
Preparation of Supported Perovskite Catalyst to Purify Membrane Concentrate of Coal Chemical Wastewater in UV-Catalytic Wet Hydrogen Peroxide Oxidation System

Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Materials
2.2. Preparation of Materials
2.3. Degradation Experiments and Analytical Method
3. Results and Discussion
3.1. Optimization of Catalyst Preparation Conditions
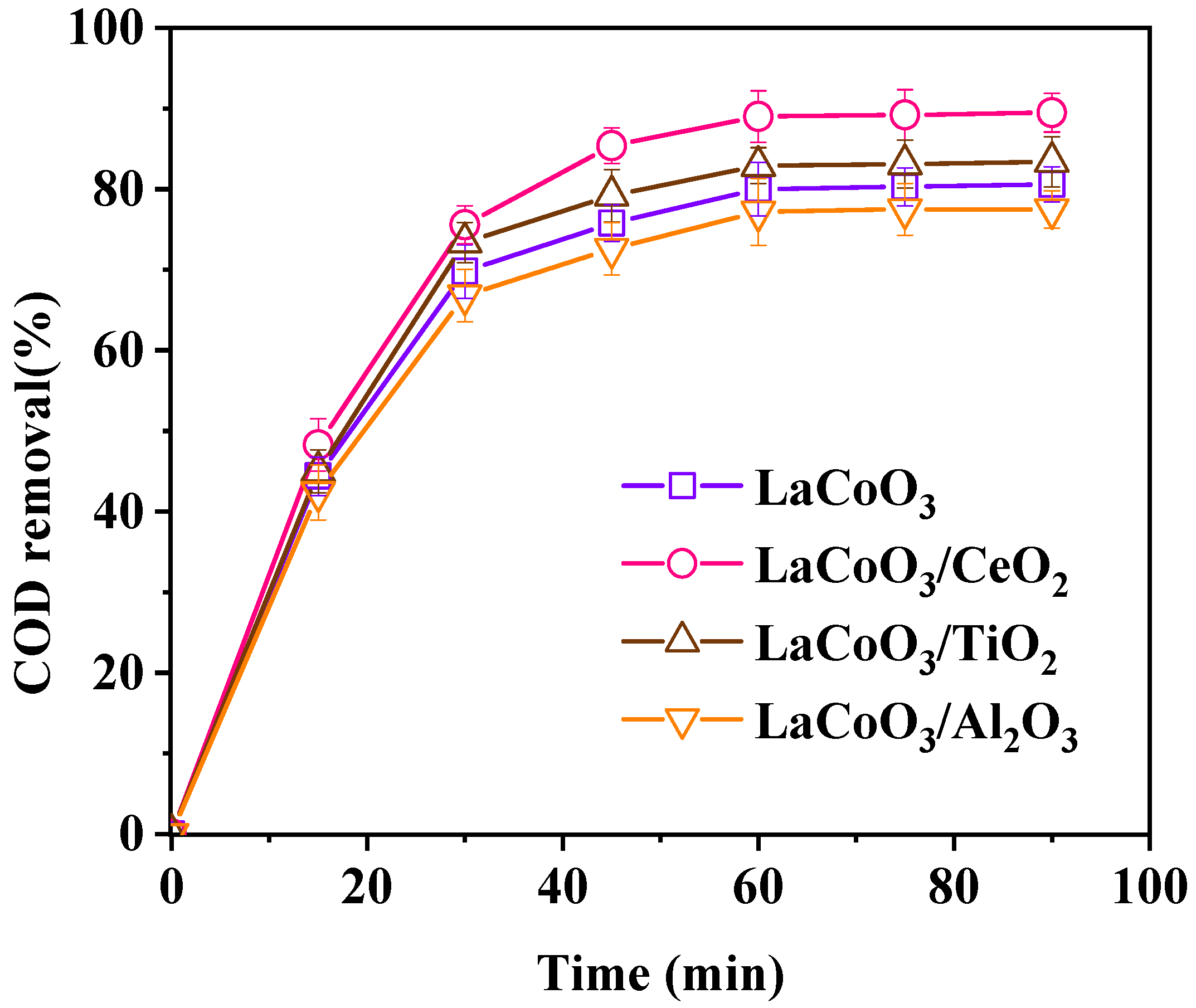
3.1.1. Effect of Catalyst Support
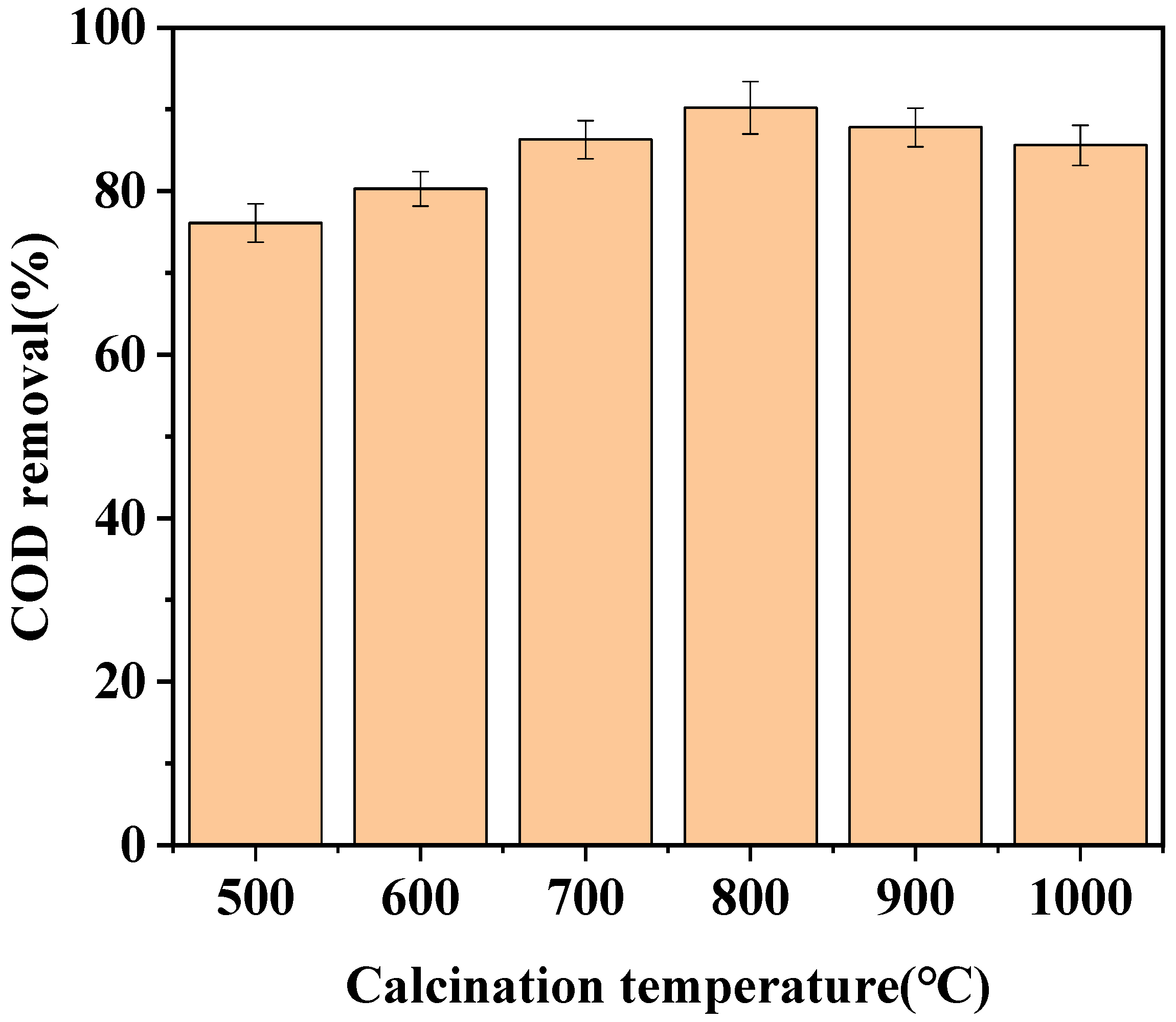
3.1.2. Effect of Calcination Temperature
3.1.3. Effect of Calcination Time
3.2. Characterization of Catalysts
3.2.1. X-ray Diffraction (XRD)
3.2.2. N2 Adsorption/Desorption Analysis
3.2.3. Scanning Electron Microscopy (SEM)
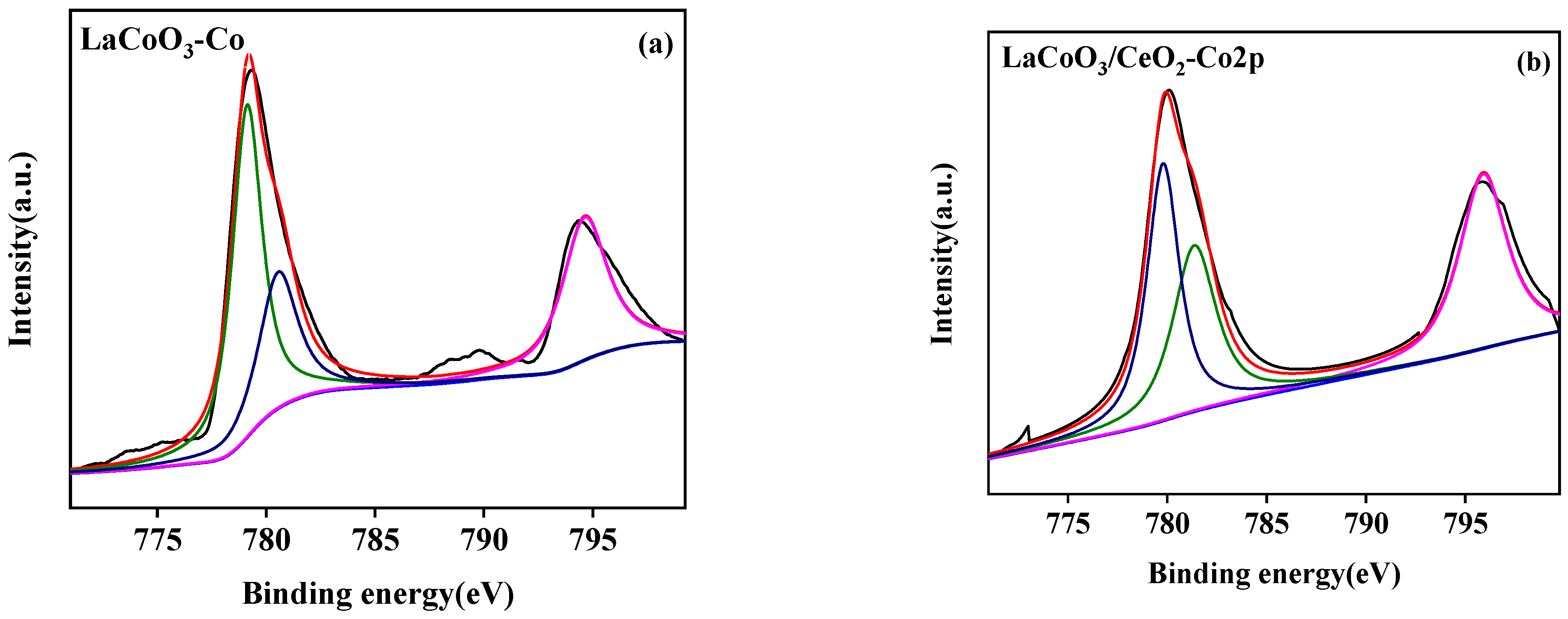
3.2.4. X-ray Photoelectron Spectroscopy (XPS)
3.3. Degradation Experiments
3.3.1. Comparison to Several Kinds of Reaction Systems
3.3.2. Effect of H2O2 Dosage on Wastewater Purification
3.3.3. Effect of Reaction Temperature on Wastewater Purification
3.3.4. Effect of Reaction Pressure on Effluent Purification
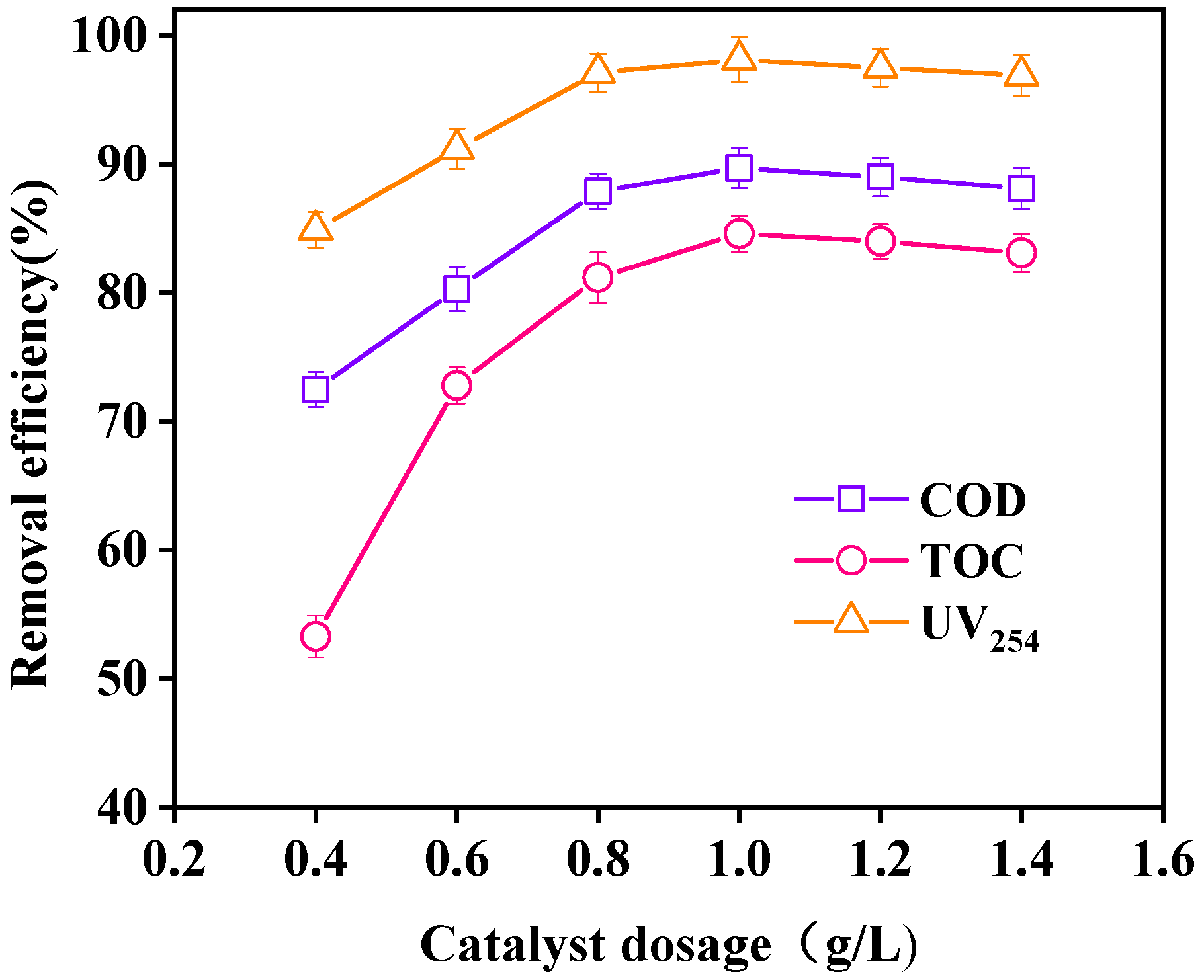
3.3.5. Effect of Catalyst Dosage on Wastewater Purification
3.4. Reusability and Stability of Catalyst
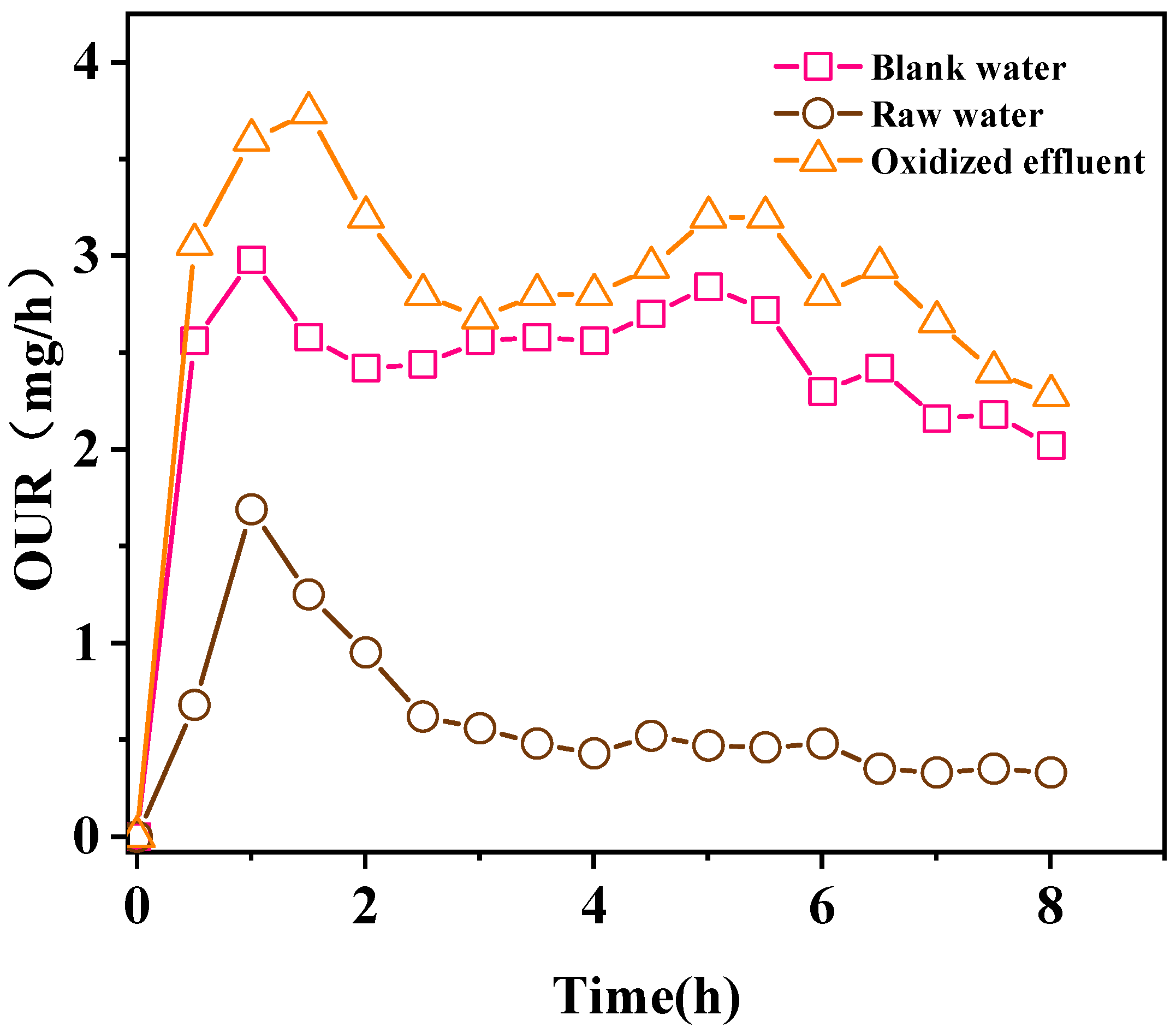
4. Biodegradability Analysis
4.1. Biodegradability
4.2. Biological Toxicity Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhao, D.; Lun, W.; Wei, J. Discussion on wastewater treatment process of coal chemical industry. In IOP Conference Series: Earth and Environmental Science; IOP Publishing: Bristol, UK, 2017; Volume 100, p. 012067. [Google Scholar]
