopes and HLAs
2.6. mRNA Vaccine Sequence Construction
2.7. Immune Simulation
2.8. Molecular Simulation
3. Results
3.1. Proteins Retrieval for Vaccine Design
3.2. Prediction and Evaluation of MHC I Epitopes
3.3. Prediction and Evaluation of MHC II Epitopes
3.4. Epitopes’ Population Coverage
3.5. Prediction and Evaluation of Linear B Cell Epitopes
3.6. Peptide-HLAs Molecular Docking
3.7. mRNA Vaccine Construction
3.8. Immune Simulations
3.9. Molecular Simulation Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lin, Z.; Wang, X.; Strong, M.J.; Concha, M.; Baddoo, M.; Xu, G.; Baribault, C.; Fewell, C.; Hulme, W.; Hedges, D. Whole-genome sequencing of the Akata and Mutu Epstein-Barr virus strains. J. Virol. 2013, 87, 1172–1182. [Google Scholar] [CrossRef] [PubMed]
- Middeldorp, J.M.; Brink, A.A.; Van den Brule, A.J.; Meijer, C.J. Pathogenic roles for Epstein–Barr virus (EBV) gene products in EBV-associated proliferative disorders. Crit. Rev. Oncol./Hematol. 2003, 45, 1–36. [Google Scholar] [CrossRef]
- Neves, M.; Marinho-Dias, J.; Ribeiro, J.; Sousa, H. Epstein-Barr virus strains and variations: Geographic or disease-specific variants? J. Med. Virol. 2017, 89, 373–387. [Google Scholar] [CrossRef]
- Farrell, P.J. Epstein–Barr virus strain variation. Epstein Barr Virus 2015, 1, 45–69. [Google Scholar]
- Young, L.S.; Rickinson, A.B. Epstein–Barr virus: 40 years on. Nat. Rev. Cancer 2004, 4, 757–768. [Google Scholar] [CrossRef]
- Rist, M.J.; Neller, M.A.; Burrows, J.M.; Burrows, S.R. T cell epitope clustering in the highly immunogenic BZLF1 antigen of Epstein-Barr virus. J. Virol. 2015, 89, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Shannon-Lowe, C.; Rickinson, A. The global landscape of EBV-associated tumors. Front. Oncol. 2019, 9, 713. [Google Scholar] [CrossRef] [PubMed]
- Ebell, M.H. Epstein-Barr virus infectious mononucleosis. Am. Fam. Physician 2004, 70, 1279–1287. [Google Scholar] [PubMed]
- Rigopoulou, E.I.; Smyk, D.S.; Matthews, C.E.; Billinis, C.; Burroughs, A.K.; Lenzi, M.; Bogdanos, D.P. Epstein-barr virus as a trigger of autoimmune liver diseases. Adv. Virol. 2012, 2012, 987471. [Google Scholar] [CrossRef] [PubMed]
- Hislop, A.D.; Taylor, G.S.; Sauce, D.; Rickinson, A.B. Cellular responses to viral infection in humans: Lessons from Epstein-Barr virus. Annu. Rev. Immunol. 2007, 25, 587–617. [Google Scholar] [CrossRef]
- Long, H.M.; Taylor, G.S.; Rickinson, A.B. Immune defence against EBV and EBV-associated disease. Curr. Opin. Immunol. 2011, 23, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Gärtner, B.C.; Preiksaitis, J. Epstein-Barr Virus. Man. Clin. Microbiol. 2015, 1738–1753. [Google Scholar]
- Van Zyl, D.G.; Mautner, J.; Delecluse, H.-J. Progress in EBV vaccines. Front. Oncol. 2019, 9, 104. [Google Scholar] [CrossRef] [PubMed]
- Sokal, E.M.; Hoppenbrouwers, K.; Vandermeulen, C.; Moutschen, M.; Léonard, P.; Moreels, A.; Haumont, M.; Bollen, A.; Smets, F.; Denis, M. Recombinant gp350 vaccine for infectious mononucleosis: A phase 2, randomized, double-blind, placebo-controlled trial to evaluate the safety, immunogenicity, and efficacy of an Epstein-Barr virus vaccine in healthy young adults. J. Infect. Dis. 2007, 196, 1749–1753. [Google Scholar] [CrossRef] [PubMed]
- Rees, L.; Tizard, E.J.; Morgan, A.J.; Cubitt, W.D.; Finerty, S.; Oyewole-Eletu, T.A.; Owen, K.; Royed, C.; Stevens, S.J.; Shroff, R.C. A phase I trial of epstein-barr virus gp350 vaccine for children with chronic kidney disease awaiting transplantation. Transplantation 2009, 88, 1025–1029. [Google Scholar] [CrossRef] [PubMed]
