
Mutational Assessment in NKX2-5 and ACTC1 Genes in Patients with Congenital Cardiac Septal Defect (CCSD) from Ethnic Kashmiri Population


Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Cases
2.3. Sample Collection/Storage
2.4. Extraction of Genomic DNA and PCR Amplification
2.5. DNA Sequencing
2.6. Putative Role of the Variants
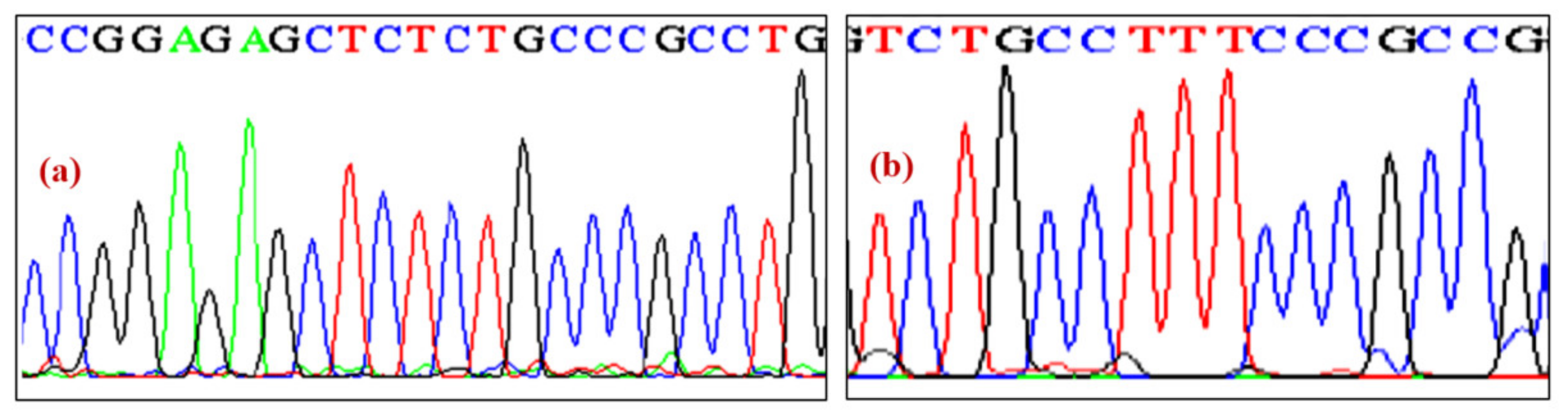
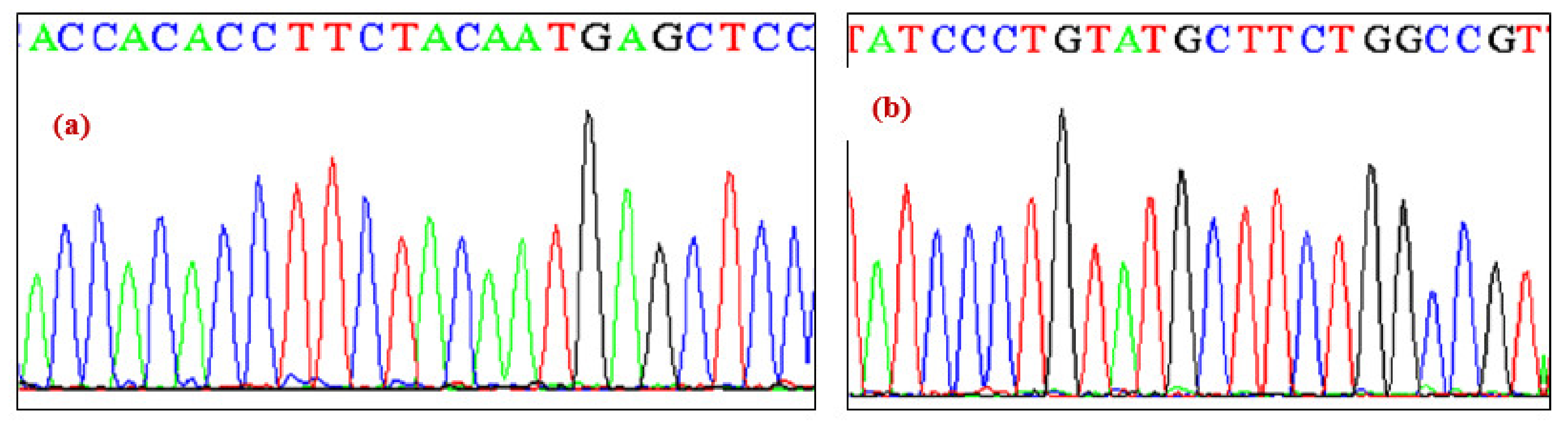
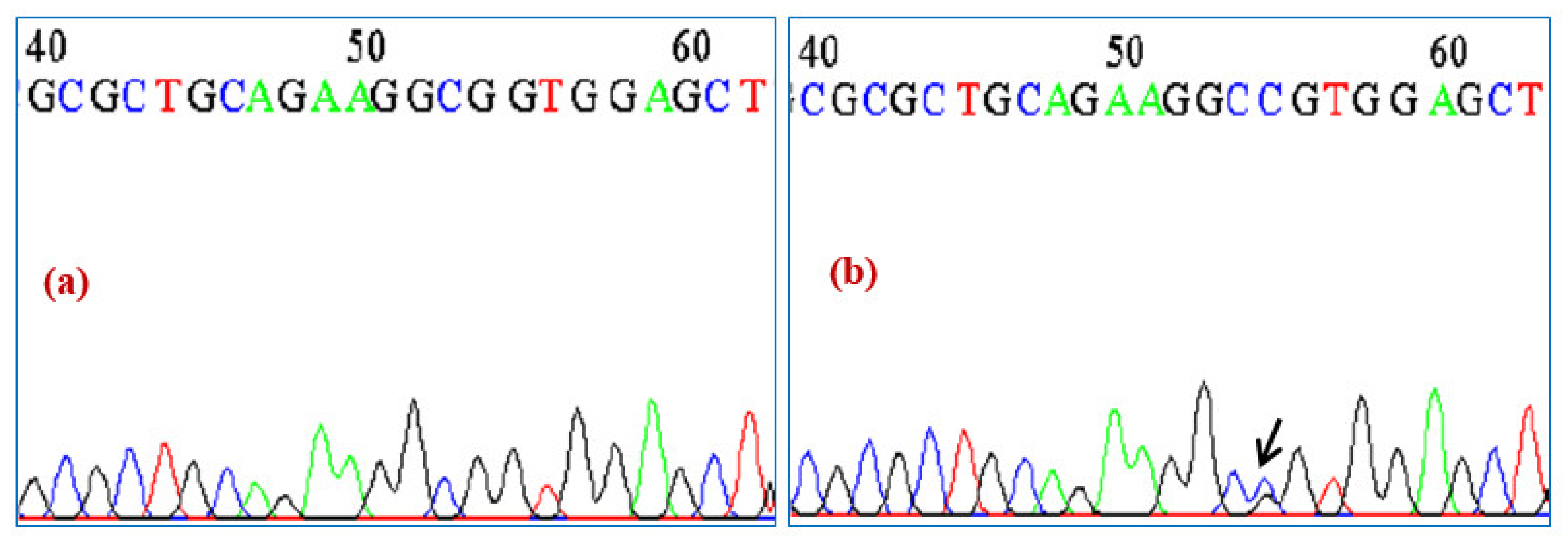
3. Results
Molecular Analysis of NKX2-5 and ACTC1 Genes
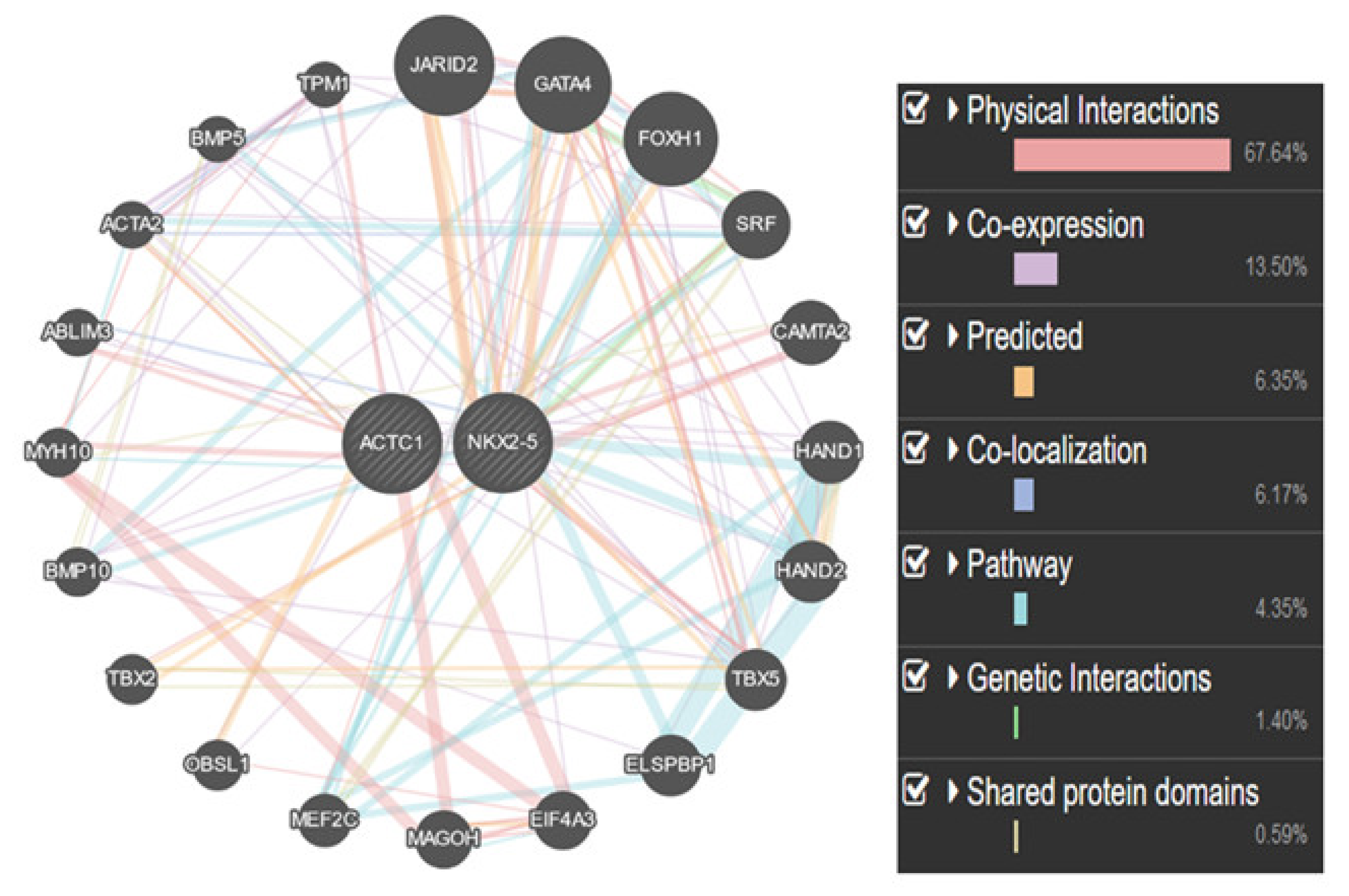
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Webber, D.M.; MacLeod, S.L.; Bamshad, M.J.; Shaw, G.M.; Finnell, R.H.; Shete, S.S.; Witte, J.S.; Erickson, S.W.; Murphy, L.D.; Hobbs, C. Developments in Our Understanding of the Genetic Basis of Birth Defects. Birth Defects Res. Part A Clin. Mol. Teratol. 2015, 103, 680–691. [Google Scholar] [CrossRef] [Green Version]
- Cogswell, M.E.; Bitsko, R.H.; Anderka, M.; Caton, A.R.; Feldkamp, M.L.; Sherlock, S.M.H.; Meyer, R.E.; Ramadhani, T.; Robbins, J.M.; Shaw, G.M. Control Selection and Participation in an Ongoing, Population-Based, Case-Control Study of Birth Defects: The National Birth Defects Prevention Study. Am. J. Epidemiol. 2009, 170, 975–985. [Google Scholar] [CrossRef]
- Leduc, R.Y.; Singh, P.; McDermid, H.E. Genetic Backgrounds and Modifier Genes of Ntd Mouse Models: An Opportunity for Greater Understanding of the Multifactorial Etiology of Neural Tube Defects. Birth Defects Res. 2017, 109, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Pierpont, M.E.; Brueckner, M.; Chung, W.K.; Garg, V.; Lacro, R.V.; McGuire, A.L.; Mital, S.; Priest, J.R.; Pu, W.T.; Roberts, A. Genetic Basis for Congenital Heart Disease: Revisited: A Scientific Statement from the American Heart Association. Circulation 2018, 138, e653–e711. [Google Scholar] [CrossRef]
- Shabana, N.A.; Shahid, S.U.; Irfan, U. Genetic Contribution to Congenital Heart Disease (CHD). Pediatric Cardiol. 2020, 41, 12–23. [Google Scholar] [CrossRef]
- Lin, A.E.; Basson, C.T.; Goldmuntz, E.; Magoulas, P.L.; McDermott, D.A.; McDonald-McGinn, D.M.; McPherson, E.; Morris, C.A.; Noonan, J.; Nowak, C. Adults with Genetic Syndromes and Cardiovascular Abnormalities: Clinical History and Management. Genet. Med. 2008, 10, 469–494. [Google Scholar] [CrossRef] [Green Version]
- Reller, M.D.; Strickland, M.J.; Riehle-Colarusso, T.; Mahle, W.T.; Correa, A. Prevalence of Congenital Heart Defects in Metropolitan Atlanta, 1998–2005. J. Pediatrics 2008, 153, 807–813. [Google Scholar] [CrossRef] [Green Version]
- Prasher, D.; Greenway, S.C.; Singh, R.B. The Impact of Epigenetics on Cardiovascular Disease. Biochem. Cell Biol. 2020, 98, 12–22. [Google Scholar] [CrossRef]
- Srivastava, D.; Olson, E.N. A Genetic Blueprint for Cardiac Development. Nature 2000, 407, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, S.; Brueckner, M. Genetics and Genomics of Congenital Heart Disease. Circ. Res. 2017, 120, 923–940. [Google Scholar] [CrossRef]
