
Association between Leptin (G2548A) and Leptin Receptor (Q223R) Polymorphisms with Plasma Leptin, BMI, Stress, Sleep and Eating Patterns among the Multiethnic Young Malaysian Adult Population from a Healthcare University

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Respondents
2.2. Questionnaire
2.3. Anthropometric Measurements
2.4. Protein Analysis
Serum Leptin, Cortisol and hsCRP
2.5. Genotyping
2.5.1. DNA Extraction
2.5.2. Polymerase Chain Reaction (PCR)
3. Statistical Analysis
4. Results
- Relationship between G2548A LEP and Q223A LepR Polymorphisms with Obesity and Plasma Leptin Levels
- Relationship between G2548A Lep and Q223A LepR Polymorphisms with Ethnicity
- Relationship between G2548A LEP and Q223A LEPR Polymorphisms with Obesity and Plasma Leptin Levels
- Relationship between LEP G2548A and LEPR Q223A Polymorphisms with Obesity and Its Associated Risk Factors
- Relationship between G2548A LEP and Q223A LEPR Polymorphisms with Group 1 (Overweight to Morbidly Obese) Cases
- G2548A LEP and Q223A LEPR polymorphism with Group 2 (Underweight to Anorexic) cases:
- G2548A LEP and Q223A LEPR polymorphisms with Group 3 with normal BMI and abnormal sleeping, eating, and stress factors:
5. Discussion
- Stage 1 (Early Stage) is when the subject gains weight but maintains adequate response to the anorectic effect of peripheral leptin:
- Stage 2 (Middle Stage) is when the subjects develop leptin insensitivity due to lack of STAT3, expressed by changes in food intake and body weight but continue to respond to leptin injections:
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ludwig, D.S. Epidemic childhood obesity: Not yet the end of the beginning. Pediatrics 2018, 141, e20174078. [Google Scholar] [CrossRef] [PubMed]
- Pozza, C.; Isidori, A.M. What’s behind the obesity epidemic. In Imaging in Bariatric Surgery; Springer: Cham, Switzerland, 2018; pp. 1–8. [Google Scholar]
- Stevens, G.A.; Singh, G.M.; Lu, Y.; Danaei, G.; Lin, J.K.; Finucane, M.M.; Bahalim, A.N.; McIntire, R.K.; Gutierrez, H.R.; Cowan, M.; et al. National, regional, and global trends in adult overweight and obesity prevalences. Popul. Health Metr. 2012, 10, 22. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.; Fleming, T.; Robinson, M.; Thomson, B.; Graetz, N.; Margono, C.; Mullany, E.C.; Biryukov, S.; Abbafati, C.; Abera, S.F.; et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2014, 384, 766–781. [Google Scholar] [CrossRef]
- Angkurawaranon, C.; Jiraporncharoen, W.; Chenthanakij, B.; Doyle, P.; Nitsch, D. Urban environments and obesity in southeast Asia: A systematic review, meta-analysis and meta-regression. PLoS ONE 2014, 9, e113547. [Google Scholar] [CrossRef] [PubMed]
- Shahid, A.; Rana, S.; Mahmood, S.; Saeed, S. Role of leptin G-2548A polymorphism in age-and gender-specific development of obesity. J. Biosci. 2015, 40, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Jan Mohamed, H.J.B.; Yap, R.W.K.; Loy, S.L.; Norris, S.A.; Biesma, R.; Aagaard-Hansen, J. Prevalence and determinants of overweight, obesity, and type 2 diabetes mellitus in adults in Malaysia. Asia Pac. J. Public Health 2015, 27, 123–135. [Google Scholar] [CrossRef]
- Malik, V.S.; Willett, W.C.; Hu, F.B. Global obesity: Trends, risk factors and policy implications. Nat. Rev. Endocrinol. 2013, 9, 13. [Google Scholar] [CrossRef]
- Alhazmi, A.H.; Al Johani, A. Prevalence and Associated Factors of Eating Disorders among Students in Taiba University, Saudi Arabia: A Cross-Sectional Study. Malays. J. Public Health Med. 2019, 19, 172–176. [Google Scholar]
- Corsi, D.J.; Finlay, J.E.; Subramanian, S. Global burden of double malnutrition: Has anyone seen it? PLoS ONE 2011, 6, e25120. [Google Scholar] [CrossRef]
- Seidell, J.C.; Verschuren, W.M.; Van Leer, E.M.; Kromhout, D. Overweight, underweight, and mortality: A prospective study of 48,287 men and women. Arch. Intern. Med. 1996, 156, 958–963. [Google Scholar] [CrossRef]
- Tebekaw, Y.; Teller, C.; Colón-Ramos, U. The burden of underweight and overweight among women in Addis Ababa, Ethiopia. BMC Public Health 2014, 14, 1126. [Google Scholar] [CrossRef] [PubMed]