- Goscé, L.; Winter, J.R.; Taylor, G.S.; Lewis, J.E.; Stagg, H.R. Modelling the dynamics of EBV transmission to inform a vaccine target product profile and future vaccination strategy. Sci. Rep. 2019, 9, 9290. [Google Scholar] [CrossRef] [PubMed]
- Haigh, T.A.; Lin, X.; Jia, H.; Hui, E.P.; Chan, A.T.; Rickinson, A.B.; Taylor, G.S. EBV latent membrane proteins (LMPs) 1 and 2 as immunotherapeutic targets: LMP-specific CD4+ cytotoxic T cell recognition of EBV-transformed B cell lines. J. Immunol. 2008, 180, 1643–1654. [Google Scholar] [CrossRef] [PubMed]
- Olotu, F.A.; Soliman, M.E. Immunoinformatics prediction of potential B-cell and T-cell epitopes as effective vaccine candidates for eliciting immunogenic responses against Epstein–Barr virus. Biomed. J. 2021, 44, 317–337. [Google Scholar] [CrossRef]
- Rappuoli, R. Bridging the knowledge gaps in vaccine design. Nat. Biotechnol. 2007, 25, 1361–1366. [Google Scholar] [CrossRef]
- Ulmer, J.B.; Mason, P.W.; Geall, A.; Mandl, C.W. RNA-based vaccines. Vaccine 2012, 30, 4414–4418. [Google Scholar] [CrossRef] [PubMed]
- Petsch, B.; Schnee, M.; Vogel, A.B.; Lange, E.; Hoffmann, B.; Voss, D.; Schlake, T.; Thess, A.; Kallen, K.-J.; Stitz, L. Protective efficacy of in vitro synthesized, specific mRNA vaccines against influenza A virus infection. Nat. Biotechnol. 2012, 30, 1210–1216. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.; Islam, S.; Chakraborty, S.; Mustafa, A.H.; Azim, K.F.; Joy, Z.F.; Hossain, M.N.; Foysal, S.H.; Hasan, M.N. Contriving a chimeric polyvalent vaccine to prevent infections caused by herpes simplex virus (type-1 and type-2): An exploratory immunoinformatic approach. J. Biomol. Struct. Dyn. 2020, 38, 2898–2915. [Google Scholar] [CrossRef]
- Nosrati, M.; Hajizade, A.; Nazarian, S.; Amani, J.; Vansofla, A.N.; Tarverdizadeh, Y. Designing a multi-epitope vaccine for cross-protection against Shigella spp: An immunoinformatics and structural vaccinology study. Mol. Immunol. 2019, 116, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Paris, R.; Bejrachandra, S.; Thongcharoen, P.; Nitayaphan, S.; Pitisuttithum, P.; Sambor, A.; Gurunathan, S.; Francis, D.; Ratto-Kim, S.; Karnasuta, C. HLA class II restriction of HIV-1 clade-specific neutralizing antibody responses in ethnic Thai recipients of the RV144 prime-boost vaccine combination of ALVAC-HIV and AIDSVAX® B/E. Vaccine 2012, 30, 832–836. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.P.; Mishra, B.N. Major histocompatibility complex linked databases and prediction tools for designing vaccines. Hum. Immunol. 2016, 77, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Consortium, U. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.V.; Lundegaard, C.; Lamberth, K.; Buus, S.; Lund, O.; Nielsen, M. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinform. 2007, 8, 424. [Google Scholar] [CrossRef] [PubMed]
- Vita, R.; Mahajan, S.; Overton, J.A.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The immune epitope database (IEDB): 2018 update. Nucleic Acids Res. 2019, 47, D339–D343. [Google Scholar] [CrossRef]
