



The Interplay of GPER1 with 17β-Aminoestrogens in the Regulation of the Proliferation of Cervical and Breast Cancer Cells: A Pharmacological Approach
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Molecular Modeling and Docking
2.3. Cell Culture
2.4. Cell Viability
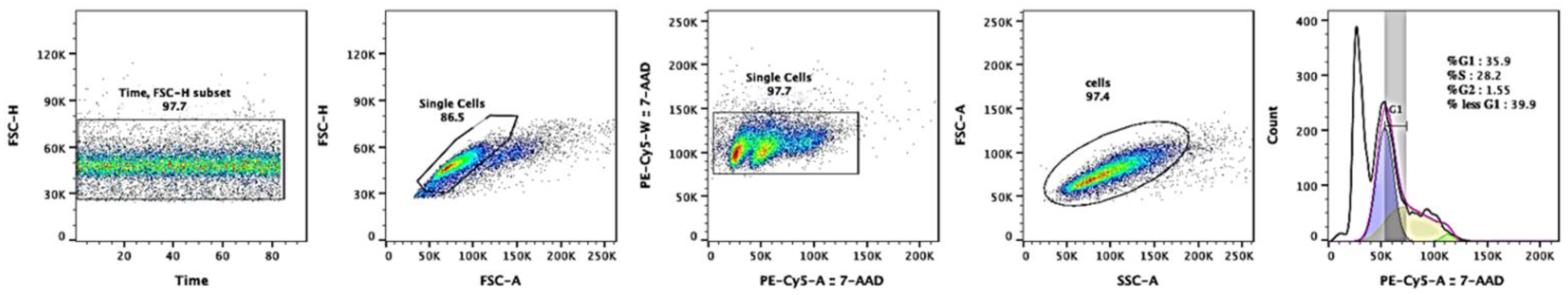
2.5. Cell-Cycle Analysis
2.6. Phosphorylation Analysis
2.7. Statistical Analysis
3. Results
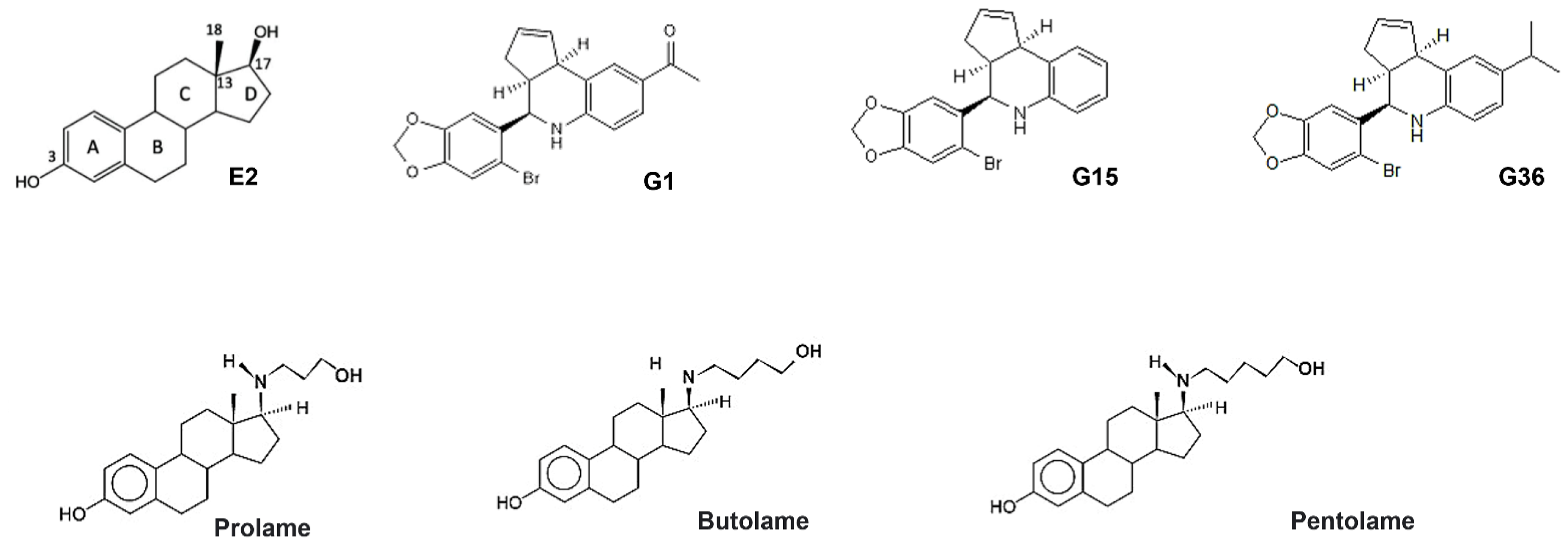
3.1. Ligand Structures
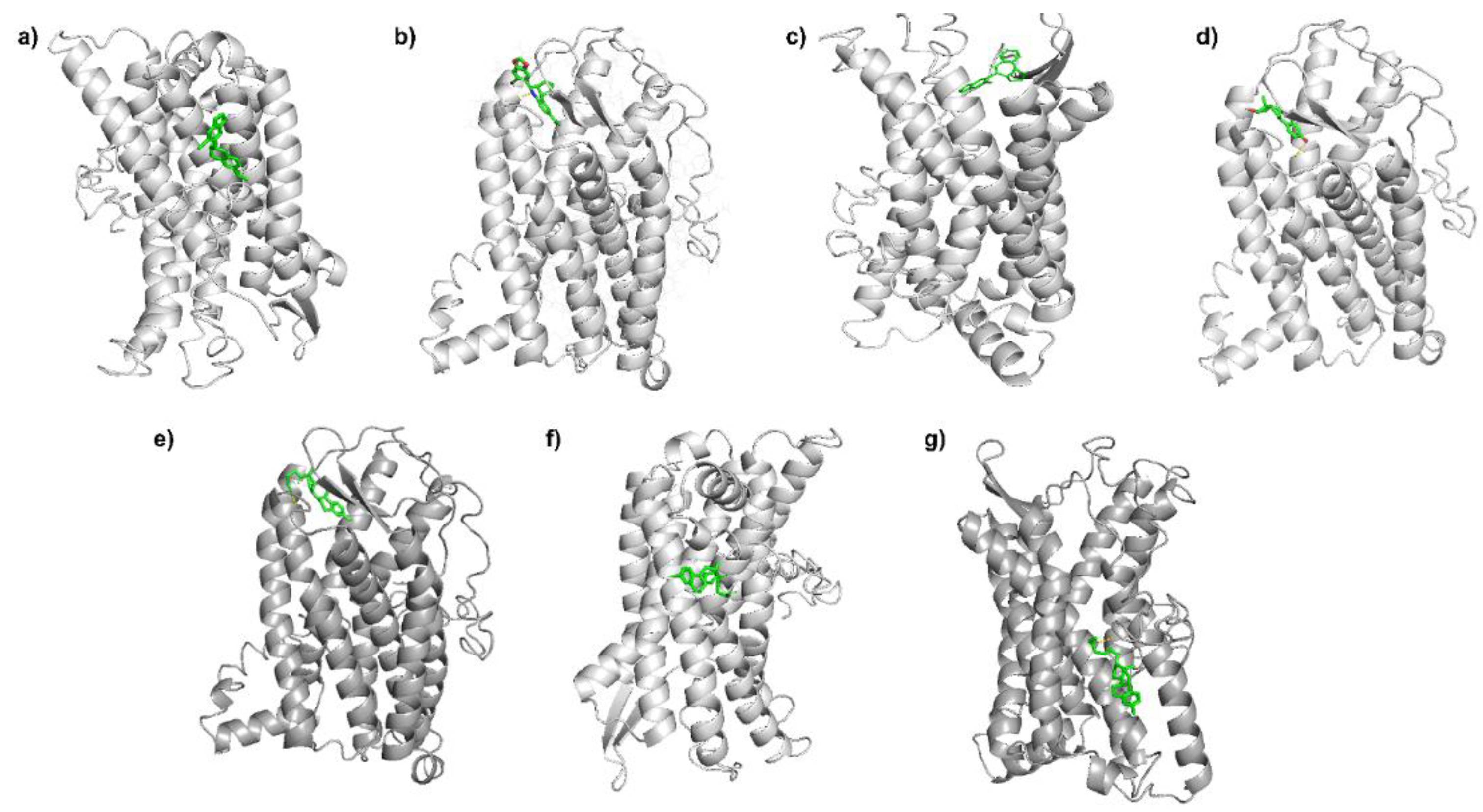
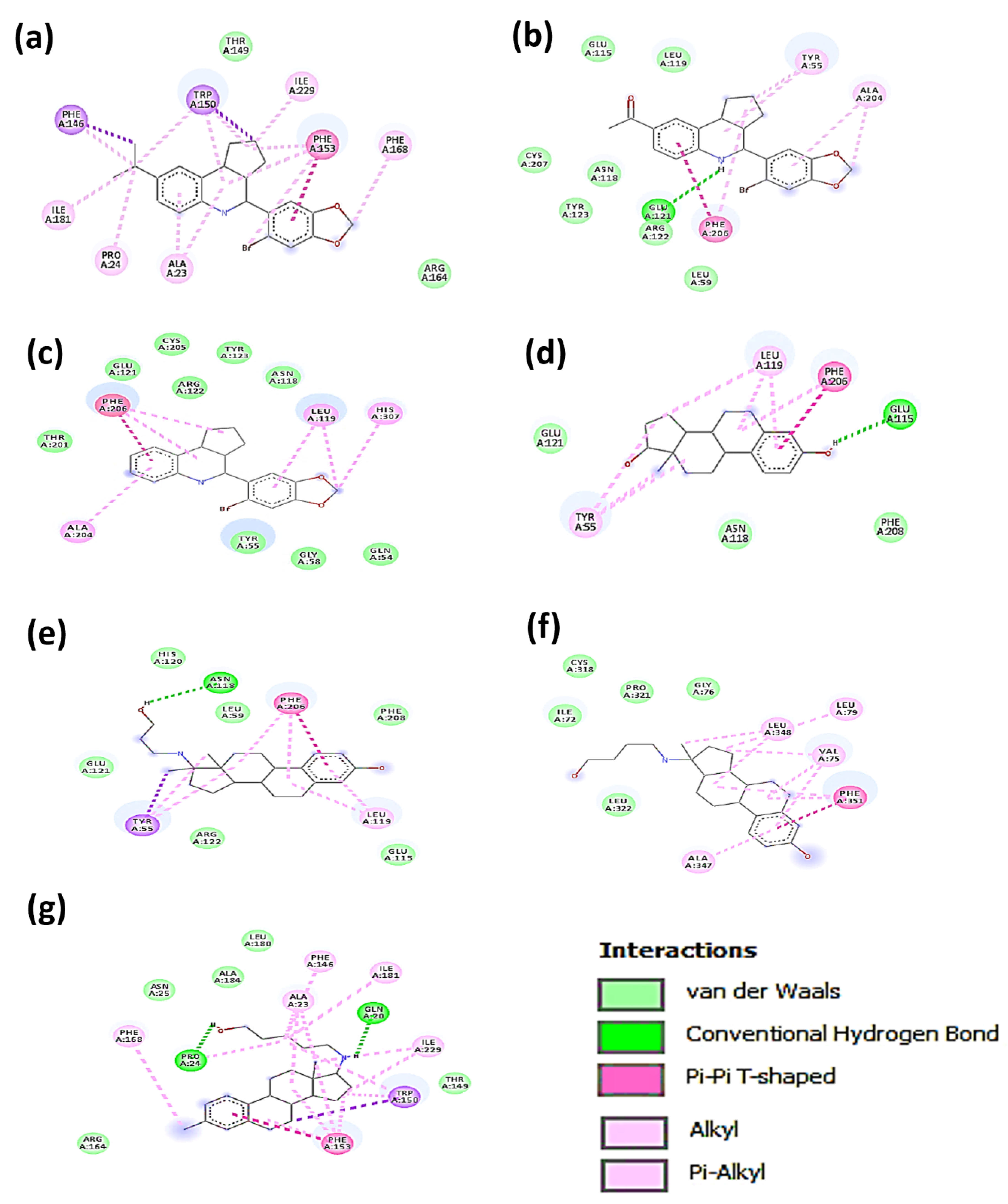
3.2. Molecular Docking Analysis
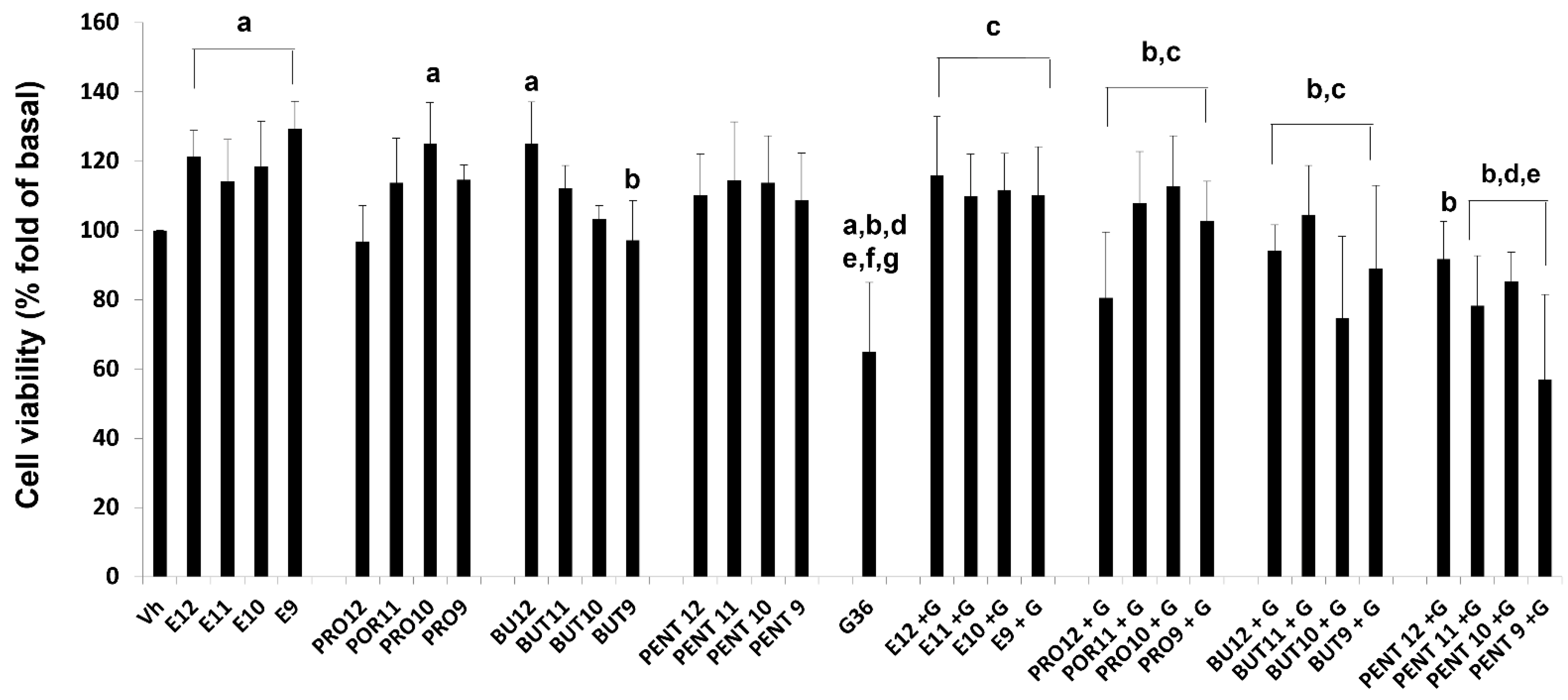
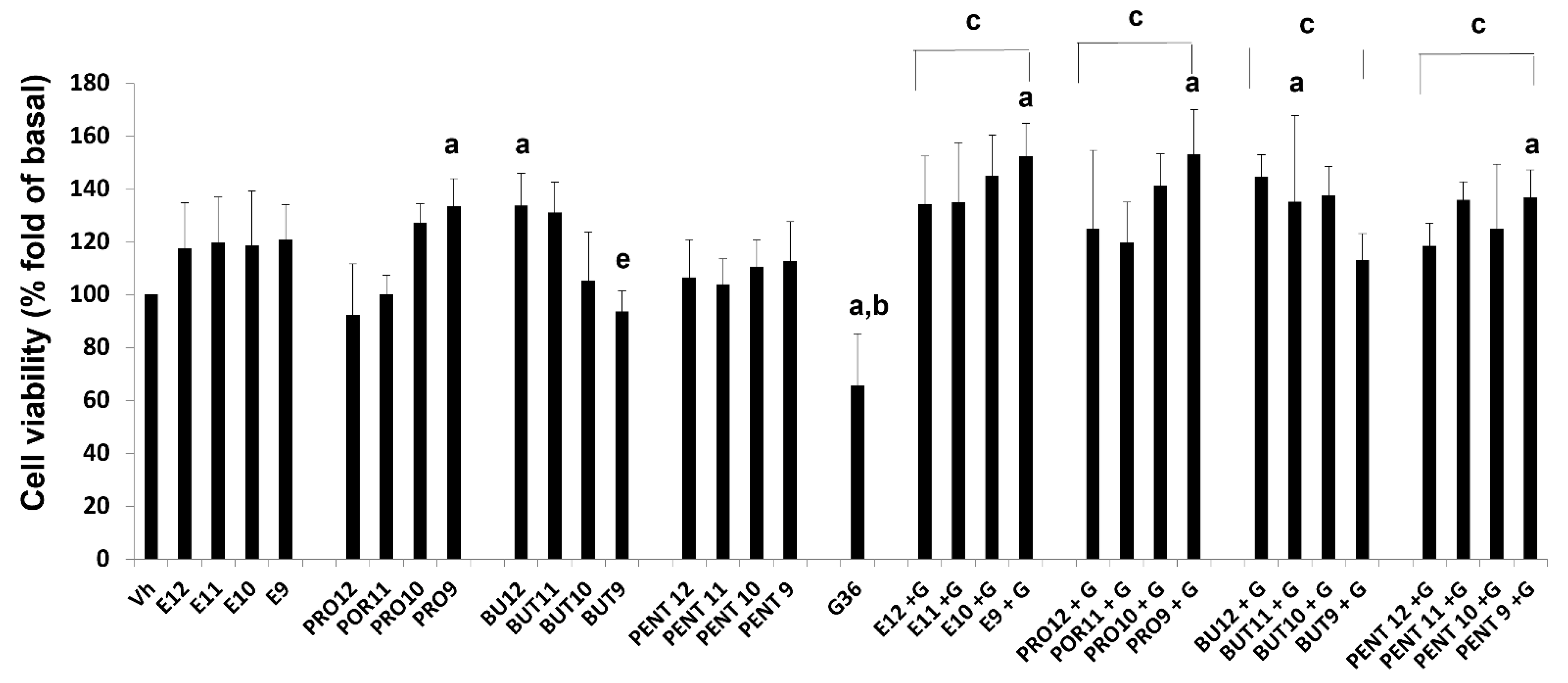
3.3. Cell Viability
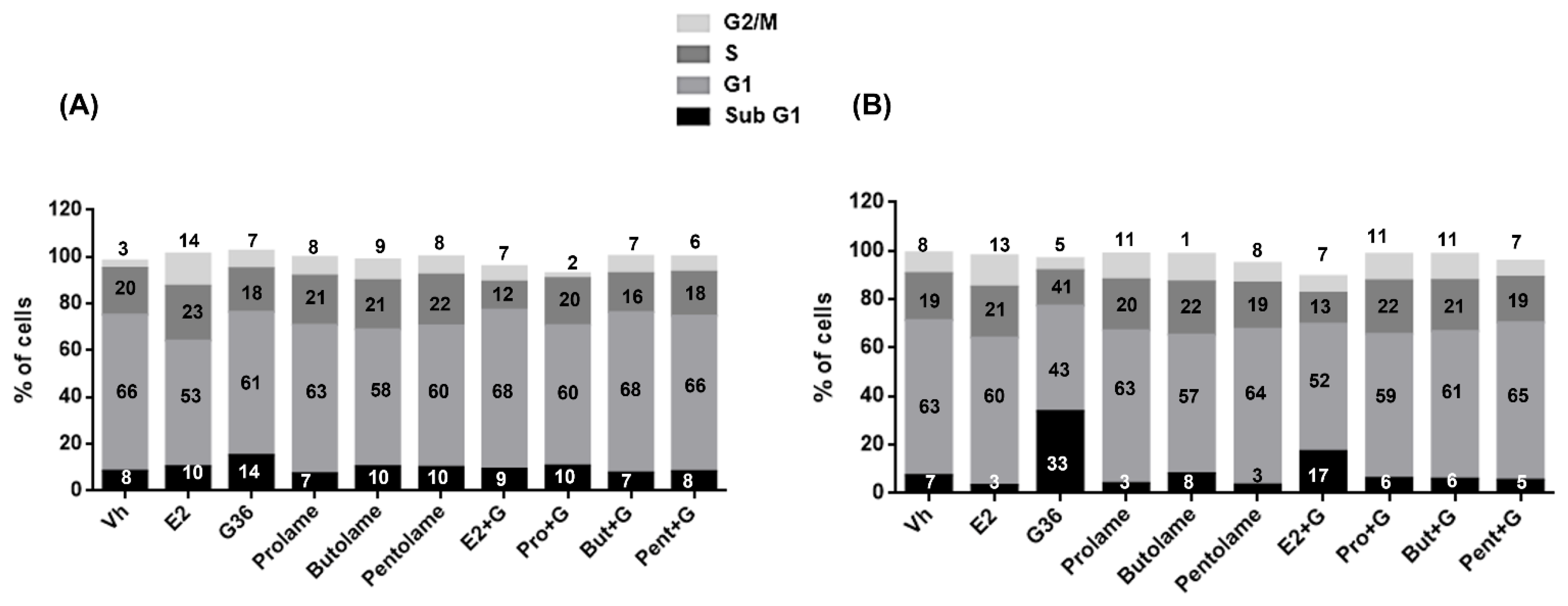
3.4. Cell Cycle
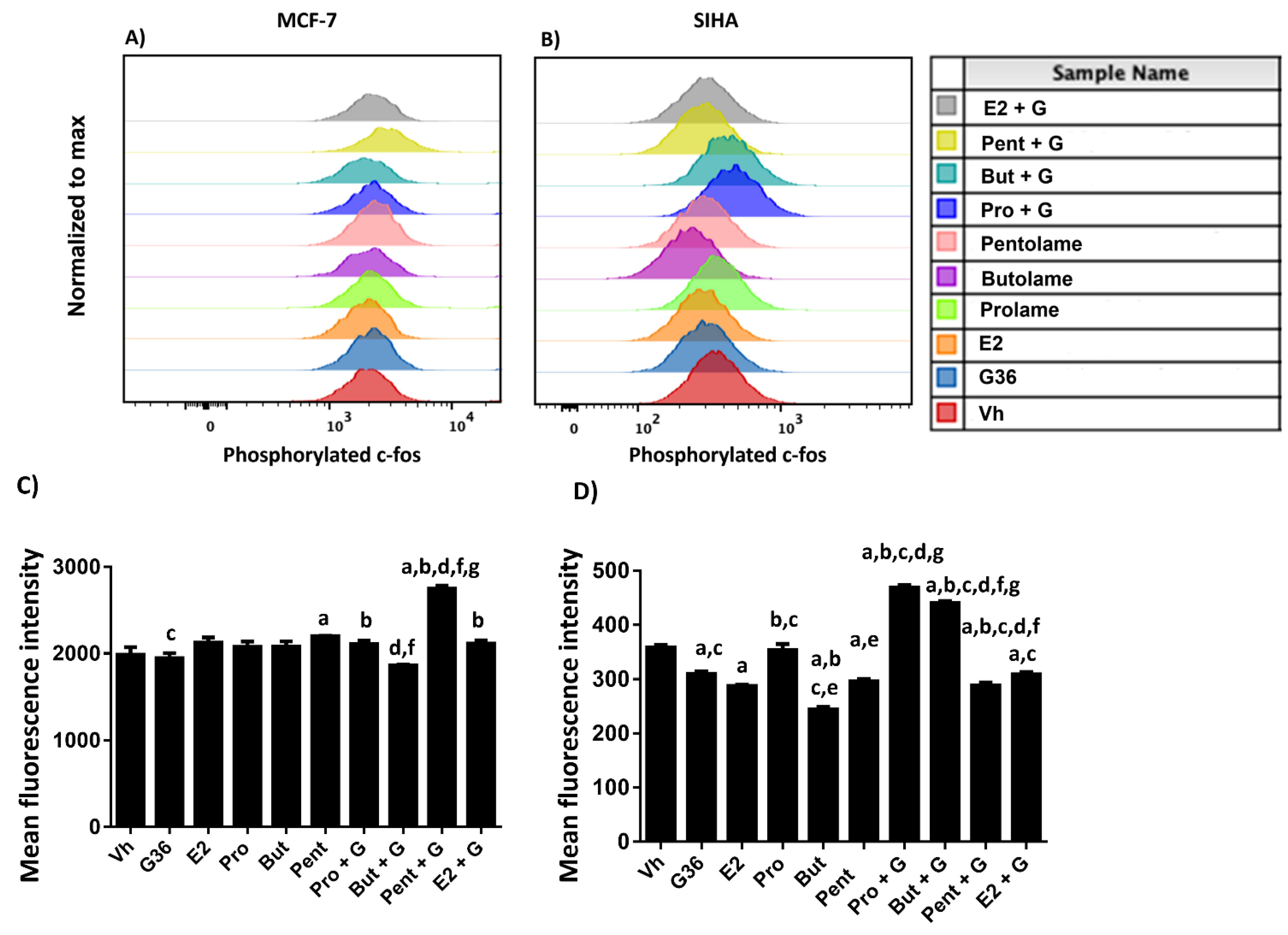
3.5. Phosphorylation Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, D.; Zhou, Q.; Labroska, V.; Qin, S.; Darbalaei, S.; Wu, Y.; Yuliantie, E.; Xie, L.; Tao, H.; Cheng, J.; et al. G protein-coupled receptors: Structure- and function-based drug discovery. Signal Transduct. Target. Ther. 2021, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Zhang, H.; Kong, Q.; Jiang, Y. Mechanisms for estrogen receptor expression in human cancer. Exp. Hematol. Oncol. 2018, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.; Wu, L.; Fu, L.; Wang, B.; Zhou, H. Unifying mechanism in the initiation of breast cancer by metabolism of estrogen (Review). Mol. Med. Rep. 2017, 16, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.; Quinn, J.; Pang, Y.; Graeber, C.; Shaw, S.; Dong, J.; Thomas, P. Activation of the novel estrogen receptor G protein-coupled receptor 30 (GPR30) at the plasma membrane. Endocrinology 2007, 148, 3236–3245. [Google Scholar] [CrossRef] [PubMed]
- Prossnitz, E.R.; Arterburn, J.B.; Sklar, L.A. GPR30: A G protein-coupled receptor for estrogen. Mol. Cell. Endocrinol. 2007, 265–266, 138–142. [Google Scholar] [CrossRef]
- Fu, X.; Simoncini, T. Extra-nuclear signaling of estrogen receptors. IUBMB Life 2008, 60, 502–510. [Google Scholar] [CrossRef]
- Meyer, M.R.; Prossnitz, E.R.; Barton, M. GPER/GPR30 and Regulation of Vascular Tone and Blood Pressure. Immunol. Endocr. Metab. Agents Med. Chem. 2011, 11, 255–261. [Google Scholar] [CrossRef]
- Notas, G.; Kampa, M.; Castanas, E. G Protein-Coupled Estrogen Receptor in Immune Cells and Its Role in Immune-Related Diseases. Front. Endocrinol (Lausanne) 2020, 11, 579420. [Google Scholar] [CrossRef]
- Sharma, G.; Prossnitz, E.R. GPER/GPR30 Knockout Mice: Effects of GPER on Metabolism. Methods Mol. Biol. 2016, 1366, 489–502. [Google Scholar]
- Prossnitz, E.R.; Sklar, L.A.; Oprea, T.I.; Arterburn, J.B. GPR30: A novel therapeutic target in estrogen-related disease. Trends Pharmacol. Sci. 2008, 29, 116–123. [Google Scholar] [CrossRef]