- Kasmini, K.; Idris, M.; Fatimah, A.; Hanafiah, S.; Iran, H.; Asmah Bee, M. Prevalence of overweight and obese school children aged between 7 to 16 years amongst the major 3 ethnic groups in Kuala Lumpur, Malaysia. Asia Pac. J. Clin. Nutr. 1997, 6, 172–174. [Google Scholar] [PubMed]
- Leigh, S.-J.; Morris, M.J. The role of reward circuitry and food addiction in the obesity epidemic: An update. Biol. Psychol. 2018, 131, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Yeh, J.; Trang, A.; Henning, S.M.; Wilhalme, H.; Carpenter, C.; Heber, D.; Li, Z. Food cravings, food addiction, and a dopamine-resistant (DRD2 A1) receptor polymorphism in Asian American college students. Asia Pac. J. Clin. Nutr. 2016, 25, 424. [Google Scholar]
- Misra, A. Revisions of cutoffs of body mass index to define overweight and obesity are needed for the Asian-ethnic groups. Int. J. Obes. 2003, 27, 1294–1296. [Google Scholar] [CrossRef]
- Ghee, L.K. A review of adult obesity research in Malaysia. Med. J. Malays. 2016, 71, 7. [Google Scholar]
- Soriano-Guillén, L.; Barrios, V.; Campos-Barros, Á.; Argente, J. Ghrelin levels in obesity and anorexia nervosa: Effect of weight reduction or recuperation. J. Pediatr. 2004, 144, 36–42. [Google Scholar] [CrossRef]
- Venner, A.A.; Lyon, M.E.; Doyle-Baker, P.K. Leptin: A potential biomarker for childhood obesity? Clin. Biochem. 2006, 39, 1047–1056. [Google Scholar] [CrossRef]
- Considine, R.V. Leptin and Other Endocrine Systems Leptin; Springer: Cham, Switzerland, 2006; pp. 103–123. [Google Scholar]
- Mammes, O.; Aubert, R.; Betoulle, D.; Pean, F.; Herbeth, B.; Visvikis, S.; Siest, G.; Fumeron, F. LEPR gene polymorphisms: Associations with overweight, fat mass and response to diet in women. Eur. J. Clin. Investig. 2001, 31, 398–404. [Google Scholar] [CrossRef]
- Mammes, O.; Betoulle, D.; Aubert, R.; Giraud, V.; Tuzet, S.; Petiet, A.; Colas-Linhart, N.; Fumeron, F. Novel polymorphisms in the 5′ region of the LEP gene: Association with leptin levels and response to low-calorie diet in human obesity. Diabetes 1998, 47, 487–489. [Google Scholar] [CrossRef]
- Gotoda, T.; Manning, B.S.; Goldstone, A.P.; Imrie, H.; Evans, A.L.; Strosberg, A.D.; McKeigue, P.M.; Scott, J.; Aitman, T.J. Leptin receptor gene variation and obesity: Lack of association in a white British male population. Hum. Mol. Genet. 1997, 6, 869–876. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, R.; Cerda, A.; Genvigir, F.D.V.; Sampaio, M.F.; Armaganijan, D.; Bernik, M.M.S.; Dorea, E.L.; Hirata, M.H.; Hinuy, H.M.; Hirata, R.D.C. Leptin receptor gene polymorphisms are associated with adiposity and metabolic alterations in Brazilian individuals. Arq. Bras. Endocrinol. Metabol. 2013, 57, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Wauters, M.; Mertens, I.; Rankinen, T.; Chagnon, M.; Bouchard, C.; Van Gaal, L. Leptin receptor gene polymorphisms are associated with insulin in obese women with impaired glucose tolerance. J. Clin. Endocrinol. Metab. 2001, 86, 3227–3232. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, C. The Genetics of Obesity; CRC Press: Boca Raton, FL, USA, 1994. [Google Scholar]
- Romao, I.; Roth, J. Genetic and environmental interactions in obesity and type 2 diabetes. J. Am. Diet. Assoc. 2008, 108, S24–S28. [Google Scholar] [CrossRef] [PubMed]
- Andreoli, M.F.; Donato, J.; Cakir, I.; Perello, M. Leptin resensitisation: A reversion of leptin-resistant states. J. Endocrinol. 2019, 241, R81–R96. [Google Scholar] [CrossRef] [PubMed]
- Ghilardi, N.; Ziegler, S.; Wiestner, A.; Stoffel, R.; Heim, M.H.; Skoda, R.C. Defective STAT signaling by the leptin receptor in diabetic mice. Proc. Natl. Acad. Sci. USA 1996, 93, 6231–6235. [Google Scholar] [CrossRef] [PubMed]
- Zarkesh-Esfahani, H.; Pockley, A.G.; Wu, Z.; Hellewell, P.G.; Weetman, A.P.; Ross, R.J. Leptin indirectly activates human neutrophils via induction of TNF-α. J. Immunol. 2004, 172, 1809–1814. [Google Scholar] [CrossRef]