- Ma, H.; Wang, H.; Tian, C.; Wang, L.; Yuan, W.; Qi, Y.; Ma, H.; Chao, Z.; Lv, W. An integrated membrane-and thermal-based system for coal chemical wastewater treatment with near-zero liquid discharge. J. Clean. Prod. 2021, 291, 125842. [Google Scholar] [CrossRef]
- Zhang, L.; Li, A.; Lu, Y.; Yan, L.; Zhong, S.; Deng, C. Characterization and removal of dissolved organic matter (DOM) from landfill leachate rejected by nanofiltration. Waste Manag. 2009, 29, 1035–1040. [Google Scholar] [CrossRef]
- Tu, S.; Zhu, X.; Du, J.; Liu, T.; Liang, H.; Hong, X. Degradation of Reactive Brilliant Red X-3B by UV-CWPO process with Cu-Zn-Ce catalys. Acta Sci. Circumstantia 2014, 34, 1696–1704. [Google Scholar]
- Garcia-Costa, A.L.; Silveira, J.E.; Zazo, J.A.; Dionysiou, D.D.; Casas, J.A. Graphite as catalyst for UV-A LED assisted catalytic wet peroxide oxidation of ibuprofen and diclofenac. Chem. Eng. J. Adv. 2021, 6, 100090. [Google Scholar] [CrossRef]
- Garcia-Muñoz, P.; Lefevre, C.; Robert, D.; Keller, N. Ti-substituted LaFeO3 perovskite as photoassisted CWPO catalyst for water treatment. Appl. Catal. B Environ. 2019, 248, 120–128. [Google Scholar] [CrossRef]
- Zazo, J.A.; Pliego, G.; García-Muñoz, P.; Casas, J.A.; Rodriguez, J.J. UV-LED assisted catalytic wet peroxide oxidation with a Fe (II)-Fe (III)/activated carbon catalyst. Appl. Catal. B Environ. 2016, 192, 350–356. [Google Scholar] [CrossRef]
- Garcia-Costa, A.L.; Sarabia, A.; Zazo, J.A.; Casas, J.A. UV-assisted Catalytic Wet Peroxide Oxidation and adsorption as efficient process for arsenic removal in groundwater. Catal. Today 2021, 361, 176–182. [Google Scholar] [CrossRef]