- Takada, Y.; Kato, C.; Kondo, S.; Korenaga, R.; Ando, J. Cloning of cDNAs encoding G protein-coupled receptor expressed in human endothelial cells exposed to fluid shear stress. Biochem. Biophys. Res. Commun. 1997, 240, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Barton, M.; Filardo, E.J.; Lolait, S.J.; Thomas, P.; Maggiolini, M.; Prossnitz, E.R. Twenty years of the G protein-coupled estrogen receptor GPER: Historical and personal perspectives. J. Steroid Biochem. Mol. Biol. 2018, 176, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.J.; Graeber, C.T.; Quinn, J.A.; Resnick, M.B.; Giri, D.; DeLellis, R.A.; Steinhoff, M.M.; Sabo, E. Distribution of GPR30, a seven membrane-spanning estrogen receptor, in primary breast cancer and its association with clinicopathologic determinants of tumor progression. Clin. Cancer Res. 2006, 12, 6359–6366. [Google Scholar] [CrossRef]
- Fujiwara, S.; Terada, S.; Kogata, Y.; Maruoka, H.; Tanaka, Y.; Tanaka, T.; Tsunetoh, S.; Sasaki, H.; Ohmichi, M. GPR30 signaling to regulate epithelial-mesenchymal transition and predict survival in ovarian cancer. J. Clin. Oncol. 2019, 37 (Suppl. 15), e17041. [Google Scholar] [CrossRef]
- Hsu, L.H.; Chu, N.M.; Lin, Y.F.; Kao, S.H. G-Protein Coupled Estrogen Receptor in Breast Cancer. Int. J. Mol. Sci. 2019, 20, 306. [Google Scholar] [CrossRef] [PubMed]
- Jacenik, D.; Beswick, E.J.; Krajewska, W.M.; Prossnitz, E.R. G protein-coupled estrogen receptor in colon function, immune regulation and carcinogenesis. World J. Gastroenterol. 2019, 25, 4092–4104. [Google Scholar] [CrossRef]
- Jung, J. Role of G Protein-Coupled Estrogen Receptor in Cancer Progression. Toxicol. Res. 2019, 35, 209–214. [Google Scholar] [CrossRef]
- Friese, K.; Kost, B.; Vattai, A.; Marme, F.; Kuhn, C.; Mahner, S.; Dannecker, C.; Jeschke, U.; Heublein, S. The G protein-coupled estrogen receptor (GPER/GPR30) may serve as a prognostic marker in early-stage cervical cancer. J. Cancer Res. Clin. Oncol. 2018, 144, 13–19. [Google Scholar] [CrossRef]
- Zhu, C.X.; Xiong, W.; Wang, M.L.; Yang, J.; Shi, H.J.; Chen, H.Q.; Niu, G. Nuclear G protein-coupled oestrogen receptor (GPR30) predicts poor survival in patients with ovarian cancer. J. Int. Med. Res. 2018, 46, 723–731. [Google Scholar] [CrossRef]
- Sriram, K.; Insel, P.A. G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol. Pharmacol. 2018, 93, 251–258. [Google Scholar] [CrossRef]