- Majumdar, U.; Yasuhara, J.; Garg, V. In Vivo and In Vitro Genetic Models of Congenital Heart Disease. Cold Spring Harb. Perspect. Biol. 2021, 13, a036764. [Google Scholar] [CrossRef] [PubMed]
- Stennard, F.A.; Costa, M.W.; Elliott, D.A.; Rankin, S.; Haast, S.J.; Lai, D.; McDonald, L.P.; Niederreither, K.; Dolle, P.; Bruneau, B.G. Cardiac T-Box Factor Tbx20 Directly Interacts with Nkx2-5, GATA4, and GATA5 in Regulation of Gene Expression in the Developing Heart. Dev. Biol. 2003, 262, 206–224. [Google Scholar] [CrossRef] [Green Version]
- George, V.; Colombo, S.; Targoff, K.L. An Early Requirement for Nkx2.5 Ensures the First and Second Heart Field Ventricular Identity and Cardiac Function into Adulthood. Dev. Biol. 2015, 400, 10–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruneau, B.G. The Developing Heart and Congenital Heart Defects: A Make or Break Situation. Clin. Genet. 2003, 63, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Lu, Y.-B.; Liang, S.-T.; Zhang, Q.-J.; Xu, J.; She, Z.-G.; Zhang, Z.-Q.; Yang, R.-F.; Mao, B.-B.; Xu, Z. SIRT1 Mediates the Protective Function of Nkx2.5 during Stress in Cardiomyocytes. Basic Res. Cardiol. 2013, 108, 364. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.-K.; Qiu, X.-B.; Yuan, F.; Wang, J.; Zhao, C.-M.; Liu, X.-Y.; Zhang, X.-L.; Li, R.-G.; Xu, Y.-J.; Hou, X.-M. A Novel NKX2.5 Loss-of-Function Mutation Associated with Congenital Bicuspid Aortic Valve. Am. J. Cardiol. 2014, 114, 1891–1895. [Google Scholar] [CrossRef] [PubMed]
