
Metagenomic Analysis of Microbial Alliances for Efficient Degradation of PHE: Microbial Community Structure and Reconstruction of Metabolic Network

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Soil Sampling and Enrichment
2.2. PHE Degradation Experiment
2.3. Extraction and Detection of PHE
2.4. Illumina MiSeq Sequencing of Microbial Community and Analysis
3. Results and Discussion
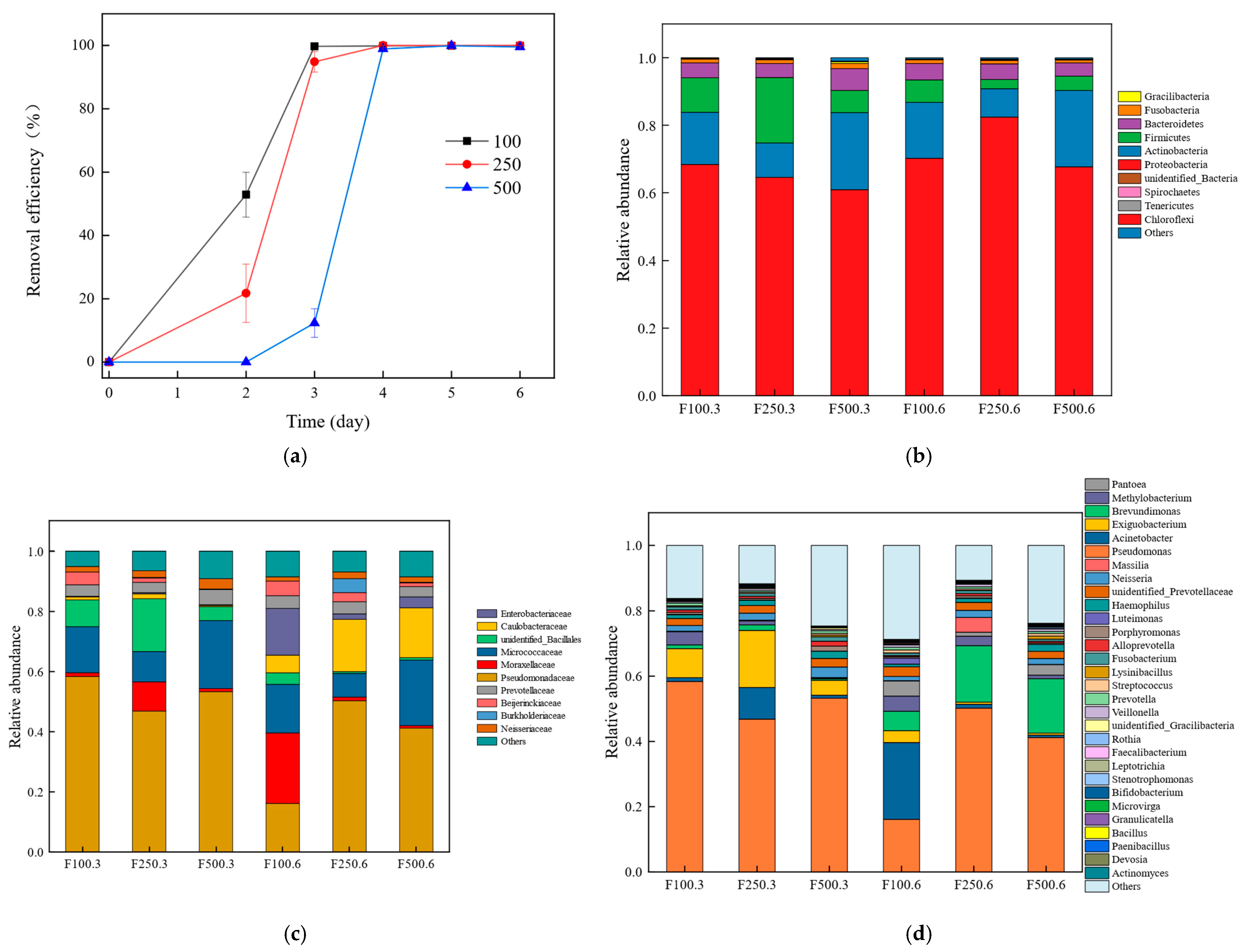
3.1. The Degradation of PHE
3.2. Microbial Community Structure of the Six Samples
3.3. Metabolic Analysis of Evolutionary Genealogy of Genes: Non-Supervised Orthologous Groups Database (eggNOG)
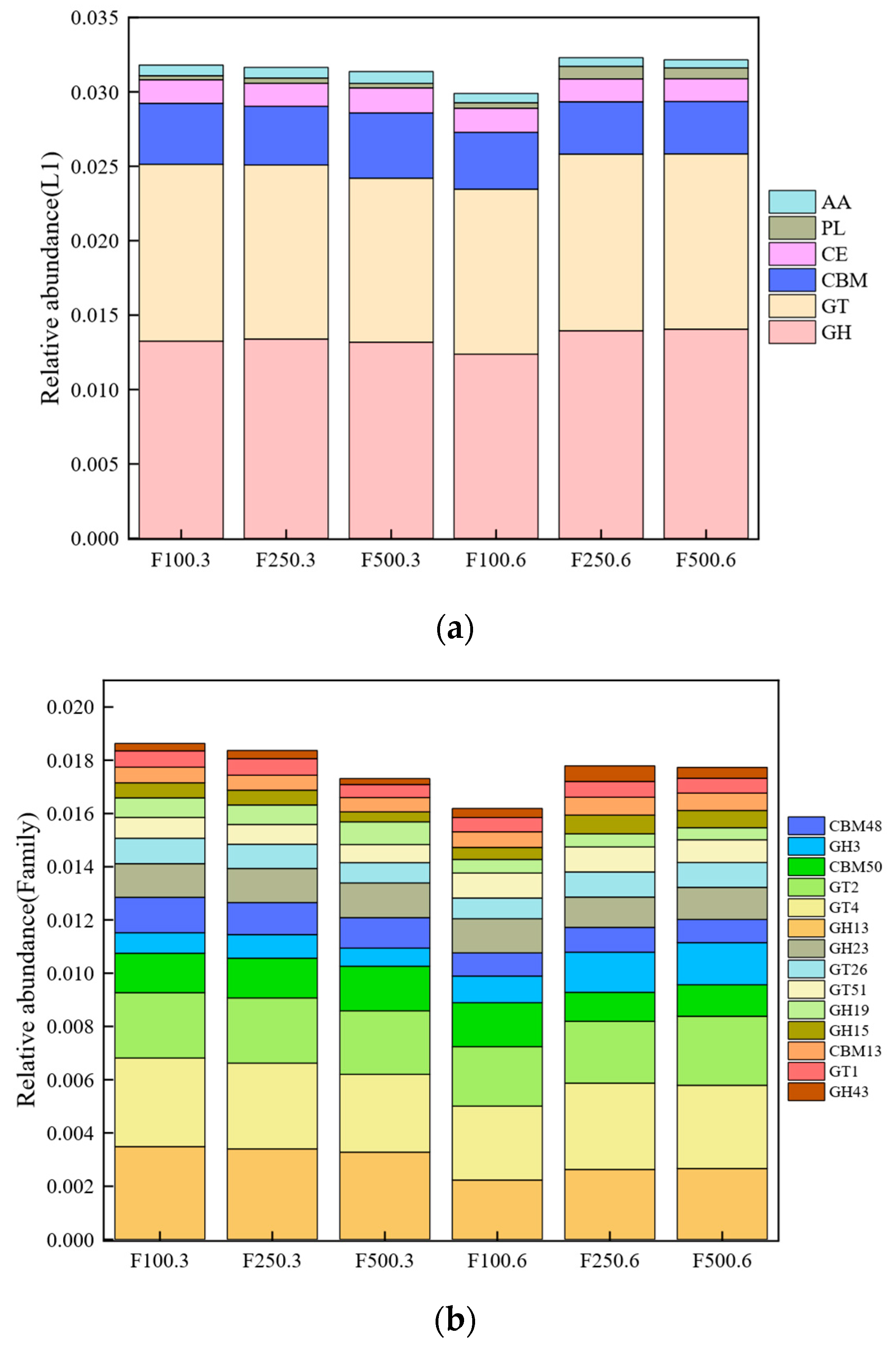
3.4. Metagenomic Analysis of Carbohydrate-Active Enzymes (CAZy)
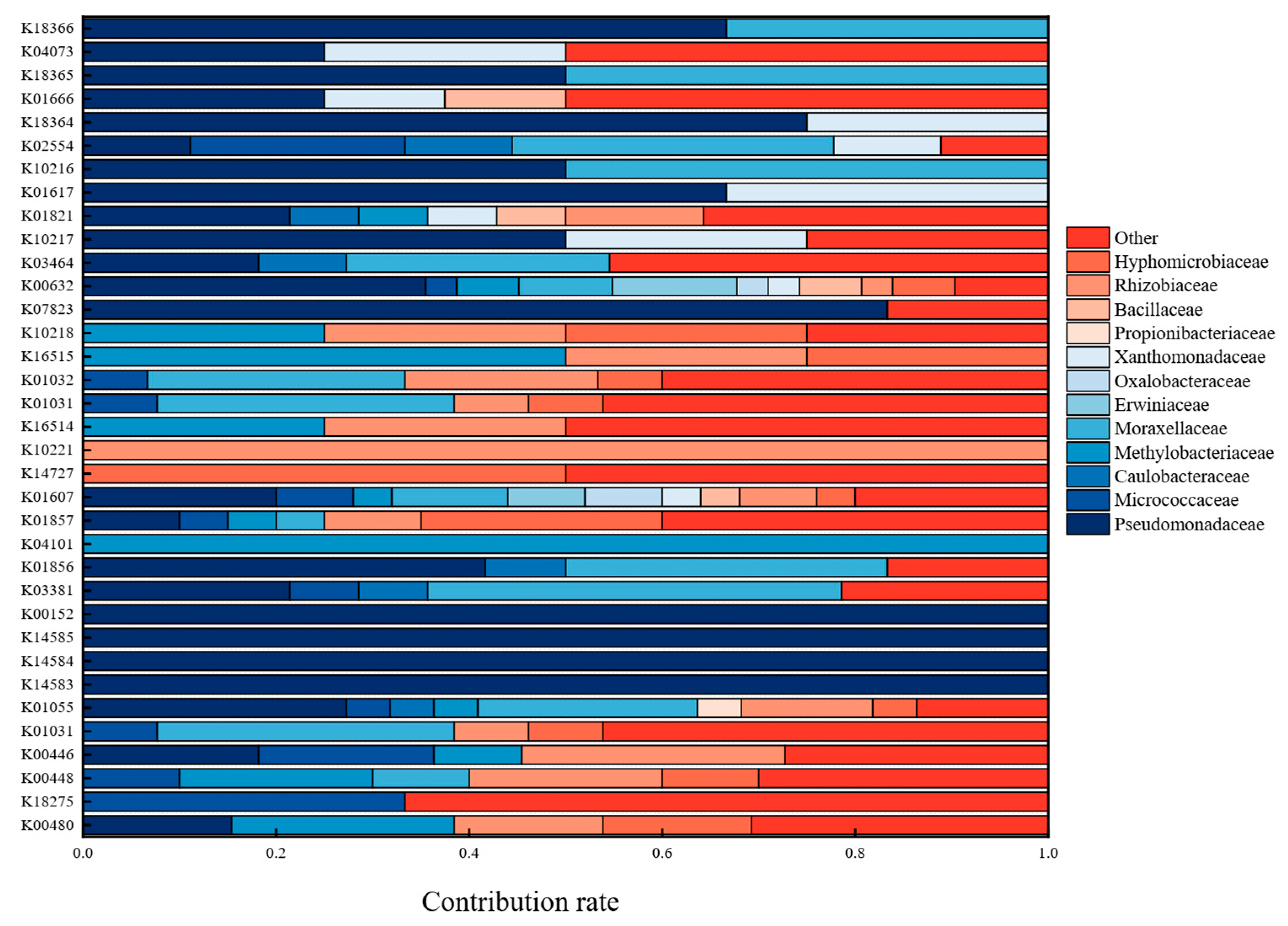
3.5. Metagenomic Analysis of Kyoto Encyclopedia of Genes and Genomes (KEGG)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Umar, Z.D.; Aziz, N.A.A.; Zulkifli, S.Z.; Mustafa, M. Rapid biodegradation of polycyclic aromatic hydrocarbons (PAHs) using effective Cronobacter sakazakii MM045 (KT933253). MethodsX 2017, 4, 104–117. [Google Scholar] [CrossRef]
- Chen, H.; Song, Q.; Diao, X.; Zhou, H. Proteomic and metabolomic analysis on the toxicological effects of Benzo[a]pyrene in pearl oyster Pinctada martensii. Aquat. Toxicol. 2016, 175, 81–89. [Google Scholar] [CrossRef]
- White, P.A.; Claxton, L. Mutagens in contaminated soil: A review. Mutat. Res. Mutat. Res. 2004, 567, 227–345. [Google Scholar] [CrossRef]
- Yu, Y.; Wang, X.; Wang, B.; Tao, S.; Liu, W.; Wang, X.; Cao, J.; Li, B.; Lu, X.; Wong, M.H. Polycyclic Aromatic Hydrocarbon Residues in Human Milk, Placenta, and Umbilical Cord Blood in Beijing, China. Environ. Sci. Technol. 2011, 45, 10235–10242. [Google Scholar] [CrossRef]
- Saikia, J.; Khare, P.; Saikia, P.; Saikia, B. Polycyclic aromatic hydrocarbons (PAHs) around tea processing industries using high-sulfur coals. Environ. Geochem. Health 2017, 39, 1101–1116. [Google Scholar] [CrossRef]
- Markiewicz, A.; Björklund, K.; Eriksson, E.; Kalmykova, Y.; Strömvall, A.-M.; Siopi, A. Emissions of organic pollutants from traffic and roads: Priority pollutants selection and substance flow analysis. Sci. Total Environ. 2017, 580, 1162–1174. [Google Scholar] [CrossRef]
- Dhar, K.; Subashchandrabose, S.R.; Venkateswarlu, K.; Krishnan, K.; Megharaj, M. Anaerobic Microbial Degradation of Polycyclic Aromatic Hydrocarbons: A Comprehensive Review. In Reviews of Environmental Contamination and Toxicology; de Voogt, P., Ed.; Springer International Publishing: Cham, Switzerland, 2020; Volume 251, pp. 25–108. [Google Scholar]