- Ghafoor, D.; Kousar, A.; Ahmed, W.; Khan, S.; Ullah, Z.; Ullah, N.; Khan, S.; Ahmed, S.; Khan, Z.; Riaz, R. Computational vaccinology guided design of multi-epitopes subunit vaccine designing against Hantaan virus and its validation through immune simulations. Infect. Genet. Evol. 2021, 93, 104950. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Ali, S.S.; Zaheer, I.; Saleem, S.; Ziaullah; Zaman, N.; Iqbal, A.; Suleman, M.; Wadood, A.; Rehman, A.U. Proteome-wide mapping and reverse vaccinology-based B and T cell multi-epitope subunit vaccine designing for immune response reinforcement against Porphyromonas gingivalis. J. Biomol. Struct. Dyn. 2020, 40, 833–847. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Khan, S.; Ali, A.; Akbar, H.; Sayaf, A.M.; Khan, A.; Wei, D.-Q. Immunoinformatics approaches to explore Helicobacter Pylori proteome (Virulence Factors) to design B and T cell multi-epitope subunit vaccine. Sci. Rep. 2019, 9, 13321. [Google Scholar] [CrossRef] [PubMed]
- Calis, J.J.; Maybeno, M.; Greenbaum, J.A.; Weiskopf, D.; De Silva, A.D.; Sette, A.; Keşmir, C.; Peters, B. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput. Biol. 2013, 9, e1003266. [Google Scholar] [CrossRef] [PubMed]
- Sormanni, P.; Aprile, F.A.; Vendruscolo, M. The CamSol method of rational design of protein mutants with enhanced solubility. J. Mol. Biol. 2015, 427, 478–490. [Google Scholar] [CrossRef]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Consortium, O.S.D.D.; Raghava, G.P. In silico approach for predicting toxicity of peptides and proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef]
- Andreatta, M.; Nielsen, M. Gapped sequence alignment using artificial neural networks: Application to the MHC class I system. Bioinformatics 2016, 32, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Patiyal, S.; Dhall, A.; Pande, A.; Arora, C.; Raghava, G.P. AlgPred 2.0: An improved method for predicting allergenic proteins and mapping of IgE epitopes. Brief. Bioinform. 2021, 22, bbaa294. [Google Scholar] [CrossRef]
- Dhanda, S.K.; Vir, P.; Raghava, G.P. Designing of interferon-gamma inducing MHC class-II binders. Biol. Direct 2013, 8, e73957. [Google Scholar] [CrossRef]
- Angelo, M.A.; Grifoni, A.; O’Rourke, P.H.; Sidney, J.; Paul, S.; Peters, B.; de Silva, A.D.; Phillips, E.; Mallal, S.; Diehl, S.A. Human CD4+ T cell responses to an attenuated tetravalent dengue vaccine parallel those induced by natural infection in magnitude, HLA restriction, and antigen specificity. J. Virol. 2017, 91, e02147-16. [Google Scholar]
- EL-Manzalawy, Y.; Dobbs, D.; Honavar, V. Predicting linear B-cell epitopes using string kernels. J. Mol. Recognit. Interdiscip. J. 2008, 21, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Lamiable, A.; Thévenet, P.; Rey, J.; Vavrusa, M.; Derreumaux, P.; Tufféry, P. PEP-FOLD3: Faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 2016, 44, W449–W454. [Google Scholar] [CrossRef] [PubMed]
- Weng, G.; Wang, E.; Wang, Z.; Liu, H.; Zhu, F.; Li, D.; Hou, T. HawkDock: A web server to predict and analyze the protein–protein complex based on computational docking and MM/GBSA. Nucleic Acids Res. 2019, 47, W322–W330. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Li, Y.; Tian, S.; Xu, L.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 4. Accuracies of MM/PBSA and MM/GBSA methodologies evaluated by various simulation protocols using PDBbind data set. Phys. Chem. Chem. Phys. 2014, 16, 16719–16729. [Google Scholar] [CrossRef] [PubMed]
- Kozak, M. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell 1986, 44, 283–292. [Google Scholar] [CrossRef]