- Bouchikhi, I.E.; Belhassan, K.; Moufid, F.Z.; Bouguenouch, L.; Samri, I.; Houssaïni, M.I.; Ouldim, K.; Atmani, S. Screening of NKX2.5 Gene in Moroccan Tetralogy of Fallot (TOF) Patients: Worldwide Mutation Rate Comparisons Show a Significant Association between R25C Variant and TOF Phenotype. Egypt J. Med. Hum. Genet. 2021, 22, 18. [Google Scholar] [CrossRef]
- Reamon-Buettner, S.M.; Borlak, J. NKX2-5: An Update on This Hypermutable Homeodomain Protein and Its Role in Human Congenital Heart Disease (CHD). Hum. Mutat. 2010, 31, 1185–1194. [Google Scholar] [CrossRef] [Green Version]
- Frank, D.; Rangrez, A.Y.; Friedrich, C.; Dittmann, S.; Stallmeyer, B.; Yadav, P.; Bernt, A.; Schulze-Bahr, E.; Borlepawar, A.; Zimmermann, W.-H. Cardiac α-Actin (ACTC1) Gene Mutation Causes Atrial-Septal Defects Associated with Late-Onset Dilated Cardiomyopathy. Circ. Genom. Precis. Med. 2019, 12, e002491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Du, X.; Zhou, Z.; Jiang, J.; Zhang, Z.; Ye, L.; Hong, H. A Gain-of-Function ACTC1 3′ UTR Mutation That Introduces a MiR-139-5p Target Site May Be Associated with a Dominant Familial Atrial Septal Defect. Sci. Rep. 2016, 6, 25404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, B.L.; Song, T.; Sadayappan, S. Myofilaments: Movers and Rulers of the Sarcomere. Compr. Physiol. 2011, 7, 675–692. [Google Scholar]
- Bang, M.-L. Animal Models of Congenital Cardiomyopathies Associated with Mutations in Z-line Proteins. J. Cell. Physiol. 2017, 232, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, A.P.; Sairam, M.; Princy, K.J.; Viswamithra, V.; Kumar, C.S.; Ali, A.; Nallari, P. Possibility of RNA Editing in Cardiomyopathies and Channelopathies. J. Adv. Life Nat. Sci. 2015, 1, 1–7. [Google Scholar]