- Daisy, B.H.; Strobel, G.A.; Castillo, U.; Ezra, D.; Sears, J.; Weaver, D.K.; Runyon, J.B. Naphthalene, an insect repellent, is produced by Muscodor vitigenus, a novel endophytic fungus. Microbiology 2002, 148 Pt 11, 3737–3741. [Google Scholar] [CrossRef]
- Johnston, J.E.; Lim, E.; Roh, H. Impact of upstream oil extraction and environmental public health: A review of the evidence. Sci. Total Environ. 2019, 657, 187–199. [Google Scholar] [CrossRef]
- Cai, Z.; Hao, X.; Sun, X.; Du, P.; Liu, W.; Fu, J. Highly active WO3@anatase-SiO2 aerogel for solar-light-driven phenanthrene degradation: Mechanism insight and toxicity assessment. Water Res. 2019, 162, 369–382. [Google Scholar] [CrossRef]
- Chauhan, H.; Rafatullah, M.; Ali, K.A.; Siddiqui, M.; Khan, M.; Alshareef, S. Metal-Based Nanocomposite Materials for Efficient Photocatalytic Degradation of Phenanthrene from Aqueous Solutions. Polymers 2021, 13, 2374. [Google Scholar] [CrossRef]
- Zhang, Z.; Sun, J.; Guo, H.; Wang, C.; Fang, T.; Rogers, M.J.; He, J.; Wang, H. Anaerobic biodegradation of phenanthrene by a newly isolated nitrate-dependent Achromobacter denitrificans strain PheN1 and exploration of the biotransformation processes by metabolite and genome analyses. Environ. Microbiol. 2021, 23, 908–923. [Google Scholar] [CrossRef]
- Rabodonirina, S.; Rasolomampianina, R.; Krier, F.; Drider, D.; Merhaby, D.; Net, S.; Ouddane, B. Degradation of fluorene and phenanthrene in PAHs-contaminated soil using Pseudomonas and Bacillus strains isolated from oil spill sites. J. Environ. Manag. 2019, 232, 1–7. [Google Scholar] [CrossRef]
- Ghosal, D.; Ghosh, S.; Dutta, T.K.; Ahn, Y. Corrigendum: Current State of Knowledge in Microbial Degradation of Polycyclic Aromatic Hydrocarbons (PAHs): A Review. Front. Microbiol. 2016, 7, 1837. [Google Scholar] [CrossRef]
- Premnath, N.; Mohanrasu, K.; Rao, R.G.R.; Dinesh, G.; Prakash, G.S.; Ananthi, V.; Ponnuchamy, K.; Muthusamy, G.; Arun, A. A crucial review on polycyclic aromatic Hydrocarbons—Environmental occurrence and strategies for microbial degradation. Chemosphere 2021, 280, 130608. [Google Scholar] [CrossRef]
- Sukhdhane, K.S.; Pandey, P.K.; Ajima, M.N.O.; Jayakumar, T.; Vennila, A.; Raut, S.M. Isolation and Characterization of Phenanthrene-Degrading Bacteria from PAHs Contaminated Mangrove Sediment of Thane Creek in Mumbai, India. Polycycl. Aromat. Compd. 2019, 39, 73–83. [Google Scholar] [CrossRef]