- Liu, Q. Comparative analysis of base biases around the stop codons in six eukaryotes. Biosystems 2005, 81, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zaro, J.L.; Shen, W.-C. Fusion protein linkers: Property, design and functionality. Adv. Drug Deliv. Rev. 2013, 65, 1357–1369. [Google Scholar] [CrossRef]
- Pandey, R.K.; Bhatt, T.K.; Prajapati, V.K. Novel immunoinformatics approaches to design multi-epitope subunit vaccine for malaria by investigating anopheles salivary protein. Sci. Rep. 2018, 8, 1125. [Google Scholar] [CrossRef]
- Shey, R.A.; Ghogomu, S.M.; Esoh, K.K.; Nebangwa, N.D.; Shintouo, C.M.; Nongley, N.F.; Asa, B.F.; Ngale, F.N.; Vanhamme, L.; Souopgui, J. In-silico design of a multi-epitope vaccine candidate against onchocerciasis and related filarial diseases. Sci. Rep. 2019, 9, 4409. [Google Scholar] [CrossRef] [PubMed]
- Velders, M.P.; Weijzen, S.; Eiben, G.L.; Elmishad, A.G.; Kloetzel, P.-M.; Higgins, T.; Ciccarelli, R.B.; Evans, M.; Man, S.; Smith, L. Defined flanking spacers and enhanced proteolysis is essential for eradication of established tumors by an epitope string DNA vaccine. J. Immunol. 2001, 166, 5366–5373. [Google Scholar] [CrossRef] [PubMed]
- Ahammad, I.; Lira, S.S. Designing a novel mRNA vaccine against SARS-CoV-2: An immunoinformatics approach. International J. Biol. Macromol. 2020, 162, 820–837. [Google Scholar] [CrossRef] [PubMed]
- Gallie, D.R. The cap and poly (A) tail function synergistically to regulate mRNA translational efficiency. Genes Dev. 1991, 5, 2108–2116. [Google Scholar] [CrossRef] [PubMed]
- Munroe, D.; Jacobson, A. mRNA poly (A) tail, a 3’enhancer of translational initiation. Mol. Cell. Biol. 1990, 10, 3441–3455. [Google Scholar]
- Zhao, Y.; Moon, E.; Carpenito, C.; Paulos, C.M.; Liu, X.; Brennan, A.L.; Chew, A.; Carroll, R.G.; Scholler, J.; Levine, B.L. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res. 2010, 70, 9053–9061. [Google Scholar] [CrossRef] [PubMed]
- Holtkamp, S.; Kreiter, S.; Selmi, A.; Simon, P.; Koslowski, M.; Huber, C.; Türeci, O.z.; Sahin, U. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood 2006, 108, 4009–4017. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, P.; Ross, J. Poly (A), poly (A) binding protein and the regulation of mRNA stability. Trends Biochem. Sci. 1989, 14, 373–377. [Google Scholar] [CrossRef]
- Wang, Z.; Day, N.; Trifillis, P.; Kiledjian, M. An mRNA stability complex functions with poly (A)-binding protein to stabilize mRNA in vitro. Mol. Cell. Biol. 1999, 19, 4552–4560. [Google Scholar] [CrossRef] [PubMed]
- Pourseif, M.M.; Parvizpour, S.; Jafari, B.; Dehghani, J.; Naghili, B.; Omidi, Y. A domain-based vaccine construct against SARS-CoV-2, the causative agent of COVID-19 pandemic: Development of self-amplifying mRNA and peptide vaccines. BioImpacts BI 2021, 11, 65. [Google Scholar] [PubMed]
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational immunology meets bioinformatics: The use of prediction tools for molecular binding in the simulation of the immune system. PLoS ONE 2010, 5, e9862. [Google Scholar] [CrossRef] [PubMed]
- Castiglione, F.; Mantile, F.; De Berardinis, P.; Prisco, A. How the interval between prime and boost injection affects the immune response in a computational model of the immune system. Comput. Math. Methods Med. 2012, 2012, 842329. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham III, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [PubMed]