- Lander, J.; Ware, S.M. Copy Number Variation in Congenital Heart Defects. Curr. Genet. Med. Rep. 2014, 2, 168–178. [Google Scholar] [CrossRef] [Green Version]
- Vecoli, C.; Pulignani, S.; Foffa, I.; Grazia Andreassi, M. Congenital Heart Disease: The Crossroads of Genetics, Epigenetics and Environment. Curr. Genom. 2014, 15, 390–399. [Google Scholar] [CrossRef] [Green Version]
- Hinton, R.B.; Ware, S.M. Heart Failure in Pediatric Patients with Congenital Heart Disease. Circ. Res. 2017, 120, 978–994. [Google Scholar] [CrossRef] [Green Version]
- Precone, V.; Krasi, G.; Guerri, G.; Madureri, A.; Piazzani, M.; Michelini, S.; Barati, S.; Maniscalchi, T.; Bressan, S.; Bertelli, M. Cardiomyopathies. Acta Bio-Med. Atenei Parm. 2019, 90, 32–43. [Google Scholar]
- Stallmeyer, B.; Fenge, H.; Nowak-Göttl, U.; Schulze-Bahr, E. Mutational Spectrum in the Cardiac Transcription Factor Gene NKX2.5 (CSX) Associated with Congenital Heart Disease. Clin. Genet. 2010, 78, 533–540. [Google Scholar] [CrossRef]
- Bartlett, H.; Veenstra, G.J.C.; Weeks, D.L. Examining the Cardiac NK-2 Genes in Early Heart Development. Pediatric Cardiol. 2010, 31, 335–341. [Google Scholar] [CrossRef] [Green Version]
- Hwang, K.-J.; Ferretti, J.A. The Vnd/NK-2 Gene from Drosophila Melanogaster: How Relatively Small Molecular Changes Can Lead to Catastrophic Genetic Anomalies. Int. Biol. Rev. 2017, 1. [Google Scholar] [CrossRef]