- Bahrenberg, G.; Behrmann, I.; Barthel, A.; Hekerman, P.; Heinrich, P.C.; Joost, H.-G.; Becker, W. Identification of the critical sequence elements in the cytoplasmic domain of leptin receptor isoforms required for Janus kinase/signal transducer and activator of transcription activation by receptor heterodimers. Mol. Endocrinol. 2002, 16, 859–872. [Google Scholar] [CrossRef]
- Kalra, S.P.; Dube, M.G.; Pu, S.; Xu, B.; Horvath, T.L.; Kalra, P.S. Interacting appetite-regulating pathways in the hypothalamic regulation of body weight. Endocr. Rev. 1999, 20, 68–100. [Google Scholar]
- Bjørbæk, C.; El-Haschimi, K.; Frantz, J.D.; Flier, J.S. The role of SOCS-3 in leptin signaling and leptin resistance. J. Biol. Chem. 1999, 274, 30059–30065. [Google Scholar] [CrossRef]
- Hamilton, B.S.; Paglia, D.; Kwan, A.Y.; Deitel, M. Increased obese mRNA expression in omental fat cells from massively obese humans. Nat. Med. 1995, 1, 953–956. [Google Scholar] [CrossRef] [PubMed]
- Barr, V.A.; Malide, D.; Zarnowski, M.J.; Taylor, S.I.; Cushman, S.W. Insulin stimulates both leptin secretion and production by rat white adipose tissue. Endocrinology 1997, 138, 4463–4472. [Google Scholar] [CrossRef] [PubMed]
- Bradley, R.L.; Cheatham, B. Regulation of ob gene expression and leptin secretion by insulin and dexamethasone in rat adipocytes. Diabetes 1999, 48, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Considine, R.V.; Sinha, M.K.; Heiman, M.L.; Kriauciunas, A.; Stephens, T.W.; Nyce, M.R.; Ohannesian, J.P.; Marco, C.C.; McKee, L.J.; Bauer, T.L.; et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N. Engl. J. Med. 1996, 334, 292–295. [Google Scholar] [CrossRef]
- Wilson, M.E.; Fisher, J.; Brown, J. Chronic subcutaneous leptin infusion diminishes the responsiveness of the hypothalamic–pituitary–adrenal (HPA) axis in female rhesus monkeys. Physiol. Behav. 2005, 84, 449–458. [Google Scholar] [CrossRef]
- Montague, C.T.; Farooqi, I.S.; Whitehead, J.P.; Soos, M.A.; Rau, H.; Wareham, N.J.; Sewter, C.P.; Digby, J.E.; Mohammed, S.N.; Hurst, J.A.; et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature 1997, 387, 903–908. [Google Scholar] [CrossRef]
- Strobel, A.; Issad, T.; Camoin, L.; Ozata, M.; Strosberg, A.D. A leptin missense mutation associated with hypogonadism and morbid obesity. Nat. Genet. 1998, 18, 213–215. [Google Scholar] [CrossRef]
- Wasim, M. Role of leptin in obesity. J. Obes. Weight Loss Ther. 2015, 5, 1–3. [Google Scholar] [CrossRef]
- Maffei, Á.; Halaas, J.; Ravussin, E.; Pratley, R.; Lee, G.; Zhang, Y.; Fei, H.; Kim, S.; Lallone, R.; Ranganathan, S.; et al. Leptin levels in human and rodent: Measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat. Med. 1995, 1, 1155–1161. [Google Scholar] [CrossRef]
- Fairbrother, U.; Kidd, E.; Malagamuwa, T.; Walley, A. Genetics of severe obesity. Curr. Diabetes Rep. 2018, 18, 1–9. [Google Scholar] [CrossRef]
- Rohde, K.; Keller, M.; la Cour Poulsen, L.; Blüher, M.; Kovacs, P.; Böttcher, Y. Genetics and epigenetics in obesity. Metabolism 2019, 92, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Lamas, O.; Marti, A.; Martinez, J.A. Obesity and immunocompetence. Eur. J. Clin. Nutr. 2002, 56, S42–S45. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.-H.; Say, Y.-H. Leptin and leptin receptor gene polymorphisms and their association with plasma leptin levels and obesity in a multi-ethnic Malaysian suburban population. J. Physiol. Anthropol. 2014, 33, 15. [Google Scholar] [CrossRef] [PubMed]
- Lucantoni, R.; Ponti, E.; Berselli, M.E.; Savia, G.; Minocci, A.; Calò, G.; De Medici, C.; Liuzzi, A.; Di Blasio, A.M. The A19G polymorphism in the 5′ untranslated region of the human obese gene does not affect leptin levels in severely obese patients. J. Clin. Endocrinol. Metab. 2000, 85, 3589–3591. [Google Scholar]
- Vigouroux, C.; Fajas, L.; Khallouf, E.; Meier, M.; Gyapay, G.; Lascols, O.; Auwerx, J.; Weissenbach, J.; Capeau, J.; Magre, J. Human peroxisome proliferator-activated receptor-gamma2: Genetic mapping, identification of a variant in the coding sequence, and exclusion as the gene responsible for lipoatrophic diabetes. Diabetes 1998, 47, 490–492. [Google Scholar] [CrossRef]