- Pearlman, D.A.; Case, D.A.; Caldwell, J.W.; Ross, W.S.; Cheatham III, T.E.; DeBolt, S.; Ferguson, D.; Seibel, G.; Kollman, P. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 1995, 91, 1–41. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Henson, B.W.; Perkins, E.M.; Cothran, J.E.; Desai, P. Self-assembly of Epstein-Barr virus capsids. J. Virol. 2009, 83, 3877–3890. [Google Scholar] [PubMed]
- Johannsen, E.; Luftig, M.; Chase, M.R.; Weicksel, S.; Cahir-McFarland, E.; Illanes, D.; Sarracino, D.; Kieff, E. Proteins of purified Epstein-Barr virus. Proc. Natl. Acad. Sci. USA 2004, 101, 16286–16291. [Google Scholar] [CrossRef] [PubMed]
- Lake, C.M.; Hutt-Fletcher, L.M. Epstein-Barr virus that lacks glycoprotein gN is impaired in assembly and infection. J. Virol. 2000, 74, 11162–11172. [Google Scholar]
- Oda, T.; Imai, S.; Chiba, S.; Takada, K. Epstein–Barr virus lacking glycoprotein gp85 cannot infect B cells and epithelial cells. Virology 2000, 276, 52–58. [Google Scholar]
- Murayama, K.; Nakayama, S.; Kato-Murayama, M.; Akasaka, R.; Ohbayashi, N.; Kamewari-Hayami, Y.; Terada, T.; Shirouzu, M.; Tsurumi, T.; Yokoyama, S. Crystal structure of Epstein-Barr virus DNA polymerase processivity factor BMRF1. J. Biol. Chem. 2009, 284, 35896–35905. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Hong, Y.; Dorsky, D.; Holley-Guthrie, E.; Zalani, S.; Elshiekh, N.A.; Kiehl, A.; Le, T.; Kenney, S. Functional and physical interactions between the Epstein-Barr virus (EBV) proteins BZLF1 and BMRF1: Effects on EBV transcription and lytic replication. J. Virol. 1996, 70, 5131–5142. [Google Scholar] [PubMed]
- Gustafson, E.A.; Chillemi, A.C.; Sage, D.R.; Fingeroth, J.D. The Epstein-Barr virus thymidine kinase does not phosphorylate ganciclovir or acyclovir and demonstrates a narrow substrate specificity compared to the herpes simplex virus type 1 thymidine kinase. Antimicrob. Agents Chemother. 1998, 42, 2923–2931. [Google Scholar] [PubMed]
- Gill, M.B.; Kutok, J.L.; Fingeroth, J.D. Epstein-Barr virus thymidine kinase is a centrosomal resident precisely localized to the periphery of centrioles. J. Virol. 2007, 81, 6523–6535. [Google Scholar] [PubMed]
- Cohen, J.I.; Fauci, A.S.; Varmus, H.; Nabel, G.J. Epstein-Barr virus: An important vaccine target for cancer prevention. Sci. Transl. Med. 2011, 3, 107fs107. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, S.A. Vaccines: The fourth century. Clin. Vaccine Immunol. 2009, 16, 1709–1719. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef]
- Awasthi, S.; Friedman, H.M. An mRNA vaccine to prevent genital herpes. Transl. Res. 2021, 242, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Bueno, L.L.; Lobo, F.P.; Morais, C.G.; Mourão, L.C.; de Ávila, R.A.M.; Soares, I.S.; Fontes, C.J.; Lacerda, M.V.; Olortegui, C.C.; Bartholomeu, D.C. Identification of a highly antigenic linear B cell epitope within Plasmodium vivax apical membrane antigen 1 (AMA-1). PLoS ONE 2011, 6, e21289. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Ali, S.S.; Khan, M.T.; Saleem, S.; Ali, A.; Suleman, M.; Babar, Z.; Shafiq, A.; Khan, M.; Wei, D.-Q. Combined drug repurposing and virtual screening strategies with molecular dynamics simulation identified potent inhibitors for SARS-CoV-2 main protease (3CLpro). J. Biomol. Struct. Dyn. 2021, 39, 4659–4670. [Google Scholar] [CrossRef]