- Qin, Y.; Shen, F.; Zhu, T.; Hong, W.; Liu, X. Catalytic oxidation of ethyl acetate over LaBO3 (B = Co, Mn, Ni, Fe) perovskites supported silver catalysts. RSC Adv. 2018, 8, 33425–33431. [Google Scholar] [CrossRef]
- Hou, X.; Ren, J.; Li, F.; Ma, C.; Zhang, X.; Feng, H. Research progress of perovskite materials as catalysts. In IOP Conference Series: Earth and Environmental Science; IOP Publishing: Bristol, UK, 2019; Volume 295, p. 032020. [Google Scholar]
- Dukkanci, M. Degradation of bisphenol-a using a sonophoto Fenton-like hybrid process over a LaFeO3 perovskite catalyst and a comparison of its activity with that of a TiO2 photocatalyst. Turk. J. Chem. 2016, 40, 784–801. [Google Scholar] [CrossRef]
- Taran, O.P.; Ayusheev, A.B.; Ogorodnikova, O.L.; Prosvirin, I.P.; Isupova, L.A.; Parmon, V.N. Perovskite-like catalysts LaBO3 (B = Cu, Fe, Mn, Co, Ni) for wet peroxide oxidation of phenol. Appl. Catal. B Environ. 2016, 180, 86–93. [Google Scholar] [CrossRef]
- Hsu, Y.C.; Chang, S.H.; Chung, W.C.; Chang, M.B. Photocatalytic removal of trichloroethylene from water with LaFeO3. Environ. Sci. Pollut. Res. 2019, 26, 26276–26285. [Google Scholar] [CrossRef]
- Palas, B.; Ersöz, G.; Atalay, S. Catalytic wet air oxidation of Reactive Black 5 in the presence of LaNiO3 perovskite catalyst as a green process for azo dye removal. Chemosphere 2018, 209, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Bhattar, S.; Abedin, M.A.; Kanitkar, S.; Spivey, J.J. A review on dry reforming of methane over perovskite derived catalysts. Catal. Today 2020, 365, 2–23. [Google Scholar] [CrossRef]