- Diallo, M.; Vural, C.; Cay, H.; Ozdemir, G. Enhanced biodegradation of crude oil in soil by a developed bacterial consortium and indigenous plant growth promoting bacteria. J. Appl. Microbiol. 2021, 130, 1192–1207. [Google Scholar] [CrossRef]
- Zada, S.; Zhou, H.; Xie, J.; Hu, Z.; Ali, S.; Sajjad, W.; Wang, H. Bacterial degradation of pyrene: Biochemical reactions and mechanisms. Int. Biodeterior. Biodegrad. 2021, 162, 105233. [Google Scholar] [CrossRef]
- Meena, M.D.; Yadav, R.K.; Narjary, B.; Yadav, G.; Jat, H.S.; Sheoran, P.; Meena, M.K.; Antil, R.S.; Meena, B.L.; Singh, H.V.; et al. Municipal solid waste (MSW): Strategies to improve salt affected soil sustainability: A review. Waste Manag. 2019, 84, 38–53. [Google Scholar] [CrossRef]
- Haas, B.J.; Gevers, D.; Earl, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E.; et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011, 21, 494–504. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Kanehisa, M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res. 2006, 34, D354–D357. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016, 44, D286–D293. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef]
- Chauhan, A.; Fazlurrahman; Oakeshott, J.G.; Jain, R.K. Bacterial metabolism of polycyclic aromatic hydrocarbons: Strategies for bioremediation. Indian J. Microbiol. 2008, 48, 95–113. [Google Scholar] [CrossRef]
- Gupta, B.; Puri, S.; Thakur, I.S.; Kaur, J. Enhanced pyrene degradation by a biosurfactant producing Acinetobacter baumannii BJ5: Growth kinetics, toxicity and substrate inhibition studies. Environ. Technol. Innov. 2020, 19, 100804. [Google Scholar] [CrossRef]
- Wang, F.; Li, C.; Wang, H.; Chen, W.; Huang, Q. Characterization of a phenanthrene-degrading microbial consortium enriched from petrochemical contaminated environment. Int. Biodeterior. Biodegrad. 2016, 115, 286–292. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, Y.; Jin, J.; Wang, T.; Wang, J.; Jiang, B. A high-efficiency phenanthrene-degrading Diaphorobacter sp. isolated from PAH-contaminated river sediment. Sci. Total Environ. 2020, 746, 140455. [Google Scholar] [CrossRef]
- Equero, G.M.; Ecassin, D.; Ebotter, M.; Eperini, L.; Eluna, G.M. Patterns of benthic bacterial diversity in coastal areas contaminated by heavy metals, polycyclic aromatic hydrocarbons (PAHs) and polychlorinated biphenyls (PCBs). Front. Microbiol. 2015, 6, 1053. [Google Scholar] [CrossRef]