- Ogawa, T.; Hirose, H.; Yamamoto, Y.; Nishikai, K.; Miyashita, K.; Nakamura, H.; Saito, I.; Saruta, T. Relationships between serum soluble leptin receptor level and serum leptin and adiponectin levels, insulin resistance index, lipid profile, and leptin receptor gene polymorphisms in the Japanese population. Metabolism 2004, 53, 879–885. [Google Scholar] [CrossRef]
- Lovibond, P.F.; Lovibond, S.H. The structure of negative emotional states: Comparison of the Depression Anxiety Stress Scales (DASS) with the Beck Depression and Anxiety Inventories. Behav. Res. Ther. 1995, 33, 335–343. [Google Scholar] [CrossRef]
- Hishan, S.S.; Jaiprakash, H.; Ramakrishnan, S.; Mohanraj, J.; Shanker, J.; Keong, L.B. Prevalence and Socio-Demographic Association of Depression, Anxiety and Stress among University Students. Int. J. Eng. Technol. 2018, 7, 688–691. [Google Scholar] [CrossRef]
- Samani, S.; Joukar, B. A study on the reliability and validity of the short form of the depression anxiety stress scale (DASS-21). J. Soc. Sci. Humanit. Shiraz Univ. 2007, 26, 65–77. [Google Scholar]
- Aboalshamat, K.; Hou, X.-Y.; Strodl, E. Psychological well-being status among medical and dental students in Makkah, Saudi Arabia: A cross-sectional study. Med. Teach. 2015, 37 (Suppl. S1), S75–S81. [Google Scholar] [CrossRef]
- Buysse, D.J.; Reynolds, C.F., III; Monk, T.H.; Berman, S.R.; Kupfer, D.J. The Pittsburgh Sleep Quality Index: A new instrument for psychiatric practice and research. Psychiatry Res. 1989, 28, 193–213. [Google Scholar] [CrossRef]
- Smyth, C. The Pittsburgh sleep quality index (PSQI). J. Gerontol. Nursing 1999, 25, 10. [Google Scholar] [CrossRef] [PubMed]
- Mollayeva, T.; Thurairajah, P.; Burton, K.; Mollayeva, S.; Shapiro, C.M.; Colantonio, A. The Pittsburgh sleep quality index as a screening tool for sleep dysfunction in clinical and non-clinical samples: A systematic review and meta-analysis. Sleep Med. Rev. 2016, 25, 52–73. [Google Scholar] [CrossRef] [PubMed]
- Brytek-Matera, A.; Rogoza, R.; Czepczor-Bernat, K. The Three-Factor Eating Questionnaire-R18 Polish version: Factor structure analysis among normal weight and obese adult women. Arch. Psychiatry Psychother. 2017, 3, 81–90. [Google Scholar] [CrossRef]
- Kavazidou, E.; Proios, M.; Liolios, I.; Doganis, G.; Petrou, K.; Tsatsoulis, A.; Tsiligiroglou-Fachantidou, A. Structure validity of the three-factor eating questionnaire-R18 in Greek population. J. Hum. Sport Exerc. 2012, 7, 1. [Google Scholar] [CrossRef]
- Grunfeld, C.; Zhao, C.; Fuller, J.; Pollack, A.; Moser, A.; Friedman, J.; Feingold, K.R. Endotoxin and cytokines induce expression of leptin, the ob gene product, in hamsters. J. Clin. Investig. 1996, 97, 2152–2157. [Google Scholar] [CrossRef]
- Moini, J.; Ahangari, R.; Miller, C.; Samsam, M. Global Health Complications of Obesity; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Plata-Salaman, C.R.; Borkoski, J.P. Chemokines/intercrines and central regulation of feeding. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 1994, 266, R1711–R1715. [Google Scholar] [CrossRef]
- Hastuti, P.; Zukhrufia, I.; Padwaswari, M.H.; Nuraini, A.; Sadewa, A.H. Polymorphism in leptin receptor gene was associated with obesity in Yogyakarta, Indonesia. Egypt. J. Med. Hum. Genet. 2016, 17, 271–276. [Google Scholar] [CrossRef]
- Mahmoudi, R.; Noori Alavicheh, B.; Mozaffari, N.; Amin, M.; Fararouei, M.; Nikseresht, M. Polymorphisms of leptin (−2548 G/A) and leptin receptor (Q223R) genes in Iranian women with breast cancer. Int. J. Genom. 2015, 2015, 132720. [Google Scholar] [CrossRef]
- Wauters, M.; Mertens, I.; Chagnon, M.; Rankinen, T.; Considine, R.; Chagnon, Y.; Van Gaal, L.F.; Bouchard, C. Polymorphisms in the leptin receptor gene, body composition and fat distribution in overweight and obese women. Int. J. Obes. 2001, 25, 714. [Google Scholar] [CrossRef]
- Chung, W.K.; Power-Kehoe, L.; Chua, M.; Chu, F.; Aronne, L.; Huma, Z.; Sothern, M.; Udall, J.N.; Kahle, B.; Leibel, R.L. Exonic and Intronic Sequence Variation in the Human Leptin Receptor Gene (LEPR); American Diabetes Association: Arlington, VA, USA, 1997. [Google Scholar]