- Han, Z.; Olson, E.N. Hand Is a Direct Target of Tinman and GATA Factors during Drosophila Cardiogenesis and Hematopoiesis. Development 2005, 132, 3525–3536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajewski, K.; Zhang, Q.; Choi, C.Y.; Fossett, N.; Dang, A.; Kim, Y.H.; Kim, Y.; Schulz, R.A. Pannier Is a Transcriptional Target and Partner of Tinman during Drosophila Cardiogenesis. Dev. Biol. 2001, 233, 425–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behrens, A.N.; Ren, Y.; Ferdous, A.; Garry, D.J.; Martin, C.M. Nkx2-5 Regulates Tdgf1 (Cripto) Early during Cardiac Development. J. Clin. Exp. Cardiol. 2012, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pashmforoush, M.; Lu, J.T.; Chen, H.; St Amand, T.; Kondo, R.; Pradervand, S.; Evans, S.M.; Clark, B.; Feramisco, J.R.; Giles, W. Nkx2-5 Pathways and Congenital Heart Disease: Loss of Ventricular Myocyte Lineage Specification Leads to Progressive Cardiomyopathy and Complete Heart Block. Cell 2004, 117, 373–386. [Google Scholar] [CrossRef] [Green Version]
- Matsson, H.; Eason, J.; Bookwalter, C.S.; Klar, J.; Gustavsson, P.; Sunnegårdh, J.; Enell, H.; Jonzon, A.; Vikkula, M.; Gutierrez, I. Alpha-Cardiac Actin Mutations Produce Atrial Septal Defects. Hum. Mol. Genet. 2008, 17, 256–265. [Google Scholar] [CrossRef]
- Monserrat, L.; Hermida-Prieto, M.; Fernandez, X.; Rodríguez, I.; Dumont, C.; Cazón, L.; Cuesta, M.G.; Gonzalez-Juanatey, C.; Peteiro, J.; Álvarez, N. Mutation in the Alpha-Cardiac Actin Gene Associated with Apical Hypertrophic Cardiomyopathy, Left Ventricular Non-Compaction, and Septal Defects. Eur. Heart J. 2007, 28, 1953–1961. [Google Scholar] [CrossRef] [Green Version]