- Lemmel, F.; Maunoury-Danger, F.; Leyval, C.; Cébron, A. DNA stable isotope probing reveals contrasted activity and phenanthrene-degrading bacteria identity in a gradient of anthropized soils. FEMS Microbiol. Ecol. 2019, 95, fiz181. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Wang, L.; Yang, S.; Xun, Y.; Zhang, T.; Wei, W. Thermally enhanced anoxic biodegradation of polycyclic aromatic hydrocarbons (PAHs) in a highly contaminated aged soil. J. Environ. Chem. Eng. 2022, 10, 107236. [Google Scholar] [CrossRef]
- Dong, Y.; Wu, S.; Fan, H.; Li, X.; Li, Y.; Xu, S.; Bai, Z.; Zhuang, X. Ecological selection of bacterial taxa with larger genome sizes in response to polycyclic aromatic hydrocarbons stress. J. Environ. Sci. 2022, 112, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zhang, G.; Liao, X. Negative role of biochars in the dissipation and vegetable uptake of polycyclic aromatic hydrocarbons (PAHs) in an agricultural soil: Cautions for application of biochars to remediate PAHs-contaminated soil. Ecotoxicol. Environ. Saf. 2021, 213, 112075. [Google Scholar] [CrossRef]
- Isaac, P.; Martínez, F.L.; Bourguignon, N.; Sánchez, L.A.; Ferrero, M.A. Improved PAHs removal performance by a defined bacterial consortium of indigenous Pseudomonas and actinobacteria from Patagonia, Argentina. Int. Biodeterior. Biodegrad. 2015, 101, 23–31. [Google Scholar] [CrossRef]
- Lee, D.W.; Lee, H.; Lee, A.H.; Kwon, B.-O.; Khim, J.S.; Yim, U.H.; Kim, B.S.; Kim, J.-J. Microbial community composition and PAHs removal potential of indigenous bacteria in oil contaminated sediment of Taean coast, Korea. Environ. Pollut. 2018, 234, 503–512. [Google Scholar] [CrossRef]
- Kirchman, D.L. The ecology of Cytophaga–Flavobacteria in aquatic environments. FEMS Microbiol. Ecol. 2002, 39, 91–100. [Google Scholar] [CrossRef]
- Gambino, E.; Toscanesi, M.; Del Prete, F.; Flagiello, F.; Falcucci, G.; Minutillo, M.; Trifuoggi, M.; Guida, M.; Nastro, R.A.; Jannelli, E. Polycyclic Aromatic Hydrocarbons (PAHs) Degradation and Detoxification of Water Environment in Single-chamber Air-cathode Microbial Fuel Cells (MFCs). Fuel Cells 2017, 17, 618–626. [Google Scholar] [CrossRef]
- Storey, S.; Ashaari, M.M.; Clipson, N.; Doyle, E.; De Menezes, A.B. Opportunistic Bacteria Dominate the Soil Microbiome Response to Phenanthrene in a Microcosm-Based Study. Front. Microbiol. 2018, 9, 2815. [Google Scholar] [CrossRef]
