
A ‘Dilute and Shoot’ Liquid Chromatography-Mass Spectrometry Method for Multiclass Drug Analysis in Pre-Cut Dried Blood Spots

 ,
,  and
and
Abstract

1. Introduction
2. Materials and Methods
2.1. Chemicals
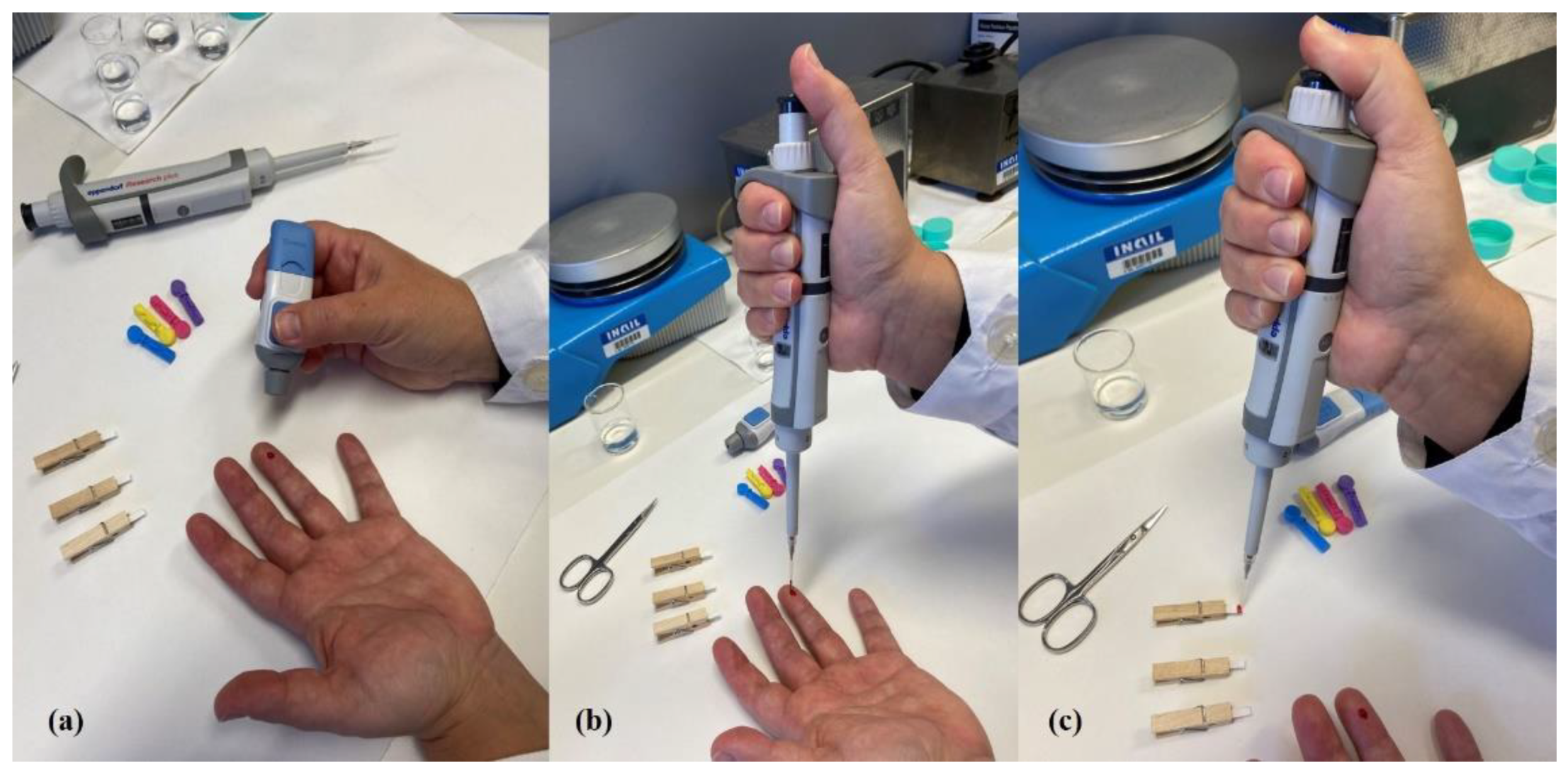
2.2. Dried Blood Spot Sampling