- Khan, A.; Junaid, M.; Kaushik, A.C.; Ali, A.; Ali, S.S.; Mehmood, A.; Wei, D.-Q. Computational identification, characterization and validation of potential antigenic peptide vaccines from hrHPVs E6 proteins using immunoinformatics and computational systems biology approaches. PLoS ONE 2018, 13, e0196484. [Google Scholar] [CrossRef]
- Khan, A.; Zia, T.; Suleman, M.; Khan, T.; Ali, S.S.; Abbasi, A.A.; Mohammad, A.; Wei, D.-Q. Higher infectivity of the SARS-CoV-2 new variants is associated with K417N/T, E484K, and N501Y mutants: An insight from structural data. J. Cell. Physiol. 2021, 236, 7045–7057. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Heng, W.; Wang, Y.; Qiu, J.; Wei, X.; Peng, S.; Saleem, S.; Khan, M.; Ali, S.S.; Wei, D.-Q. In silico and in vitro evaluation of kaempferol as a potential inhibitor of the SARS-CoV-2 main protease (3CLpro). Phytother. Res. PTR 2021, 35, 2841–2845. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Wei, D.-Q.; Kousar, K.; Abubaker, J.; Ahmad, S.; Ali, J.; Al-Mulla, F.; Ali, S.S.; Nizam-Uddin, N.; Sayaf, A.M. Preliminary Structural Data Revealed that the SARS-CoV-2 B. 1.617 Variant’s RBD binds to ACE2 receptor stronger than the Wild Type to Enhance the Infectivity. ChemBioChem 2021, 22, 2641–2649. [Google Scholar] [CrossRef] [PubMed]
- Moutaftsi, M.; Peters, B.; Pasquetto, V.; Tscharke, D.C.; Sidney, J.; Bui, H.-H.; Grey, H.; Sette, A. A consensus epitope prediction approach identifies the breadth of murine T CD8+-cell responses to vaccinia virus. Nat. Biotechnol. 2006, 24, 817–819. [Google Scholar] [CrossRef]
- Rosendahl Huber, S.; van Beek, J.; de Jonge, J.; Luytjes, W.; van Baarle, D. T cell responses to viral infections–opportunities for peptide vaccination. Front. Immunol. 2014, 5, 171. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, U.; Peterson, K.E.; Messer, R.; Stromnes, I.M.; Race, B.; Hasenkrug, K.J. Role of interleukin-4 (IL-4), IL-12, and gamma interferon in primary and vaccine-primed immune responses to Friend retrovirus infection. J. Virol. 2001, 75, 654–660. [Google Scholar] [CrossRef]
- Rojas, J.M.; Avia, M.; Martín, V.; Sevilla, N. IL-10: A multifunctional cytokine in viral infections. J. Immunol. Res. 2017, 2017, 6104054. [Google Scholar] [CrossRef] [PubMed]
- Luckheeram, R.V.; Zhou, R.; Verma, A.D.; Xia, B. CD4+ T cells: Differentiation and functions. Clin. Dev. Immunol. 2012, 2012, 925135. [Google Scholar] [CrossRef] [PubMed]
- Kaech, S.M.; Wherry, E.J. Heterogeneity and cell-fate decisions in effector and memory CD8+ T cell differentiation during viral infection. Immunity 2007, 27, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Kaech, S.M.; Ahmed, R. Memory CD8+ T cell differentiation: Initial antigen encounter triggers a developmental program in naive cells. Nat. Immunol. 2001, 2, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Batista, F.D.; Iber, D.; Neuberger, M.S. B cells acquire antigen from target cells after synapse formation. Nature 2001, 411, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Hodgkin, P.D.; Castle, B.E.; Kehry, M.R. B cell differentiation induced by helper T cell membranes: Evidence for sequential isotype switching and a requirement for lymphokines during proliferation. Eur. J. Immunol. 1994, 24, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Mitchison, A. Latent help to and from H-2 antigens. Eur. J. Immunol. 1992, 22, 123–127. [Google Scholar] [CrossRef]
- Forthal, D.N. Functions of antibodies. Microbiol. Spectr. 2014, 2, 2–4. [Google Scholar] [CrossRef]