- Liu, G.; Wang, J.M. Effects of Nano-Copper(II) Oxide and Nano-Magnesium Oxide Particles on Activated Sludge. Water Environ. Res. 2012, 84, 569–576. [Google Scholar] [CrossRef]
- Sudhakaran, M.S.P.; Hossain, M.; Gnanasekaran, G.; Mok, Y.S. Dry reforming of propane over γ-Al2O3 and nickel foam supported novel SrNiO3 perovskite catalyst. Catalysts 2019, 9, 68. [Google Scholar]
- Solís, R.R.; Rivas, F.J.; Gimeno, O.; Pérez-Bote, J.L. Synergism between peroxymonosulfate and LaCoO3-TiO2 photocatalysis for oxidation of herbicides. Oper. Var. Catal. Charact. Assess. J. Chem. Technol. Biotechnol. 2017, 92, 2159–2170. [Google Scholar] [CrossRef]
- Hammouda, S.B.; Zhao, F.; Safaei, Z.; Babu, I.; Ramasamy, D.L.; Sillanpää, M. Reactivity of novel Ceria–Perovskite composites CeO2-LaMO3 (MCu, Fe) in the catalytic wet peroxidative oxidation of the new emergent pollutant ‘Bisphenol F’: Characterization, kinetic and mechanism studies. Appl. Catal. B Environ. 2017, 218, 119–136. [Google Scholar] [CrossRef]
- Wang, X.; Zuo, J.; Luo, Y.; Jiang, L. New route to CeO2/LaCoO3 with high oxygen mobility for total benzene oxidation. Appl. Surf. Sci. 2017, 396, 95–101. [Google Scholar] [CrossRef]
- Yang, X.; Gao, Q.; Zhao, Z.; Guo, Y.; Guo, Y.; Wang, L.; Wang, Y.; Zhan, W. Surface tuning of noble metal doped perovskite oxide by synergistic effect of thermal treatment and acid etching: A new path to high-performance catalysts for methane combustion. Appl. Catal. B Environ. 2018, 239, 373–382. [Google Scholar] [CrossRef]