- Bell, T.H.; Yergeau, E.; Martineau, C.; Juck, D.; Whyte, L.G.; Greer, C.W. Identification of Nitrogen-Incorporating Bacteria in Petroleum-Contaminated Arctic Soils by Using [15N]DNA-Based Stable Isotope Probing and Pyrosequencing. Appl. Environ. Microbiol. 2011, 77, 4163–4171. [Google Scholar] [CrossRef] [Green Version]
- Medina, R.; Fernández-González, A.J.; García-Rodríguez, F.M.; Villadas, P.J.; Rosso, J.A.; Fernández-López, M.; Del Panno, M.T. Exploring the effect of composting technologies on the recovery of hydrocarbon contaminated soil post chemical oxidative treatment. Appl. Soil Ecol. 2019, 150, 103459. [Google Scholar] [CrossRef]
- Liu, Y.; Hu, H.; Zanaroli, G.; Xu, P.; Tang, H. A Pseudomonas sp. strain uniquely degrades PAHs and heterocyclic derivatives via lateral dioxygenation pathways. J. Hazard. Mater. 2021, 403, 123956. [Google Scholar] [CrossRef]
- Zang, T.; Wu, H.; Zhang, Y.; Wei, C. The response of polycyclic aromatic hydrocarbon degradation in coking wastewater treatment after bioaugmentation with biosurfactant-producing bacteria Pseudomonas aeruginosa S5. Water Sci. Technol. 2021, 83, 1017–1027. [Google Scholar] [CrossRef]
- Safahieh, A. Isolation and characterization of Pseudomonas resistant to heavy metals and poly aromatics hydrocarbons (PAHs) from Persian Gulf sediments. Afr. J. Biotechnol. 2012, 11, 4418–4423. [Google Scholar] [CrossRef]
- Ma, J.; Xu, L.; Jia, L. Characterization of pyrene degradation by Pseudomonas sp. strain Jpyr-1 isolated from active sewage sludge. Bioresour. Technol. 2013, 140, 15–21. [Google Scholar] [CrossRef]
- Edlund, A.; Jansson, J.K. Use of bromodeoxyuridine immunocapture to identify psychrotolerant phenanthrene-degrading bacteria in phenanthrene-enriched polluted Baltic Sea sediments. FEMS Microbiol. Ecol. 2008, 65, 513–525. [Google Scholar] [CrossRef]
- Muangchinda, C.; Srisuwankarn, P.; Boubpha, S.; Chavanich, S.; Pinyakong, O. The effect of bioaugmentation with Exiguobacterium sp. AO-11 on crude oil removal and the bacterial community in sediment microcosms, and the development of a liquid ready-to-use inoculum. Chemosphere 2020, 250, 126303. [Google Scholar] [CrossRef]
- Wu, M.; Guo, X.; Wu, J.; Chen, K. Effect of compost amendment and bioaugmentation on PAH degradation and microbial community shifting in petroleum-contaminated soil. Chemosphere 2020, 256, 126998. [Google Scholar] [CrossRef]