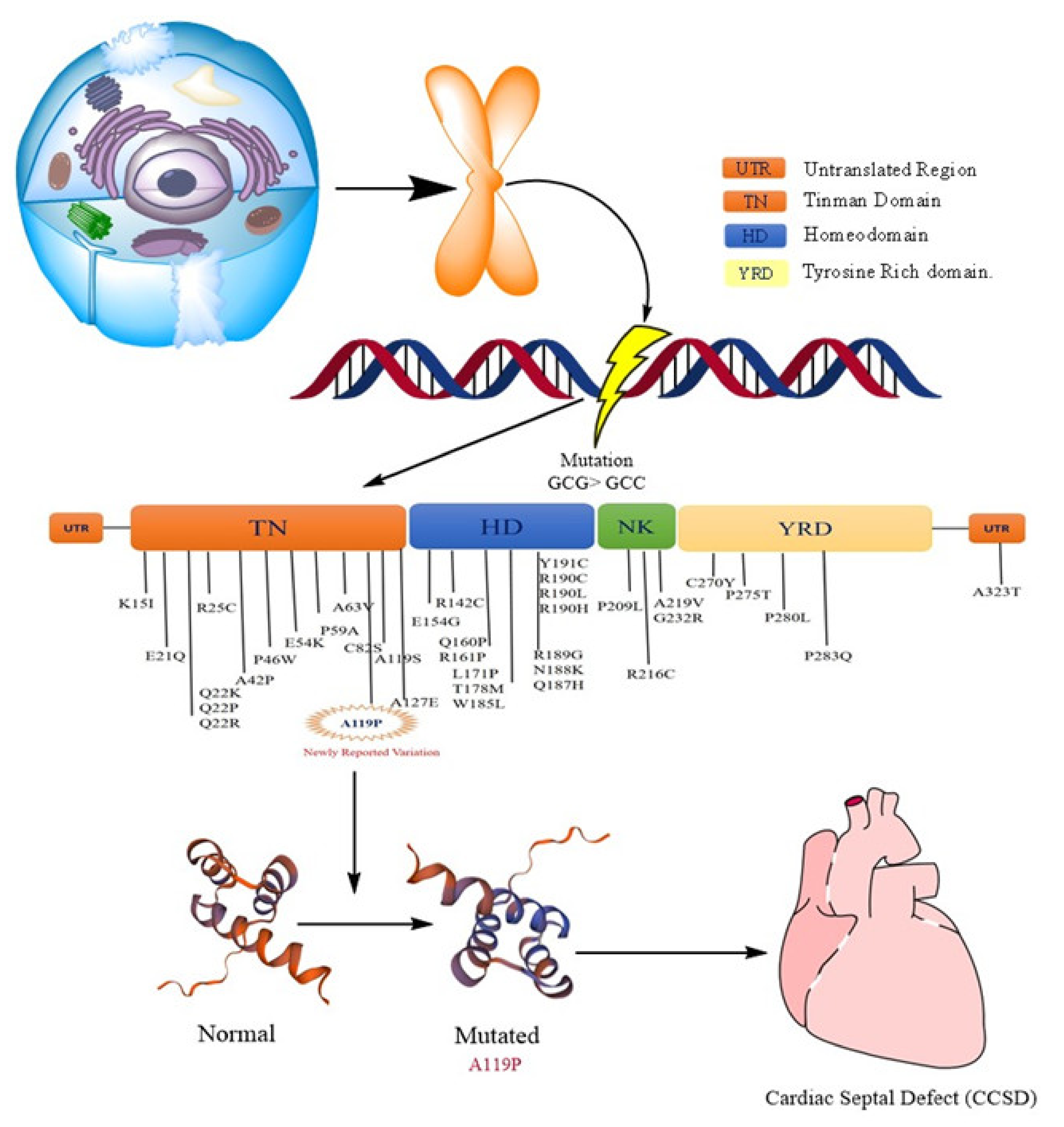

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Types | Male (Number and % Age) | Female (Number and % Age) |
| ASD | (n = 03, 10%) | (n = 09, 30%) |
| ASD with PAPVC | Nil | (n = 01, 3.33%) |
| ASD with VSD | (n = 01, 3.33%) | Nil |
| VSD | (n = 02, 6.66%) | (n = 04, 13.33%) |
| VSD with RSOV | (n = 01, 3.33%) | Nil |
| TOF | (n = 01, 3.33%) | (n = 01, 3.33%) |
| TA with ASD and VSD | (n = 02, 6.66%) | (n = 01, 3.33%) |
| DCRV with VSD | (n = 02, 6.66%) | (n = 02, 6.66%) |
| Gender distribution | 39.97% | 60.03% |
| Age Distribution of Patients | ||
| Age | Male (Number and % Age) | Female (Number and % Age) |
| 0–10 years | (n = 04, 13.3%) | (n = 03, 10%) |
| 11–20 years | (n = 03, 10%) | (n = 04, 13.33%) |
| 21–30 years | (n = 03, 10%) | (n = 04, 13.3%) |
| 31–40 years | (n = 02, 6.66%) | (n = 03, 10%) |
| 41–50 years | (n = 01, 3.33%) | (n = 02, 6.66%) |
| 51–60 years | (n = 01, 3.33%) | Nil |
| Consanguinity, Disease Pathologies, Other Clinical Symptoms of Patients | ||
| Consanguineous marriage | (n = 04, 13.33%) | (n = 02, 6.66%) |
| Recurrent chest infection | (n = 6, 20%) | (n = 13, 43.33%) |
| Palpitation | (n = 9, 30% | (n = 15, 50%) |
| Breathlessness | (n = 7, 23.33%) | (n = 12, 40%) |
| Fatigue | (n = 11, 36.66%) | (n = 13, 43.33%) |
| Cough | (n = 7, 23.33%) | (n = 10, 33.33%) |
| Poor weight gain | (n = 7, 23.33%) | (n = 7, 23.33%) |
| Recurrent fever | (n = 6, 20%) | (n = 8, 26.66%) |