- Wang, S.; Xu, X.; Zhu, J.; Tang, D.; Zhao, Z. Effect of preparation method on physicochemical properties and catalytic performances of LaCoO3 perovskite for CO oxidation. J. Rare Earths 2019, 37, 970–977. [Google Scholar] [CrossRef]
- Ruder, H. Synthesis of LaCoO3 powder by a combined mechanical/thermal process. Z. Nat. B 2018, 73, 719–724. [Google Scholar]
- Tepech-Carrillo, L.; Escobedo-Morales, A.; Perez-Centeno, A.; Chigo-Anota, E.; Sanchez-Ramirez, J.F.; Lopez-Apreza, E.; Gutierrez-Gutierrez, J. Preparation of nanosized LaCoO3 through calcination of a hydrothermally synthesized precursor. J. Nanomater. 2016, 2016, 6917950. [Google Scholar] [CrossRef]
- Sun, J.; Zhao, Z.; Li, Y.; Yu, X.; Zhao, L.; Li, J.; Wei, Y.; Liu, J. Synthesis and catalytic performance of macroporous La1-xCexCoO3 perovskite oxide catalysts with high oxygen mobility for catalytic combustion of soot. J. Rare Earths 2020, 38, 584–593. [Google Scholar] [CrossRef]
- Alifanti, M.; Blangenois, N.; Florea, M.; Delmon, B. Supported Co-based perovskites as catalysts for total oxidation of methane. Appl. Catal. A Gen. 2005, 280, 255–265. [Google Scholar] [CrossRef]
- Yang, Z.C.; Yu, A.Q.; Shan, C.; Gao, G.D.; Pan, B.C. Enhanced Fe(III)-mediated Fenton oxidation of atrazine in the presence of functionalized multi-walled carbon nanotubes. Water Res. 2018, 137, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.X.; Wang, Y.; Jia, A.P.; Liu, H.H.; Luo, M.F.; Lu, J.Q. Enhanced activity for catalytic oxidation of 1, 2-dichloroethane over Al-substituted LaMnO3 perovskite catalysts. Appl. Surf. Sci. 2014, 307, 178–188. [Google Scholar] [CrossRef]