- Nie, Y.; Tang, Y.Q.; Li, Y.; Chi, C.Q.; Cai, M.; Wu, X.L. The Genome Sequence of PolymorphumgilvumSL003B- 26A1T Reveals Its Genetic Basis for Crude Oil Degradation and Adaptation to the Saline Soil. PLoS ONE 2012, 7, e31261. [Google Scholar] [CrossRef]
- Oshkin, I.Y.; Miroshnikov, K.K.; Grouzdev, D.S.; Dedysh, S.N. Pan-Genome-Based Analysis as a Framework for Demarcating Two Closely Related Methanotroph Genera Methylocystis and Methylosinus. Microorganisms 2020, 8, 768. [Google Scholar] [CrossRef]
- Maddocks, S.E.; Oyston, P.C.F. Structure and function of the LysR-type transcriptional regulator (LTTR) family proteins. Microbiology 2008, 154, 3609–3623. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cui, R.; Shi, G.; Dai, Y.; Dong, J.; Wu, Q.; Zhang, H.; Dai, J. Dioxin-like polychlorinated biphenyl 126 (PCB126) disrupts gut microbiota-host metabolic dysfunction in mice via aryl hydrocarbon receptor activation. Ecotoxicol. Environ. Saf. 2022, 236, 113448. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.; Kan, J.; Hu, J.; Hu, Z. Genes and novel sRNAs involved in PAHs degradation in marine bacteria Rhodococcus sp. P14 revealed by the genome and transcriptome analysis. 3 Biotech 2020, 10, 140. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Li, Y.; He, Q.; Li, B.; Yuan, Y.; Wen, J. Effects of different surfactants on the degradation of petroleum hydrocarbons by mixed-bacteria. J. Chem. Technol. Biotechnol. 2022, 97, 208–217. [Google Scholar] [CrossRef]
- Wang, X.; Yao, B.; Su, X. Linking Enzymatic Oxidative Degradation of Lignin to Organics Detoxification. Int. J. Mol. Sci. 2018, 19, 3373. [Google Scholar] [CrossRef]
- Roth, C.; Weizenmann, N.; Bexten, N.; Saenger, W.; Zimmermann, W.; Maier, T.; Sträter, N. Amylose recognition and ring-size determination of amylomaltase. Sci. Adv. 2017, 3, e1601386. [Google Scholar] [CrossRef]
- Thompson, C.E.; Beys-Da-Silva, W.O.; Santi, L.; Berger, M.; Vainstein, M.H.; Guimarães, J.A.; Vasconcelos, A.T. A potential source for cellulolytic enzyme discovery and environmental aspects revealed through metagenomics of Brazilian mangroves. AMB Express 2013, 3, 65. [Google Scholar] [CrossRef]
- Cockburn, D.; Nielsen, M.M.; Christiansen, C.; Andersen, J.M.; Rannes, J.B.; Blennow, A.; Svensson, B. Surface binding sites in amylase have distinct roles in recognition of starch structure motifs and degradation. Int. J. Biol. Macromol. 2015, 75, 338–345. [Google Scholar] [CrossRef]