- Bruce Thompson, D.; Ravussin, E.; Bennett, P.H.; Bogardus, C. Structure and sequence variation at the human leptin receptor gene in lean and obese Pima Indians. Hum. Mol. Genet. 1997, 6, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Wasim, M.; Awan, F.R.; Najam, S.S.; Khan, A.R.; Khan, H.N. Role of leptin deficiency, inefficiency, and leptin receptors in obesity. Biochem. Genet. 2016, 54, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Peltz, G.; Sanderson, M.; Pérez, A.; Sexton, K.; Casares, D.O.; Fadden, M.K. Serum leptin concentration, adiposity, and body fat distribution in Mexican-Americans. Arch. Med. Res. 2007, 38, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Gundlach, A.L. Galanin/GALP and galanin receptors: Role in central control of feeding, body weight/obesity and reproduction? Eur. J. Pharmacol. 2002, 440, 255–268. [Google Scholar] [CrossRef]
- Seth, A.; Stanley, S.; Jethwa, P.; Gardiner, J.; Ghatei, M.; Bloom, S. Galanin-like peptide stimulates the release of gonadotropin-releasing hormone in vitro and may mediate the effects of leptin on the hypothalamo-pituitary-gonadal axis. Endocrinology 2004, 145, 743–750. [Google Scholar] [CrossRef][Green Version]
- Kuan, P.; Ho, H.; Shuhaili, M.; Siti, A.; Gudum, H. Gender differences in body mass index, body weight perception and weight loss strategies among undergraduates in Universiti Malaysia Sarawak. Malays. J. Nutr. 2011, 17, 67–75. [Google Scholar]
- Rampal, L.; Rampal, S.; Khor, G.L.; Zain, A.M.; Ooyub, S.B.; Rahmat, R.B.; Ghani, S.N.; Krishnan, J. A national study on the prevalence of obesity among 16,127 Malaysians. Asia Pac. J. Clin. Nutr. 2007, 16, 561–566. [Google Scholar]
- Montague, C.T.; Prins, J.B.; Sanders, L.; Digby, J.E.; O’Rahilly, S. Depot-and sex-specific differences in human leptin mRNA expression: Implications for the control of regional fat distribution. Diabetes 1997, 46, 342–347. [Google Scholar] [CrossRef]
- Rosenbaum, M.; Leibel, R.L. Role of gonadal steroids in the sexual dimorphisms in body composition and circulating concentrations of leptin. J. Clin. Endocrinol. Metab. 1999, 84, 1784–1789. [Google Scholar] [CrossRef]
- Williams, L.B.; Fawcett, R.L.; Waechter, A.S.; Zhang, P.; Kogon, B.E.; Jones, R.; Inman, M.; Huse, J.; Considine, R.V. Leptin production in adipocytes from morbidly obese subjects: Stimulation by dexamethasone, inhibition with troglitazone, and influence of gender. J. Clin. Endocrinol. Metab. 2000, 85, 2678–2684. [Google Scholar] [CrossRef]
- Enriori, P.J.; Evans, A.E.; Sinnayah, P.; Cowley, M.A. Leptin resistance and obesity. Obesity 2006, 14 (Suppl. S8), 254S–258S. [Google Scholar] [CrossRef] [PubMed]
- Ng, Z.Y.; Veerapen, M.K.; Hon, W.M.; Lim, R.L.H. Association of leptin/receptor and TNF-α gene variants with adolescent obesity in Malaysia. Pediatr. Int. 2014, 56, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Hassink, S.G.; Sheslow, D.V.; de Lancey, E.; Opentanova, I.; Considine, R.V.; Caro, J.F. Serum Leptin in Children with Obesity: Relationship to Gender and Development. Pediatrics 1996, 98, 201–203. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, S.; Ganeshkumar, P.; Prakash, M.; Amalraj, V. Prevalence of overweight/obesity among the medical students, Malaysia. Med. J. Malays. 2012, 67, 442–444. [Google Scholar]
- Ismail, M.; Chee, S.; Nawawi, H.; Yusoff, K.; Lim, T.; James, W. Obesity in Malaysia. Obes. Rev. 2002, 3, 203–208. [Google Scholar] [CrossRef]
- Van Rossum, C.T.; Hoebee, B.; Van Baak, M.A.; Mars, M.; Saris, W.H.; Seidell, J.C. Genetic variation in the leptin receptor gene, leptin, and weight gain in young Dutch adults. Obes. Res. 2003, 11, 377–386. [Google Scholar] [CrossRef]
- Pejovic, S.; Vgontzas, A.N.; Basta, M.; Tsaoussoglou, M.; Zoumakis, E.; Vgontzas, A.; Bixler, E.O.; Chrousos, G.P. Leptin and hunger levels in young healthy adults after one night of sleep loss. J. Sleep Res. 2010, 19, 552–558. [Google Scholar] [CrossRef]
- Engineer, D.R.; Garcia, J.M. Leptin in anorexia and cachexia syndrome. Int. J. Pept. 2012, 2012, 287457. [Google Scholar] [CrossRef]
- Bagchi, D.; Preuss, H.G. Obesity: Epidemiology, Pathophysiology, and Prevention; CRC Press: Boca Raton, FL, USA, 2012. [Google Scholar]