- O’Connor, B.P.; Cascalho, M.; Noelle, R.J. Short-lived and long-lived bone marrow plasma cells are derived from a novel precursor population. J. Exp. Med. 2002, 195, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Zhao, J.; Chen, Z. Insights into the molecular mechanisms of protein-ligand interactions by molecular docking and molecular dynamics simulation: A case of oligopeptide binding protein. Comput. Math. Methods Med. 2018, 2018, 3502514. [Google Scholar] [CrossRef] [PubMed]
- Kozak, M. Circumstances and mechanisms of inhibition of translation by secondary structure in eucaryotic mRNAs. Mol. Cell. Biol. 1989, 9, 5134–5142. [Google Scholar] [PubMed]
- Kou, Y.; Xu, Y.; Zhao, Z.; Liu, J.; Wu, Y.; You, Q.; Wang, L.; Gao, F.; Cai, L.; Jiang, C. Tissue plasminogen activator (tPA) signal sequence enhances immunogenicity of MVA-based vaccine against tuberculosis. Immunol. Lett. 2017, 190, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Kreiter, S.; Selmi, A.; Diken, M.; Sebastian, M.; Osterloh, P.; Schild, H.; Huber, C.; Türeci, Ö.; Sahin, U. Increased antigen presentation efficiency by coupling antigens to MHC class I trafficking signals. J. Immunol. 2008, 180, 309–318. [Google Scholar] [CrossRef]
- Fang, D.; Cui, K.; Mao, K.; Hu, G.; Li, R.; Zheng, M.; Riteau, N.; Reiner, S.L.; Sher, A.; Zhao, K. Transient T-bet expression functionally specifies a distinct T follicular helper subset. J. Exp. Med. 2018, 215, 2705–2714. [Google Scholar] [CrossRef]
- Snapper, C.M.; Paul, W.E. Interferon-gamma and B cell stimulatory factor-1 reciprocally regulate Ig isotype production. Science 1987, 236, 944–947. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S.NO | Protein | UniProt ID | Antigenic Score | Amino Acid Count | Function | Reference |
|---|---|---|---|---|---|---|
| 1 | Major capsid protein | P03226 | 0.42 | 1381 | Major Capsid Protein plays a key role in capsid self-assembly of EBV. | [63,64] |
| 2 | Envelope glycoprotein M | P03215 | 0.48 | 405 | Viral self-assembly. It also aids gpL and gpH in correctly incorporating into the virus membrane. | [65] |
| 3 | Envelope glycoprotein H | P03231 | 0.53 | 706 | It aids viral entry into host cells. | [66] |
| 4 | DNA polymerase processivity factor BMRF1 | P03191 | 0.6 | 404 | It has a critical role in lytic DNA replication. It also Interacts with DNA binding proteins of EBV. | [67,68] |
| 5 | Protein BDLF2 | P03225 | 0.52 | 420 | Interacts with protein BMRF2, a key role in the rearrangement of cellular actin to promote cell-to-cell of EBV in the epithelial cells. | [64] |
| 6 | Thymidine kinase | P03177 | 0.4 | 607 | Activates and re-activates the replication of viral DNA. | [69,70] |
| Protein | ID | Peptide | Comb | Antigenicity | Immunogenicity | Solubility | Toxicity |
|---|---|---|---|---|---|---|---|
| Major caps protein | 541 | ELHPLYDIY | 0.95 | 1.05 | 0.08 | 1.24 | −0.88 |
| 799 | YVDEGHADV | 0.95 | 0.43 | 0.21 | 1.91 | −0.69 | |
| Envelope glycoprotein m | 49 | LVDYGALNL | 0.8 | 1.24 | 0.15 | 1.32 | −0.68 |
| Thymine kinase | 534 | RADAVLLEV | 0.94 | 0.6 | 0.12 | 1.39 | −0.94 |
| Protein bdlf2 | 402 | LTDRSFPAY | 3.36 | 0.97 | 0.02 | 1.53 | −1.1 |
| DNA polymerase processivity factor BMRF1 | 83 | AVSFRNLAY | 2.25 | 1.71 | 0.12 | 1.36 | −1.33 |
| Proteins | Start–End | Peptide | Percentile Rank | Antigenicity | IFN Epitope | Allergenicity | Solubility | Toxicity |
|---|---|---|---|---|---|---|---|---|
| Major Caps Protein | 876–890 | PTVGMIRDLSASFMT | 0.51 | 0.43 | Positive | 0.27 | 1.22 | −0.95 |