| Feeding problems | (n = 4, 13.33%) | (n = 9, 30%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nazeer, N.U.; Bhat, M.A.; Rah, B.; Bhat, G.R.; Wani, S.I.; Yousuf, A.; Dar, A.M.; Afroze, D. Mutational Assessment in NKX2-5 and ACTC1 Genes in Patients with Congenital Cardiac Septal Defect (CCSD) from Ethnic Kashmiri Population. Int. J. Environ. Res. Public Health 2022, 19, 9884. https://doi.org/10.3390/ijerph19169884
Nazeer NU, Bhat MA, Rah B, Bhat GR, Wani SI, Yousuf A, Dar AM, Afroze D. Mutational Assessment in NKX2-5 and ACTC1 Genes in Patients with Congenital Cardiac Septal Defect (CCSD) from Ethnic Kashmiri Population. International Journal of Environmental Research and Public Health. 2022; 19(16):9884. https://doi.org/10.3390/ijerph19169884
Chicago/Turabian StyleNazeer, Nadeem Ul, Mohammad Akbar Bhat, Bilal Rah, Gh Rasool Bhat, Shadil Ibrahim Wani, Adfar Yousuf, Abdul Majeed Dar, and Dil Afroze. 2022. "Mutational Assessment in NKX2-5 and ACTC1 Genes in Patients with Congenital Cardiac Septal Defect (CCSD) from Ethnic Kashmiri Population" International Journal of Environmental Research and Public Health 19, no. 16: 9884. https://doi.org/10.3390/ijerph19169884
APA StyleNazeer, N. U., Bhat, M. A., Rah, B., Bhat, G. R., Wani, S. I., Yousuf, A., Dar, A. M., & Afroze, D. (2022). Mutational Assessment in NKX2-5 and ACTC1 Genes in Patients with Congenital Cardiac Septal Defect (CCSD) from Ethnic Kashmiri Population. International Journal of Environmental Research and Public Health, 19(16), 9884. https://doi.org/10.3390/ijerph19169884