- Ma, C.; Lo, P.K.; Xu, J.; Li, M.; Jiang, Z.; Li, G.; Zhu, Q.; Li, X.; Leong, S.Y.; Li, Q. Molecular mechanisms underlying lignocellulose degradation and antibiotic resistance genes removal revealed via metagenomics analysis during different agricultural wastes composting. Bioresour. Technol. 2020, 314, 123731. [Google Scholar] [CrossRef]
- Dou, R.; Sun, J.; Lu, J.; Deng, F.; Yang, C.; Lu, G.; Dang, Z. Bacterial communities and functional genes stimulated during phenanthrene degradation in soil by bio-microcapsules. Ecotoxicol. Environ. Saf. 2021, 212, 111970. [Google Scholar] [CrossRef]
- Mattei, P.; Cincinelli, A.; Martellini, T.; Natalini, R.; Pascale, E.; Renella, G. Reclamation of river dredged sediments polluted by PAHs by co-composting with green waste. Sci. Total Environ. 2016, 566–567, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, S.; He, F.; Qin, X.; Zhang, X.; Yang, Y. Characterization of a manganese peroxidase from white-rot fungus Trametes sp.48424 with strong ability of degrading different types of dyes and polycyclic aromatic hydrocarbons. J. Hazard. Mater. 2016, 320, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; He, Y.; Cai, H.; Van Nostrand, J.D.; He, Z.; Zhou, J.; Krumholz, L.R.; Jiang, H.-L. Interconnection of Key Microbial Functional Genes for Enhanced Benzo[a]pyrene Biodegradation in Sediments by Microbial Electrochemistry. Environ. Sci. Technol. 2017, 51, 8519–8529. [Google Scholar] [CrossRef]
- Feehily, C.; Karatzas, K. Role of glutamate metabolism in bacterial responses towards acid and other stresses. J. Appl. Microbiol. 2013, 114, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Badejo, A.C.; Choi, C.-W.; Badejo, A.O.; Shin, K.-H.; Hyun, J.-H.; Lee, Y.-G.; Kim, S.-I.; Park, K.-S.; Kim, S.H.; Jung, K.H.; et al. A global proteome study of Mycobacterium gilvum PYR-GCK grown on pyrene and glucose reveals the activation of glyoxylate, shikimate and gluconeogenetic pathways through the central carbon metabolism highway. Biogeochemistry 2013, 24, 741–752. [Google Scholar] [CrossRef]
- Li, X.; Song, Y.; Bian, Y.; Gu, C.; Yang, X.; Wang, F.; Jiang, X. Insights into the mechanisms underlying efficient Rhizodegradation of PAHs in biochar-amended soil: From microbial communities to soil metabolomics. Environ. Int. 2020, 144, 105995. [Google Scholar] [CrossRef]
- Deeba, F.; Pruthi, V.; Negi, Y.S. Aromatic hydrocarbon biodegradation activates neutral lipid biosynthesis in oleaginous yeast. Bioresour. Technol. 2018, 255, 273–280. [Google Scholar] [CrossRef]