| 172–186 | LERGLINTVLSVKLR | 0.78 | 0.73 | Positive | 0.3 | 1.07 | −1.42 | |
| Envelope Glycoprotein M | 98–112 | GEVALIKARKKVSGL | 0.04 | 0.93 | Positive | 0.3 | 1.93 | −1.35 |
| Thymine Kinase | 404–418 | LMLLRSQLLSYSDFI | 0.77 | 0.63 | Positive | 0.23 | 1.16 | −1.81 |
| Protein | ID | Peptide | Score | Antigenicity | Allergenicity |
|---|---|---|---|---|---|
| Major caps protein | 475 | NAAPAPRDRRETYSLQHRRP | 0.99 | 1.35 | 0.28 |
| 638 | PLVSLCINTYWERSGRLAFV | 0.96 | 0.7 | 0.26 | |
| Envelope glycoprotein M | 193 | FLWWVVFYLKPVVTNLYLGC | 0.82 | 0.92 | 0.3 |
| Envelope glycoprotein H | 131 | FYYIGTMLPNTRPHSYVFYQ | 0.94 | 0.5 | 0.28 |
| 272 | LEMKGGCREPELDTETLTTM | 0.91 | 0.66 | 0.29 | |
| Thymine kinase | 89 | AVTSNTGNSPGSRHTSCPFT | 1 | 0.5 | −0.2 |
| 199 | HSALKQKNGGKGKPSGLFEH | 0.99 | 0.64 | −0.1 | |
| Protein BDLF2 | 117 | EMDDTMASSGGQRGAPISAD | 0.95 | 0.72 | 0.29 |
| DNA polymerase processivity factor BMRF1 | 380 | KRTSSEARQKQKHPKKVKQA | 0.96 | 0.72 | 0.25 |
| 146 | MPYMPPASDRLRNEQMIGQV | 0.77 | 0.48 | 0.3 |
| Protein | HLA | PDB ID | Peptide | Affinity | MHC |
|---|---|---|---|---|---|
| Major Caps Protein | HLA-A*02:01 | 4L29 | ELHPLYDIY | 0.57 | MHC-I |
| HLA-A*02:06 | 3OXR | YVDEGHADV | 0.75 | MHC-I | |
| Envelope Glycoprotein M | HLA-A*02:06 | 3OXR | LVDYGALNL | 0.22 | MHC-I |
| Thymine Kinase | HLA-A*02:06 | 3OXR | RADAVLLEV | 0.55 | MHC-I |
| Protein BDLF2 | HLA-A*01:01 | 4NQX | LTDRSFPAY | 0.99 | MHC-I |
| DNA Polymerase Processivity Factor BMRF1 | HLA-B*15:01 | 5V4M | AVSFRNLAY | 0.7 | MHC-I |
| Major Caps Protein | HLA-DRB3*01:01 | 2Q6W | PTVGMIRDLSASFMT | 0.51 | MHC II |
| HLA-DRB1*07:01 | 6BIJ | LERGLINTVLSVKLR | 0.78 | MHC II | |
| Envelope Glycoprotein M | HLA-DRB5*01:01 | 1H15 | GEVALIKARKKVSGL | 0.04 | MHC II |
| Thymine Kinase | HLA-DRB1*15:01 | 1BX2 | LMLLRSQLLSYSDFI | 0.77 | MHC II |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Althurwi, H.N.; Alharthy, K.M.; Albaqami, F.F.; Altharawi, A.; Javed, M.R.; Muhseen, Z.T.; Tahir ul Qamar, M. mRNA-Based Vaccine Designing against Epstein-Barr Virus to Induce an Immune Response Using Immunoinformatic and Molecular Modelling Approaches. Int. J. Environ. Res. Public Health 2022, 19, 13054. https://doi.org/10.3390/ijerph192013054
Althurwi HN, Alharthy KM, Albaqami FF, Altharawi A, Javed MR, Muhseen ZT, Tahir ul Qamar M. mRNA-Based Vaccine Designing against Epstein-Barr Virus to Induce an Immune Response Using Immunoinformatic and Molecular Modelling Approaches. International Journal of Environmental Research and Public Health. 2022; 19(20):13054. https://doi.org/10.3390/ijerph192013054
Chicago/Turabian StyleAlthurwi, Hassan N., Khalid M. Alharthy, Faisal F. Albaqami, Ali Altharawi, Muhammad Rizwan Javed, Ziyad Tariq Muhseen, and Muhammad Tahir ul Qamar. 2022. "mRNA-Based Vaccine Designing against Epstein-Barr Virus to Induce an Immune Response Using Immunoinformatic and Molecular Modelling Approaches" International Journal of Environmental Research and Public Health 19, no. 20: 13054. https://doi.org/10.3390/ijerph192013054
APA StyleAlthurwi, H. N., Alharthy, K. M., Albaqami, F. F., Altharawi, A., Javed, M. R., Muhseen, Z. T., & Tahir ul Qamar, M. (2022). mRNA-Based Vaccine Designing against Epstein-Barr Virus to Induce an Immune Response Using Immunoinformatic and Molecular Modelling Approaches. International Journal of Environmental Research and Public Health, 19(20), 13054. https://doi.org/10.3390/ijerph192013054