- Liang, H.; Mou, Y.; Zhang, H.; Li, S.; Yao, C.; Yu, X. Sulfur resistance and soot combustion for La0.8K0.2Co1−yMnyO3 catalyst. Catal. Today 2017, 281, 477–481. [Google Scholar] [CrossRef]
- Li, X.; Gao, H. Influence of Ce doping on catalytic oxidation of NO on LaCoO3 (011) surface: A DFT study. Appl. Surf. Sci. 2020, 499, 143866. [Google Scholar] [CrossRef]
- Guo, Y.Q.; Yin, Y.M.; Tong, Z.; Yin, J.W.; Xiong, M.W.; Ma, Z.F. Impact of synthesis technique on the structure and electrochemical characteristics of Pr0.6Sr0.4Co0.2Fe0.8O3(PSCF) cathode material. Solid State Ion. 2011, 193, 18–22. [Google Scholar] [CrossRef]
- Wang, Y.; Zheng, Y.; Wang, H.; Li, K.; Zhu, X.; Wei, Y.; Wang, Y.; Jiang, L.; Tian, D. Enhanced activity for CO preferential oxidation via the CeO2-LaCoO3 interaction. Ind. Eng. Chem. Res 2019, 58, 4710. [Google Scholar] [CrossRef]
- Afzal, S.; Quan, X.; Zhang, J. High surface area mesoporous nanocast LaMO3(M=Mn, Fe) perovskites for efficient catalytic ozonation and an insight into probable catalytic mechanism. Appl. Catal. B Environ. 2017, 206, 692–703. [Google Scholar] [CrossRef]
- Li, W.; Qiang, Z.; Zhang, T.; Cao, F. Kinetics and mechanism of pyruvic acid degradation by ozone in thepresence of PdO/CeO2. Appl. Catal. B Environ. 2012, 113, 290–295. [Google Scholar] [CrossRef]