- Lemini, C.; Rubio-Poo, C.; Silva, G.; Garcia-Mondragon, J.; Zavala, E.; Mendoza-Patino, N.; Castro, D.; Cruz-Almanza, R.; Mandoki, J.J. Anticoagulant and estrogenic effects of two new 17 beta-aminoestrogens, butolame [17 beta-(4-hydroxy-1-butylamino)-1,3,5(10)-estratrien-3-ol] and pentolame [17 beta-(5-hydroxy-1-pentylamino)-1,3,5(10)-estratrien-3-ol]. Steroids 1993, 58, 457–461. [Google Scholar] [CrossRef]
- Gialeraki, A.; Valsami, S.; Pittaras, T.; Panayiotakopoulos, G.; Politou, M. Oral Contraceptives and HRT Risk of Thrombosis. Clin. Appl. Thromb. Hemost. 2018, 24, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Lemini, C.; Franco, Y.; Avila, M.E.; Jaimez, R. Estrogenic effects of 17 beta-aminoestrogens assessed in uteri of rats and mice. Eur. J. Pharmacol. 2005, 510, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Lemini, C.; Canchola, E. Effects of 17beta-aminoestrogens on the sexual behavior of female rats. Physiol. Behav. 2009, 96, 662–666. [Google Scholar] [CrossRef]
- Lemini, C.; Garcia-Albor, E.; Cruz-Lopez, B.; Matamoros-Trejo, G.; Martinez-Mota, L. Differential effect of the 17beta-aminoestrogens prolame, butolame and pentolame in anxiety and depression models in rats. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 64, 102–108. [Google Scholar] [CrossRef]
- Nissen, I.; Estrada, F.S.; Nava-Kopp, A.T.; Irles, C.; de-la-Pena-Diaz, A.; Fernandez, G.J.; Govezensky, T.; Zhang, L. Prolame ameliorates anxiety and spatial learning and memory impairment induced by ovariectomy in rats. Physiol. Behav. 2012, 106, 278–284. [Google Scholar] [CrossRef]
- James, C.D.; Morgan, I.M.; Bristol, M.L. The Relationship between Estrogen-Related Signaling and Human Papillomavirus Positive Cancers. Pathogens 2020, 9, 403. [Google Scholar] [CrossRef]
- Chung, S.H.; Franceschi, S.; Lambert, P.F. Estrogen and ERalpha: Culprits in cervical cancer? Trends Endocrinol. Metab. 2010, 21, 504–511. [Google Scholar] [CrossRef]
- Bronowicka-Klys, D.E.; Lianeri, M.; Jagodzinski, P.P. The role and impact of estrogens and xenoestrogen on the development of cervical cancer. Biomed. Pharmacother. 2016, 84, 1945–1953. [Google Scholar] [CrossRef]
- Brake, T.; Lambert, P.F. Estrogen contributes to the onset, persistence, and malignant progression of cervical cancer in a human papillomavirus-transgenic mouse model. Proc. Natl. Acad. Sci. USA 2005, 102, 2490–2495. [Google Scholar] [CrossRef]
- Läsche, M.; Gallwas, J.; Gründker, C. Like Brothers in Arms: How Hormonal Stimuli and Changes in the Metabolism Signaling Cooperate, Leading HPV Infection to Drive the Onset of Cervical Cancer. Int. J. Mol. Sci. 2022, 23, 5050. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.J.; Quinn, J.A.; Bland, K.I.; Frackelton, A.R., Jr. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol. Endocrinol. 