- Haleem, D.J. Investigations into the involvement of leptin in responses to stress. Behav. Pharmacol. 2014, 25, 384–397. [Google Scholar] [CrossRef]
- Haleem, D.J.; Haider, S.; Perveen, T.; Haleem, M.A. Serum leptin and cortisol, related to acutely perceived academic examination stress and performance in female university students. Appl. Psychophysiol. Biofeedback 2015, 40, 305–312. [Google Scholar] [CrossRef]
- Shireen, E.; Haider, S.; Haleem, D.J. Perception of academic examination stress: Effects on serum leptin, cortisol, appetite and performance. J. Ayub Med. Coll. Abbottabad 2011, 23, 97–99. [Google Scholar]
- Herrick, J.E.; Panza, G.S.; Gollie, J.M. Leptin, leptin soluble receptor, and the free leptin index following a diet and physical activity lifestyle intervention in obese males and females. J. Obes. 2016, 2016, 8375828. [Google Scholar] [CrossRef] [PubMed]
- Van Harmelen, V.; Reynisdottir, S.; Eriksson, P.; Thörne, A.; Hoffstedt, J.; Lönnqvist, F.; Arner, P. Leptin secretion from subcutaneous and visceral adipose tissue in women. Diabetes 1998, 47, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, N.; Ogawa, Y.; Hosoda, K.; Matsuda, J.; Masuzaki, H.; Miyawaki, T.; Azuma, N.; Natsui, K.; Nishimura, H.; Yoshimasa, Y.; et al. Human leptin receptor gene in obese Japanese subjects: Evidence against either obesity-causing mutations or association of sequence variants with obesity. Diabetologia 1997, 40, 1204–1210. [Google Scholar] [CrossRef]
- Voight, B.F.; Kudaravalli, S.; Wen, X.; Pritchard, J.K. A map of recent positive selection in the human genome. PLoS Biol. 2006, 4, e72. [Google Scholar]
- Wang, T.N.; Huang, M.C.; Chang, W.T.; Ko, A.M.S.; Tsai, E.M.; Liu, C.S.; Lee, C.H.; Ko, Y.C. G-2548A polymorphism of the leptin gene is correlated with extreme obesity in Taiwanese aborigines. Obesity 2006, 14, 183–187. [Google Scholar] [CrossRef]
- Fairbrother, U.L.; Tankó, L.B.; Walley, A.J.; Christiansen, C.; Froguel, P.; Blakemore, A.I. Leptin receptor genotype at Gln223Arg is associated with body composition, BMD, and vertebral fracture in postmenopausal Danish women. J. Bone Miner. Res. 2007, 22, 544–550. [Google Scholar] [CrossRef]
- Mattevi, V.; Zembrzuski, V.; Hutz, M. Association analysis of genes involved in the leptin-signaling pathway with obesity in Brazil. Int. J. Obes. 2002, 26, 1179–1185. [Google Scholar] [CrossRef]
- Yiannakouris, N.; Yannakoulia, M.; Melistas, L.; Chan, J.L.; Klimis-Zacas, D.; Mantzoros, C.S. The Q223R polymorphism of the leptin receptor gene is significantly associated with obesity and predicts a small percentage of body weight and body composition variability. J. Clin. Endocrinol. Metab. 2001, 86, 4434–4439. [Google Scholar] [CrossRef]
- Sahoo, S.; Khess, C.R. Prevalence of depression, anxiety, and stress among young male adults in India: A dimensional and categorical diagnoses-based study. J. Nerv. Ment. Dis. 2010, 198, 901–904. [Google Scholar] [CrossRef]
- Blüher, S.; Mantzoros, C.S. Leptin in humans: Lessons from translational research. Am. J. Clin. Nutr. 2009, 89, 991S–997S. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, I.S. Genetic and hereditary aspects of childhood obesity. Best Pract. Res. Clin. Endocrinol. Metab. 2005, 19, 359–374. [Google Scholar] [CrossRef] [PubMed]
- El-Haschimi, K.; Pierroz, D.D.; Hileman, S.M.; Bjørbæk, C.; Flier, J.S. Two defects contribute to hypothalamic leptin resistance in mice with diet-induced obesity. J. Clin. Investig. 2000, 105, 1827–1832. [Google Scholar] [CrossRef]
- Lin, S.; Thomas, T.; Storlien, L.; Huang, X. Development of high fat diet-induced obesity and leptin resistance in C57Bl/6J mice. Int. J. Obes. 2000, 24, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Prpic, V.; Watson, P.M.; Frampton, I.C.; Sabol, M.A.; Jezek, G.E.; Gettys, T.W. Differential mechanisms and development of leptin resistance in A/J versus C57BL/6J mice during diet-induced obesity. Endocrinology 2003, 144, 1155–1163. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, I.S.; Matarese, G.; Lord, G.M.; Keogh, J.M.; Lawrence, E.; Agwu, C.; Sanna, V.; Jebb, S.A.; Perna, F.; Fontana, S.; et al. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J. Clin. Investig. 2002, 110, 1093–1103. [Google Scholar] [CrossRef]