- Pajić, N.D.; Djinović, P.; Dražić, G.; Grdadolnik, J.; Šket, P.; Cerkovnik, J.; Pintar, A. Structural stabilization and characterization of active peroxo species on TiO2-nanotube based materials in mild catalytic wet peroxide oxidation process. Appl. Catal. A Gen. 2018, 562, 276–283. [Google Scholar] [CrossRef]
- Bulca, Ö.; Palas, B.; Atalay, S.; Ersöz, G. Performance investigation of the hybrid methods of adsorption or catalytic wet air oxidation subsequent to electrocoagulation in treatment of real textile wastewater and kinetic modelling. J. Water Process Eng. 2021, 40, 101821. [Google Scholar] [CrossRef]
- Zhang, J.; Tan, D.; Meng, Q.; Weng, X.; Wu, Z. Structural modification of LaCoO3 perovskite for oxidation reactions: The synergistic effect of Ca2+ and Mg2+ co-substitution on phase formation and catalytic performance. Appl. Catal. B Environ. 2015, 172, 18–26. [Google Scholar] [CrossRef]
- Samoila, P.; Cojocaru, C.; Mahu, E.; Ignat, M.; Harabagiu, V. Boosting catalytic wet-peroxide-oxidation performances of cobalt ferrite by doping with lanthanides for organic pollutants degradation. J. Environ. Chem. Eng. 2021, 9, 104961. [Google Scholar] [CrossRef]
- Brar, S.K.; Verma, M.; Tyagi, R.D.; Surampalli, R.Y. Engineered nanoparticles in wastewater and wastewater sludge–Evidence and impacts. Waste Manag. 2010, 30, 504–520. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Specific Surface Area (m2/g) | Pore Volume (cm3/g) | Pore Diameter (nm) |
|---|---|---|---|
| LaCoO3 | 2.6749 | 0.0063 | 4.9501 |
| LaCoO3/CeO2 | 13.0729 | 0.0524 | 4.0569 |
| Catalyst | Co3+/Co2+ | OL/OS |
|---|---|---|
| LaCoO3 | 1.88 | 0.80 |
| LaCoO3/CeO2 | 1.15 | 1.48 |