- Hebert, D.N.; Molinari, M. In and Out of the ER: Protein Folding, Quality Control, Degradation, and Related Human Diseases. Physiol. Rev. 2007, 87, 1377–1408. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Diebold, L.P.; Kong, H.; Schieber, M.; Huang, H.; Hensley, C.T.; Mehta, M.M.; Wang, T.; Santos, J.H.; Woychik, R.; et al. TCA Cycle and Mitochondrial Membrane Potential Are Necessary for Diverse Biological Functions. Mol. Cell 2016, 61, 199–209. [Google Scholar] [CrossRef]
- Thomas, F.; Corre, E.; Cébron, A. Stable isotope probing and metagenomics highlight the effect of plants on uncultured phenanthrene-degrading bacterial consortium in polluted soil. ISME J. 2019, 13, 1814–1830. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.-N.; Yao, J.; Zhang, Q.-Y.; Yu, C.; Chen, P.; Liu, W.-J.; Peng, D.-N.; Choi, M.M. An integrated approach of bioassay and molecular docking to study the dihydroxylation mechanism of pyrene by naphthalene dioxygenase in Rhodococcus sp. ustb-1. Chemosphere 2015, 128, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhao, X.; Liang, Y.; Li, G.; Zhou, J. Microbial functional genes reveal selection of microbial community by PAHs in polluted soils. Environ. Chem. Lett. 2013, 11, 11–17. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, P.; Chen, X.; Li, K.; Meng, R.; Pu, Y. Metagenomic Analysis of Microbial Alliances for Efficient Degradation of PHE: Microbial Community Structure and Reconstruction of Metabolic Network. Int. J. Environ. Res. Public Health 2022, 19, 12039. https://doi.org/10.3390/ijerph191912039
Xu P, Chen X, Li K, Meng R, Pu Y. Metagenomic Analysis of Microbial Alliances for Efficient Degradation of PHE: Microbial Community Structure and Reconstruction of Metabolic Network. International Journal of Environmental Research and Public Health. 2022; 19(19):12039. https://doi.org/10.3390/ijerph191912039
Chicago/Turabian StyleXu, Pan, Xiaoxiao Chen, Kai Li, Rong Meng, and Yuewu Pu. 2022. "Metagenomic Analysis of Microbial Alliances for Efficient Degradation of PHE: Microbial Community Structure and Reconstruction of Metabolic Network" International Journal of Environmental Research and Public Health 19, no. 19: 12039. https://doi.org/10.3390/ijerph191912039
APA StyleXu, P., Chen, X., Li, K., Meng, R., & Pu, Y. (2022). Metagenomic Analysis of Microbial Alliances for Efficient Degradation of PHE: Microbial Community Structure and Reconstruction of Metabolic Network. International Journal of Environmental Research and Public Health, 19(19), 12039. https://doi.org/10.3390/ijerph191912039