2000, 14, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez de Valdivia, E.; Broselid, S.; Kahn, R.; Olde, B.; Leeb-Lundberg, L.M.F. G protein-coupled estrogen receptor 1 (GPER1)/GPR30 increases ERK1/2 activity through PDZ motif-dependent and -independent mechanisms. J. Biol. Chem. 2017, 292, 9932–9943. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Shui, Y.; Wang, X.; Sheng, L.; Yang, Z.; Xue, D.; Wei, Q. EGFR and HER2 expression in primary cervical cancers and corresponding lymph node metastases: Implications for targeted radiotherapy. BMC Cancer 2008, 8, 232. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.; Pang, Y.; Filardo, E.J.; Dong, J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology 2005, 146, 624–632. [Google Scholar] [CrossRef]
- Girgert, R.; Emons, G.; Grundker, C. Inactivation of GPR30 reduces growth of triple-negative breast cancer cells: Possible application in targeted therapy. Breast Cancer Res. Treat. 2012, 134, 199–205. [Google Scholar] [CrossRef]
- Mo, Z.; Liu, M.; Yang, F.; Luo, H.; Li, Z.; Tu, G.; Yang, G. GPR30 as an initiator of tamoxifen resistance in hormone-dependent breast cancer. Breast Cancer Res. 2013, 15, R114. [Google Scholar] [CrossRef]
- Molina, L.; Bustamante, F.; Ortloff, A.; Ramos, I.; Ehrenfeld, P.; Figueroa, C.D. Continuous Exposure of Breast Cancer Cells to Tamoxifen Upregulates GPER-1 and Increases Cell Proliferation. Front. Endocrinol. (Lausanne) 2020, 11, 563165. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Maggiolini, M. Mechanisms of estrogen signaling and gene expression via GPR30. Mol. Cell. Endocrinol. 2009, 308, 32–38. [Google Scholar] [CrossRef]
- Grande, F.; Occhiuzzi, M.A.; Lappano, R.; Cirillo, F.; Guzzi, R.; Garofalo, A.; Jacquot, Y.; Maggiolini, M.; Rizzuti, B. Computational Approaches for the Discovery of GPER Targeting Compounds. Front. Endocrinol. (Lausanne) 2020, 11, 517. [Google Scholar] [CrossRef]
- Bologa, C.G.; Revankar, C.M.; Young, S.M.; Edwards, B.S.; Arterburn, J.B.; Kiselyov, A.S.; Parker, M.A.; Tkachenko, S.E.; Savchuck, N.P.; Sklar, L.A.; et al. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat. Chem. Biol. 2006, 2, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Dennis, M.K.; Burai, R.; Ramesh, C.; Petrie, W.K.; Alcon, S.N.; Nayak, T.K.; Bologa, C.G.; Leitao, A.; Brailoiu, E.; Deliu, E.; et al. In vivo effects of a GPR30 antagonist. Nat. Chem. Biol. 2009, 5, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Dennis, M.K.; Field, A.S.; Burai, R.; Ramesh, C.; Petrie, W.K.; Bologa, C.G.; Oprea, T.I.; Yamaguchi, Y.; Hayashi, S.; Sklar, L.A.; et al. Identification of a GPER/GPR30 antagonist with improved estrogen receptor counterselectivity. J. Steroid Biochem. Mol. Biol. 2011, 127, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, J.M.; Rubio-Arroyo, M.F.; Soriano-Garcia, M.; Toscano, R.A.; Perez-Cesar, M.C. Synthesis and molecular structure of prolame, N-(3-hydroxy-1,3,5(10)-estratrien-17 beta-yl)-3-hydroxypropylamine; an amino-estrogen with prolonged anticoagulant and brief estrogenic effects. Steroids 1985, 45, 151–157. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Y. GPCRRD: G protein-coupled receptor spatial restraint database for 3D structure modeling and function annotation. Bioinformatics 2010, 26, 3004–3005. [Google Scholar] [CrossRef]
- Frisch, M.J.T.; Schlegel, G.W.; Scuseria, H.B.; Robb, G.E.; Cheeseman, M.A.; Zakrzewski, J.R.; Montgomery, V.G.; Stratmann, J.A., Jr.; Burant, R.E.; Dapprich, J.C.; et al. Gaussian 98, Revision A.9; Gaussian, Inc.: Pittsburgh, PA, USA, 1998. [Google Scholar]
- Orellana, E.A.; Kasinski, A.L. Sulforhodamine B (SRB) Assay in Cell Culture to Investigate Cell Proliferation. Bio Protoc. 2016, 6, e1984. [Google Scholar] [CrossRef]
- Stanczyk, F.Z.; Clarke, N.J. Measurement of Estradiol—Challenges Ahead. J. Clin. Endocrinol. Metab. 2014, 99, 56–58. [Google Scholar] [CrossRef]
- Vargiu, V.; Amar, I.D.; Rosati, A.; Dinoi, G.; Turco, L.C.; Capozzi, V.A.; Scambia, G.; Villa, P. Hormone replacement therapy and cervical cancer: A systematic review of the literature. Climacteric 2021, 24, 120–127. [Google Scholar] [CrossRef]
- Roura, E.; Travier, N.; Waterboer, T.; de Sanjose, S.; Bosch, F.X.; Pawlita, M.; Pala, V.; Weiderpass, E.; Margall, N.; Dillner, J.; et al. The Influence of Hormonal Factors on the Risk of Developing Cervical Cancer and Pre-Cancer: Results from the EPIC Cohort. PLoS ONE 2016, 11, e0147029. [Google Scholar]
- Breast cancer and hormone replacement therapy: Collaborative reanalysis of data from 51 epidemiological studies of 52,705 women with breast cancer and 108,411 women without breast cancer. Collaborative Group on Hormonal Factors in Breast Cancer. Lancet 1997, 350, 1047–1059. [CrossRef]
- Vinogradova, Y.; Coupland, C.; Hippisley-Cox, J. Use of hormone replacement therapy and risk of breast cancer: Nested case-control studies using the QResearch and CPRD databases. BMJ 2020, 371, m3873. [Google Scholar] [CrossRef] [PubMed]
- Soriano-Ursua, M.A.; Trujillo-Ferrara, J.G.; Correa-Basurto, J.; Vilar, S. Recent structural advances of beta1 and beta2 adrenoceptors yield keys for ligand recognition and drug design. J. Med. Chem. 2013, 56, 8207–8223. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Luna, D.; Martinez-Archundia, M.; Maroun, R.C.; Ceballos-Reyes, G.; Fragoso-Vazquez, M.J.; Gonzalez-Juarez, D.E.; Correa-Basurto, J. Deciphering the GPER/GPR30-agonist and antagonists interactions using molecular modeling studies, molecular dynamics, and docking simulations. J. Biomol. Struct. Dyn. 2015, 33, 2161–2172. [Google Scholar] [CrossRef] [PubMed]