| Sample | Respiratory Depression Rate | Toxicity |
|---|---|---|
| Raw water | 83.66% | High toxic |
| Oxidation discharge | No inhibition | Non-toxic |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, W.; Liu, Z.; Chen, P.; Zhou, G.; Liu, Z.; Xu, Y. Preparation of Supported Perovskite Catalyst to Purify Membrane Concentrate of Coal Chemical Wastewater in UV-Catalytic Wet Hydrogen Peroxide Oxidation System. Int. J. Environ. Res. Public Health 2021, 18, 4906. https://doi.org/10.3390/ijerph18094906
Zhang W, Liu Z, Chen P, Zhou G, Liu Z, Xu Y. Preparation of Supported Perovskite Catalyst to Purify Membrane Concentrate of Coal Chemical Wastewater in UV-Catalytic Wet Hydrogen Peroxide Oxidation System. International Journal of Environmental Research and Public Health. 2021; 18(9):4906. https://doi.org/10.3390/ijerph18094906
Chicago/Turabian StyleZhang, Wenwen, Zhenxue Liu, Pei Chen, Guangzhen Zhou, Zhiying Liu, and Yanhua Xu. 2021. "Preparation of Supported Perovskite Catalyst to Purify Membrane Concentrate of Coal Chemical Wastewater in UV-Catalytic Wet Hydrogen Peroxide Oxidation System" International Journal of Environmental Research and Public Health 18, no. 9: 4906. https://doi.org/10.3390/ijerph18094906
APA StyleZhang, W., Liu, Z., Chen, P., Zhou, G., Liu, Z., & Xu, Y. (2021). Preparation of Supported Perovskite Catalyst to Purify Membrane Concentrate of Coal Chemical Wastewater in UV-Catalytic Wet Hydrogen Peroxide Oxidation System. International Journal of Environmental Research and Public Health, 18(9), 4906. https://doi.org/10.3390/ijerph18094906