- Licinio, J.; Caglayan, S.; Ozata, M.; Yildiz, B.O.; De Miranda, P.B.; O’Kirwan, F.; Whitby, R.; Liang, L.; Cohen, P.; Bhasin, S.; et al. Phenotypic effects of leptin replacement on morbid obesity, diabetes mellitus, hypogonadism, and behavior in leptin-deficient adults. Proc. Natl. Acad. Sci. USA 2004, 101, 4531–4536. [Google Scholar] [CrossRef]
- Chou, S.H.; Chamberland, J.P.; Liu, X.; Matarese, G.; Gao, C.; Stefanakis, R.; Brinkoetter, M.T.; Gong, H.; Arampatzi, K.; Mantzoros, C.S. Leptin is an effective treatment for hypothalamic amenorrhea. Proc. Natl. Acad. Sci. USA 2011, 108, 6585–6590. [Google Scholar] [CrossRef]


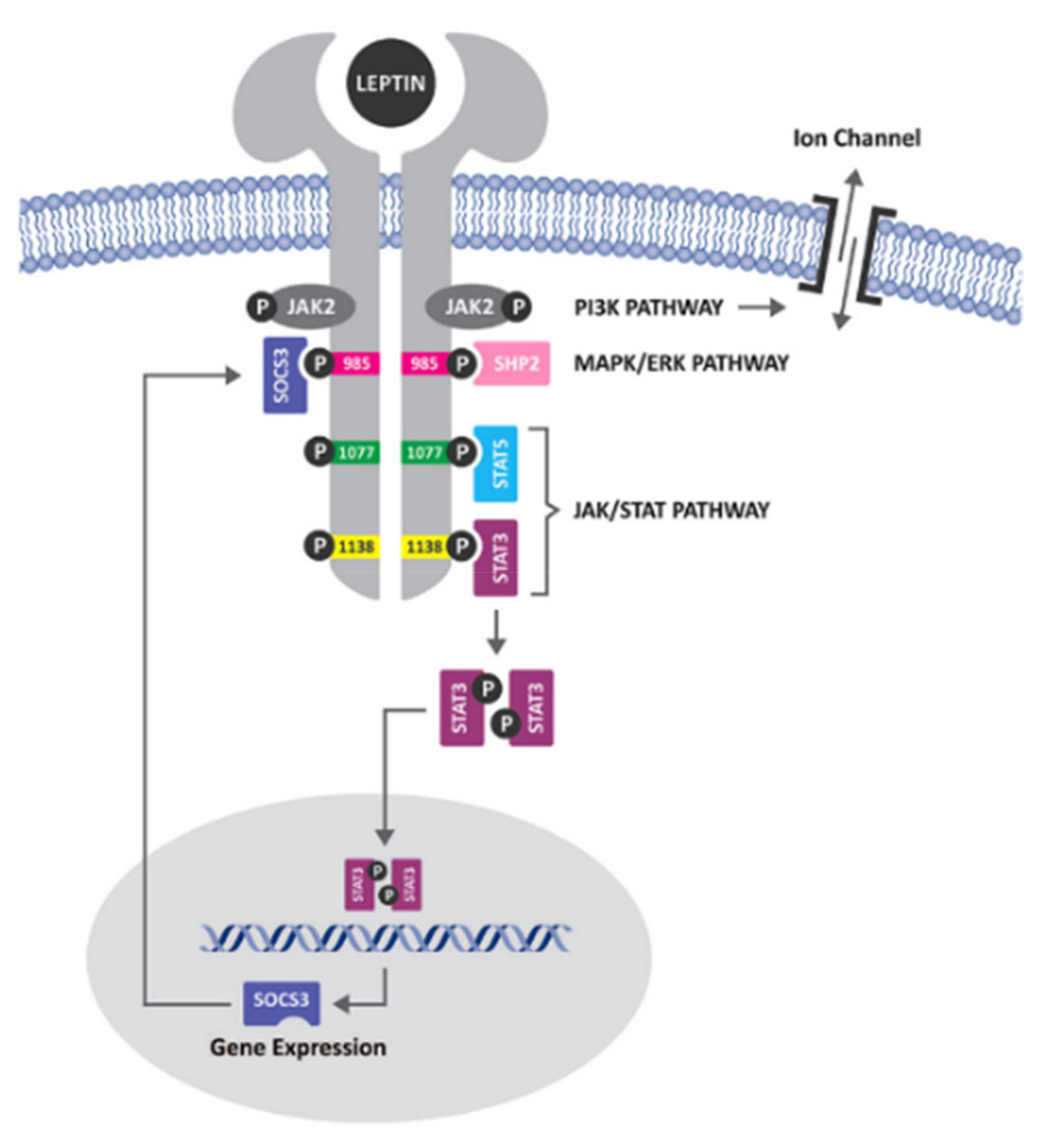{kind=link}
| Gender | Mean | Std. Deviation | F | Sig. | ||
|---|---|---|---|---|---|---|
| Gender | Male | Cases (25) | 2955.13 | 731.23 | 22.175 | 0.000 |
| Controls (29) | 2184.09 | 458.41 | ||||
| Female | Cases (64) | 3392.66 | 378.75 | 11.527 | 0.001 | |
| Controls (67) | 3009.46 | 823.43 | ||||
| Ethnicity | Malay | Cases (23) | 3322.19 | 570.03 | 0.195 | 0.660 |
| Controls (38) | 3241.81 | 750.19 | ||||
| Chinese | Cases (21) | 3113.42 | 606.56 | 10.016 | 0.003 | |
| Controls (24) | 2555.61 | 574.94 | ||||
| Indian | Cases (32) | 3294.17 | 512.29 | 18.267 | 0.000 | |
| Controls (24) | 2529.84 | 822.21 | ||||
| Others | Cases (13) | 3369.45 | 402.55 | 45.295 | 0.000 | |
| Controls (10) | 1973.31 | 592.86 | ||||
| BMI | Underweight (29) | 2880.96 | 574.43 | 2.71 | 0.01 | |
| Normal (100) | 2877.77 | 793.31 | ||||
| Overweight (29) | 3174.01 | 726.22 | ||||
| Grade 1 Obesity (19) | 3321.97 | 649.65 | ||||
| Grade 2 Obesity (3) | 3655.30 | 267.10 | ||||
| Grade 3 Obesity (5) | 3705.04 | 221.08 | ||||
| Gender [Mean (SD)] | Ethnicity [Mean (SD)] | |||||
|---|---|---|---|---|---|---|
|
Male (n = 29) |
Female (n = 34) |
Malay (n = 20) |
Chinese (n = 20) |
Indian (n = 18) |
Others (n = 5) | |
| BMI | 26.43 (9.73) | 23.08 (6.16) | 25.71 (5.66) | 23.78 (5.86) | 26.15 (11.8) | 18.12 (5.6) |
| Leptin (pg/mL) | 2568.54 (717.7) | 3296.47 (461.3) | 3048.26 (786.4) | 2814.48 (620.2) | 2988.71 (726.0) | 3103.28 (504.97) |
| LEP G2548A % (n) | ||||||
| Ala/Ala | 48.3 (14) | 44.1 (15) | 45 (9) | 60 (12) | 33.3 (6) | 40 (2) |
| Ala/Gly | 34.5 (10) | 41.2 (14) | 30 (6) | 40 (8) | 44.4 (8) | 40 (2) |
| Gly/Gly | 17.2 (5) | 14.7 (5) | 25 (5) | 0 (0) | 22.2 (4) | 20 (1) |
| LEPR Q223R % (n) | ||||||
| Arg/Arg | 6.9 (2) | 8.8 (3) | 0 (0) | 10 (2) | 11.1 (2) | 20 (1) |
| Arg/Gln | 37.9 (11) | 35.3 (12) | 60 (12) | 20 (4) | 33.3 (6) | 20 (1) |
| Gln/Gln | 55.9 (16) | 36.5 (19) | 40 (8) | 70 (14) | 55.6 (10) | 60 (3) |
| Genotype | Ethnicity | p-Value (χ2) | |||
|---|---|---|---|---|---|
| Malay | Chinese | Indian | Others | ||
| LEP G2548A | |||||
| Ala/Ala | 9 (31%) | 12 (41.4%) | 6 (20.7%) | 2 (6.9%) | 0.338 (df = 6) |
| Ala/Gly | 6 (7.6%) | 8 (33.3%) | 8 (33.3%) | 2 (8.3%) | |
| Gly/Gly | 5 (50%) | 0 (0%) | 4 (40%) | 1 (10%) | |
| LEPR Q223R | |||||
| Arg/Arg | 0 (0%) | 2 (40%) | 2 (40%) | 1 (20%) | 0.151 (df = 6) |
| Arg/Gln | 12 (52.2%) | 4 (17.4%) | 6 (26.1%) | 1 (4.3%) | |
| Gln/Gln | 8 (22.9%) | 14 (40%) | 10 (28.6%) | 3 (8.6%) | |
| Genotypes | Number | Leptin Conc. 1 | p-Value | BMI 2 | p-Value |
|---|---|---|---|---|---|
| LEP G2548A | |||||
| Ala/Ala | 29 | 2794.19 ± 649.18 | 0.196 (F = 1.67) | 22.46 ± 4.7 | 0.117 (F = 2.22) |
| Ala/Gly | 24 | 3074.30 ± 685.07 | 27.0 ± 10.6 | ||
| Gly/Gly | 10 | 3175.28 ± 786.74 | 27.9 ± 7.9 | ||
| LEPR Q223R | |||||
| Gln/Gln | 35 | 2929.59 ± 692.58 | 0.453 (0.801) | 24.1 ± 6.8 | 0.469 (F = 0.768) |
| Arg/Gln | 23 | 3074.55 ± 729.77 | 26.0 ± 10 | ||
| Arg/Arg | 5 | 2663.44 ± 816.27 | 21.5 ± 6.9 | ||
| Parameter (n = 63) | Plasma Leptin p-Value (F Score) | LEP G2548A p-Value (F Score) | LEPR Q223R p-Value (F Score) |
|---|---|---|---|
| PSQI | 0.2170 (1.53) | 0.219 (1.55) | 0.797 (0.22) |
| Stress | 0.000 (23.89) | 0.078 (2.22) | 0.959 (0.15) |
| Depression | 0.001 (5.19) | 0.492 (0.86) | 0.841 (0.35) |