- Yong, L.; Huang, M.; Wei, Y.; Xu, J.; Yi, Z. Investigating the interaction between three perfluorinated carboxylic acids and the G protein-coupled estrogen receptor: Spectroscopic analyses and computational simulations. Anal. Methods 2020, 12, 3944–3953. [Google Scholar] [CrossRef] [PubMed]
- Lappano, R.; Rosano, C.; Santolla, M.F.; Pupo, M.; De Francesco, E.M.; De Marco, P.; Ponassi, M.; Spallarossa, A.; Ranise, A.; Maggiolini, M. Two novel GPER agonists induce gene expression changes and growth effects in cancer cells. Curr. Cancer Drug Targets 2012, 12, 531–542. [Google Scholar] [CrossRef]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef]
- Pinzi, L.; Rastelli, G. Molecular Docking: Shifting Paradigms in Drug Discovery. Int. J. Mol. Sci. 2019, 20, 4331. [Google Scholar] [CrossRef]
- Lappano, R.; Rosano, C.; Pisano, A.; Santolla, M.F.; De Francesco, E.M.; De Marco, P.; Dolce, V.; Ponassi, M.; Felli, L.; Cafeo, G.; et al. A calixpyrrole derivative acts as an antagonist to GPER, a G-protein coupled receptor: Mechanisms and models. Dis. Model Mech. 2015, 8, 1237–1246. [Google Scholar]
- Zhang, Q.; Wu, Y.Z.; Zhang, Y.M.; Ji, X.H.; Hao, Q. Activation of G-protein coupled estrogen receptor inhibits the proliferation of cervical cancer cells via sustained activation of ERK1/2. Cell Biochem. Funct. 2015, 33, 134–142. [Google Scholar] [CrossRef]
- Notas, G.; Kampa, M.; Pelekanou, V.; Castanas, E. Interplay of estrogen receptors and GPR30 for the regulation of early membrane initiated transcriptional effects: A pharmacological approach. Steroids 2012, 77, 943–950. [Google Scholar] [CrossRef]
- Tian, J.M.; Ran, B.; Zhang, C.L.; Yan, D.M.; Li, X.H. Estrogen and progesterone promote breast cancer cell proliferation by inducing cyclin G1 expression. Braz. J. Med. Biol. Res. 2018, 51, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Scaling, A.L.; Prossnitz, E.R.; Hathaway, H.J. GPER mediates estrogen-induced signaling and proliferation in human breast epithelial cells and normal and malignant breast. Horm. Cancer 2014, 5, 146–160. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liao, Y.; Fan, S.; Fu, X.; Xiong, J.; Zhou, S.; Zou, M.; Wang, J. G-Protein-Coupled Estrogen Receptor Antagonist G15 Decreases Estrogen-Induced Development of Non-Small Cell Lung Cancer. Oncol. Res. 2019, 27, 283–292. [Google Scholar] [CrossRef]
- Lewis, J.S.; Vijayanathan, V.; Thomas, T.J.; Pestell, R.G.; Albanese, C.; Gallo, M.A.; Thomas, T. Activation of cyclin D1 by estradiol and spermine in MCF-7 breast cancer cells: A mechanism involving the p38 MAP kinase and phosphorylation of ATF-2. Oncol. Res. 2005, 15, 113–128. [Google Scholar] [CrossRef]
- Maggiolini, M.; Vivacqua, A.; Fasanella, G.; Recchia, A.G.; Sisci, D.; Pezzi, V.; Montanaro, D.; Musti, A.M.; Picard, D.; Ando, S. The G protein-coupled receptor GPR30 mediates c-fos up-regulation by 17beta-estradiol and phytoestrogens in breast cancer cells. J. Biol. Chem. 2004, 279, 27008–27016. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Shen, Q.; DuPré, E.; Kim, H.; Hilsenbeck, S.; Brown, P.H. cFos is critical for MCF-7 breast cancer cell growth. Oncogene 2005, 24, 6516–6524. [Google Scholar] [CrossRef]
- Ruutu, M.; Wahlroos, N.; Syrjanen, K.; Johansson, B.; Syrjanen, S. Effects of 17beta-estradiol and progesterone on transcription of human papillomavirus 16 E6/E7 oncogenes in CaSki and SiHa cell lines. Int. J. Gynecol. Cancer 2006, 16, 1261–1268. [Google Scholar] [CrossRef]
- Liu, Y.; Tian, L.B.; Yang, H.Y.; Zhang, H.P. Effects of estradiol and progesterone on the growth of HeLa cervical cancer cells. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 3959–3965. [Google Scholar]
- Tian, W.J.; Huang, M.L.; Qin, Q.F.; Chen, Q.; Fang, K.; Wang, P.L. Prognostic Impact of Epidermal Growth Factor Receptor Overexpression in Patients with Cervical Cancer: A Meta-Analysis. PLoS ONE 2016, 11, e0158787. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Interaction | ||||
|---|---|---|---|---|
| Compound | Van der Waals | Hydrogen Bonds | Pi-Interactions | Alkyl |
| G36 | ARG: 164 THR: 149 | ALA: 23 PRO: 24 PHE: 146 TRP: 150 PHE: 153 ILE: 181 | PHE: 168 ILE: 229 | |
| G1 | LEU: 59 GLU: 115 LEU: 119 TM HELIX 2 ASN: 118 ARG: 122 TYR: 123 CYS: 207 | GLU: 121 | ALA: 204 PHE: 206 | TYR: 55 |
| G15 | GLN: 54 TYR: 55 GLY: 58 ASN: 118 GLU: 121 ARG: 122 TYR: 123 THR: 201 CYS: 205 | LEU: 119 PHE: 206 | HIS: 307 TM IV | |
| E2 | ASN: 118 GLU: 121 PHE: 208 | GLU: 115 | LEU: 119 PHE: 206 | TYR: 55 |
| Prolame | LEU: 59 GLU: 115 HIS: 120 GLU: 121 ARG: 122 PHE: 208 | ASN: 118 | TYR: 55 LEU: 119 PHE: 206 | |
| Butolame | ILE: 72 GLY: 76 CIS: 318 PRO: 321 LEU: 322 | ALA: 347 PHE: 351 | VAL: 75 LEU: 79 LEU: 348 | |
| Pentolame | ASN: 25 THR: 149 ARG: 164 LEU: 180 ALA: 184 | GLN: 20 PRO: 24 | TRP: 150 PHE: 153 | ALA: 23 PHE: 146 PHE: 168 ILE: 181 ILE: 229 |
| Compound | ΔG (kcal/mol) |
|---|---|
| G36 | −9.2 |
| G1 | −8.9 |
| G15 | −8.6 |
| E2 | −8.5 |
| Prolame | −8 |
| Pentolame | −7.4 |
| Butolame | −7.3 |
| Sub G1 | G1 | S | G2/M | |
|---|---|---|---|---|
| Vh | 8.2 ± 0.3 | 66.7 ± 6.9 | 20.0 ± 4.8 | 3.06 ± 0.2 |
| E2 | 10.0 ± 3.0 | 53.7 ± 5.0 | 23.5 ± 5.0 | 13.9 ± 4.0 a |
| G36 | 14.9 ± 1.7 a,e | 61.2 ± 2.8 | 18.6 ± 2.8 | 7.5 ± 0.6 b |