| Anxiety | 0.000 (8.70) | 0.279 (1.30) | 0.457 (0.92) |
| TFEQ (Cognitive Restraint) | 0.3250 (1.16) | 0.703 (0.47) | 0.947 (0.12) |
| TFEQ (Uncontrolled Eating) | 0.0200 (3.35) | 0.554 (0.59) | 0.512 (0.67) |
| TFEQ (Emotional Eating) | 0.0100 (3.73) | 0.590 (0.64) | 0.529 (0.74) |
| Genotypes | Controls % (n) | Cases % (n) | Odds Ratio | 95% CI | p-Value |
|---|---|---|---|---|---|
| LEP G2548A | |||||
| Ala/Ala | 50 (15) | 28.6 (4) | 1 * | ||
| Ala/Gly | 30 (9) | 57.1 (8) | 0.3 | 0.06–1.28 | 0.11 |
| Gly/Gly | 20 (6) | 14.3 (2) | 0.8 | 0.11–5.50 | 0.83 |
| Alleles | |||||
| Ala-Allele | 72.2 (39) | 61.5 (16) | 0.6 | 0.22–1.65 | 0.33 |
| Gly-Allele | 27.8 (15) | 38.5 (10) | |||
| LEPR Q223R | |||||
| Gln/Gln | 53.3 (16) | 57.1 (8) | 1 * | ||
| Arg/Gln | 40 (12) | 42.9 (6) | 1 | 0.27–3.6 | 1 |
| Arg/Arg | 6.7 (2) | 0 (0) | 2.5 | 0.11–59.9 | 0.5 |
| Alleles | |||||
| Gln-Allele | 75.9 (44) | 78.6 (22) | 1.6 | 0.39–3.45 | 0.78 |
| Arg-Allele | 22.2 (14) | 21.4 (6) | |||
| Genotypes | Controls % (n) | Cases % (n) | Odds Ratio | 95% CI | p-Value |
|---|---|---|---|---|---|
| LEP G2548A | |||||
| Ala/Ala | 50 (15) | 63.6 (7) | 1 * | ||
| Ala/Gly | 30 (9) | 27.2 (3) | 0.3 | 0.06–1.28 | 0.11 |
| Gly/Gly | 20 (6) | 9.1 (1) | 0.8 | 0.11–5.50 | 0.83 |
| Alleles | |||||
| Ala-Allele | 72.2 (39) | 77.3 (17) | 0.6 | 0.22–1.65 | 0.33 |
| Gly-Allele | 27.8 (15) | 22.7 (5) | |||
| LEPR Q223R | |||||
| Gln/Gln | 53.3 (16) | 54.5 (6) | 1 * | ||
| Arg/Gln | 40 (12) | 45.5 (5) | 1 | 0.27–3.6 | 1 |
| Arg/Arg | 6.7 (2) | 0 (0) | 2.5 | 0.11–59.9 | 0.5 |
| Alleles | |||||
| Gln-Allele | 75.9 (44) | 68.2 (15) | 1.6 | 0.39–3.45 | 0.78 |
| Arg-Allele | 22.2 (14) | 31.8 (14) | |||
| Genotypes | Controls % (n) | Cases % (n) | Odds Ratio | 95% CI | p-Value |
|---|---|---|---|---|---|
| LEP G2548A | |||||
| Ala/Ala | 50 (15) | 37.5 (3) | 1 * | ||
| Ala/Gly | 30 (9) | 50 (4) | 0.3 | 0.06–1.28 | 0.11 |
| Gly/Gly | 20 (6) | 12.5 (1) | 0.8 | 0.11–5.50 | 0.83 |
| Alleles | |||||
| Ala-Allele | 72.2 (39) | 62.5 (10) | 0.6 | 0.22–1.65 | 0.33 |
| Gly-Allele | 27.8 (15) | 37.5 (6) | |||
| LEPR Q223R | |||||
| Gln/Gln | 53.3 (16) | 62.5 (5) | 1 * | ||
| Arg/Gln | 40 (12) | 25 (2) | 1 | 0.27–3.6 | 1 |
| Arg/Arg | 6.7 (2) | 12.5 (1) | 2.5 | 0.11–59.9 | 0.5 |
| Alleles | |||||
| Gln-Allele | 75.9 (44) | 75 (12) | 1.6 | 0.39–3.45 | 0.78 |
| Arg-Allele | 22.2 (14) | 25 (4) | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohanraj, J.; D’Souza, U.J.A.; Fong, S.Y.; Karkada, I.R.; Jaiprakash, H. Association between Leptin (G2548A) and Leptin Receptor (Q223R) Polymorphisms with Plasma Leptin, BMI, Stress, Sleep and Eating Patterns among the Multiethnic Young Malaysian Adult Population from a Healthcare University. Int. J. Environ. Res. Public Health 2022, 19, 8862. https://doi.org/10.3390/ijerph19148862
Mohanraj J, D’Souza UJA, Fong SY, Karkada IR, Jaiprakash H. Association between Leptin (G2548A) and Leptin Receptor (Q223R) Polymorphisms with Plasma Leptin, BMI, Stress, Sleep and Eating Patterns among the Multiethnic Young Malaysian Adult Population from a Healthcare University. International Journal of Environmental Research and Public Health. 2022; 19(14):8862. https://doi.org/10.3390/ijerph19148862
Chicago/Turabian StyleMohanraj, Jaiprakash, Urban J. A. D’Souza, Siat Yee Fong, Ivan Rolland Karkada, and Heethal Jaiprakash. 2022. "Association between Leptin (G2548A) and Leptin Receptor (Q223R) Polymorphisms with Plasma Leptin, BMI, Stress, Sleep and Eating Patterns among the Multiethnic Young Malaysian Adult Population from a Healthcare University" International Journal of Environmental Research and Public Health 19, no. 14: 8862. https://doi.org/10.3390/ijerph19148862
APA StyleMohanraj, J., D’Souza, U. J. A., Fong, S. Y., Karkada, I. R., & Jaiprakash, H. (2022). Association between Leptin (G2548A) and Leptin Receptor (Q223R) Polymorphisms with Plasma Leptin, BMI, Stress, Sleep and Eating Patterns among the Multiethnic Young Malaysian Adult Population from a Healthcare University. International Journal of Environmental Research and Public Health, 19(14), 8862. https://doi.org/10.3390/ijerph19148862