| Prolame | 7.0 ± 1.0 c | 63.6 ± 8.5 | 21.0 ± 8.5 | 8.0 ± 2.7 |
| Butolame | 10.2 ± 0.9 | 58.3 ± 4.1 | 21.0 ± 4.1 | 8.9 ± 1.4 |
| Pentolame | 9.8 ± 1.0 | 60.4 ± 4.2 | 21.7 ± 4.2 | 7.9 ± 1.1 |
| E2 + G | 9.0 ± 0.7 | 67.9 ± 9.5 | 12.0 ± 9.5 | 6.7 ± 1.4 b |
| Pro + G | 10.3 ± 2.2 | 60.0 ± 4.7 | 20.1 ± 4.7 | 2.2 ± 1.3 b,d |
| But + G | 7.3 ± 0.1 | 68.6 ± 14.0 | 16.6 ± 14.0 | 7.4 ± 2.6 b |
| Pent + G | 8.0 ± 0.8 | 66.4 ± 9.7 | 18.7 ± 9.7 | 6.6 ± 2.3 b |
| Sub G1 | G1 | S | G2/M | |
|---|---|---|---|---|
| Vh | 7.1 ± 2.3 c | 63.8 ± 8.1 c | 19.4 ± 4.1 | 8.5 ± 9.1 |
| E2 | 3.04 ± 3.4 c | 60.7 ± 2.4 c | 21.1 ± 5.7 | 12.9 ± 6.5 |
| G36 | 33.6 ± 8.0 a,b,d,e,f,g | 43.4 ± 5.6 | 14.5 ± 2.2 | 4.9 ± 3.5 |
| Prolame | 3.8 ± 2.3 c | 63.0 ± 1.2 c | 20.9 ± 6.4 | 10.7 ± 5.9 |
| Butolame | 7.8 ± 2.3 c | 57.3 ± 1.2 | 21.7 ± 6.4 | 11.4 ± 5.9 |
| Pentolame | 3.3 ± 0.3 c | 64.0 ± 6.2 c | 19.1 ± 4.3 | 8.2 ± 8.9 |
| E2 + G | 17.0 ± 1.2 b,c | 52.4 ± 10.0 | 12.8 ± 3.3 | 6.9 ± 9.2 |
| Pro + G | 5.8 ± 5.7 c | 59.5 ± 3.1 | 22.0 ± 5.1 | 11.0 ± 4.5 |
| But + G | 5.6 ± 5.6 a,c | 61.0 ± 4.5 c | 20.9 ± 4.4 | 10.8 ± 5.6 |
| Pent + G | 5.2 ± 3.5 c | 64.8 ± 5.6 c | 18.8 ± 3.7 | 6.6 ± 2.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Segovia-Mendoza, M.; Mirzaei, E.; Prado-Garcia, H.; Miranda, L.D.; Figueroa, A.; Lemini, C. The Interplay of GPER1 with 17β-Aminoestrogens in the Regulation of the Proliferation of Cervical and Breast Cancer Cells: A Pharmacological Approach. Int. J. Environ. Res. Public Health 2022, 19, 12361. https://doi.org/10.3390/ijerph191912361
Segovia-Mendoza M, Mirzaei E, Prado-Garcia H, Miranda LD, Figueroa A, Lemini C. The Interplay of GPER1 with 17β-Aminoestrogens in the Regulation of the Proliferation of Cervical and Breast Cancer Cells: A Pharmacological Approach. International Journal of Environmental Research and Public Health. 2022; 19(19):12361. https://doi.org/10.3390/ijerph191912361
Chicago/Turabian StyleSegovia-Mendoza, Mariana, Elahe Mirzaei, Heriberto Prado-Garcia, Luis D. Miranda, Alejandra Figueroa, and Cristina Lemini. 2022. "The Interplay of GPER1 with 17β-Aminoestrogens in the Regulation of the Proliferation of Cervical and Breast Cancer Cells: A Pharmacological Approach" International Journal of Environmental Research and Public Health 19, no. 19: 12361. https://doi.org/10.3390/ijerph191912361
APA StyleSegovia-Mendoza, M., Mirzaei, E., Prado-Garcia, H., Miranda, L. D., Figueroa, A., & Lemini, C. (2022). The Interplay of GPER1 with 17β-Aminoestrogens in the Regulation of the Proliferation of Cervical and Breast Cancer Cells: A Pharmacological Approach. International Journal of Environmental Research and Public Health, 19(19), 12361. https://doi.org/10.3390/ijerph191912361