2.3. Dilute and Shoot Procedure
2.4. Liquid Chromatography and Mass Spectrometry
2.5. Method Validation
3. Results and Discussion
3.1. Haematocrit (HCT) Management
3.2. Pre-Cut Spots, Support Materials and Dilution
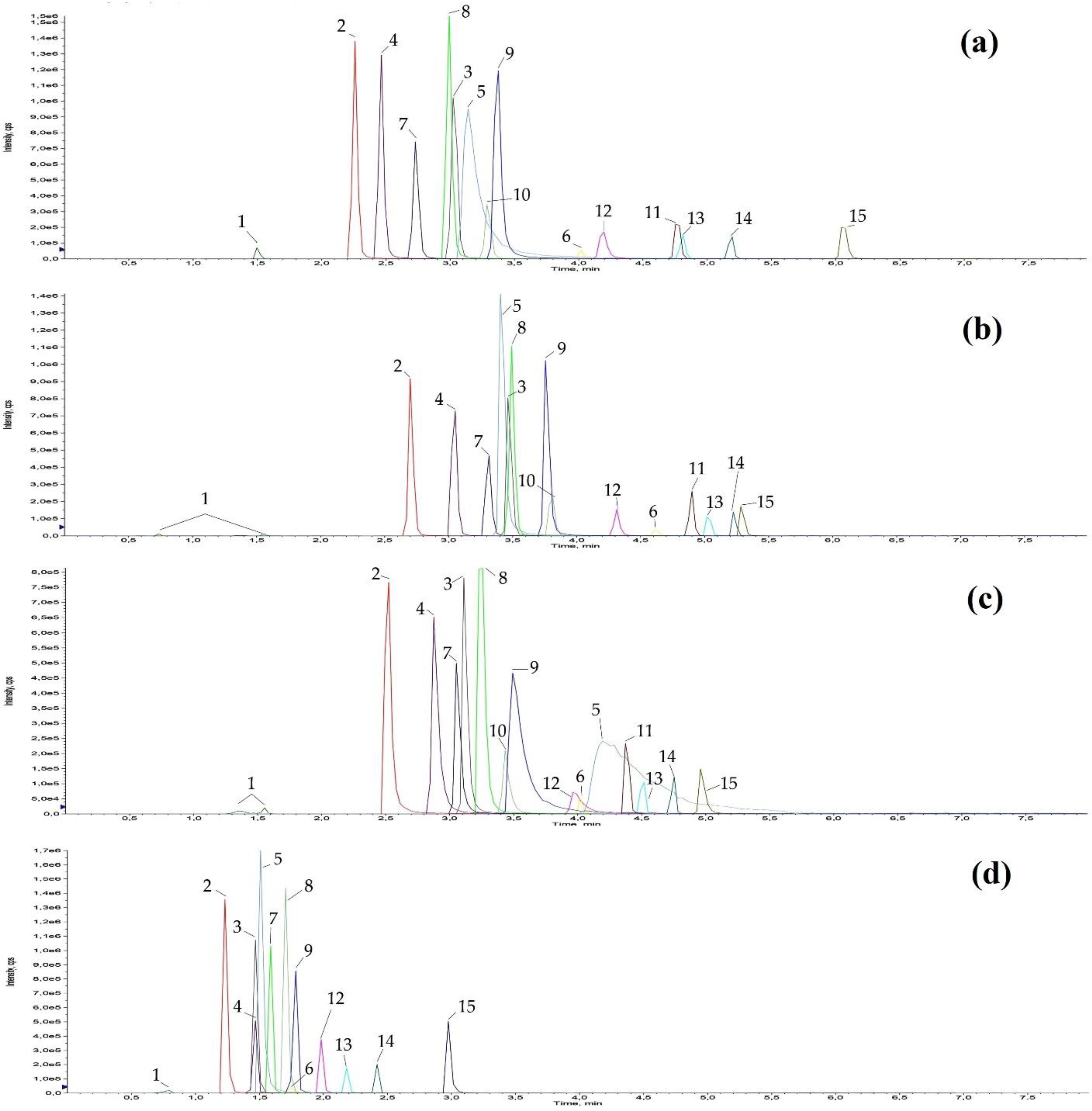
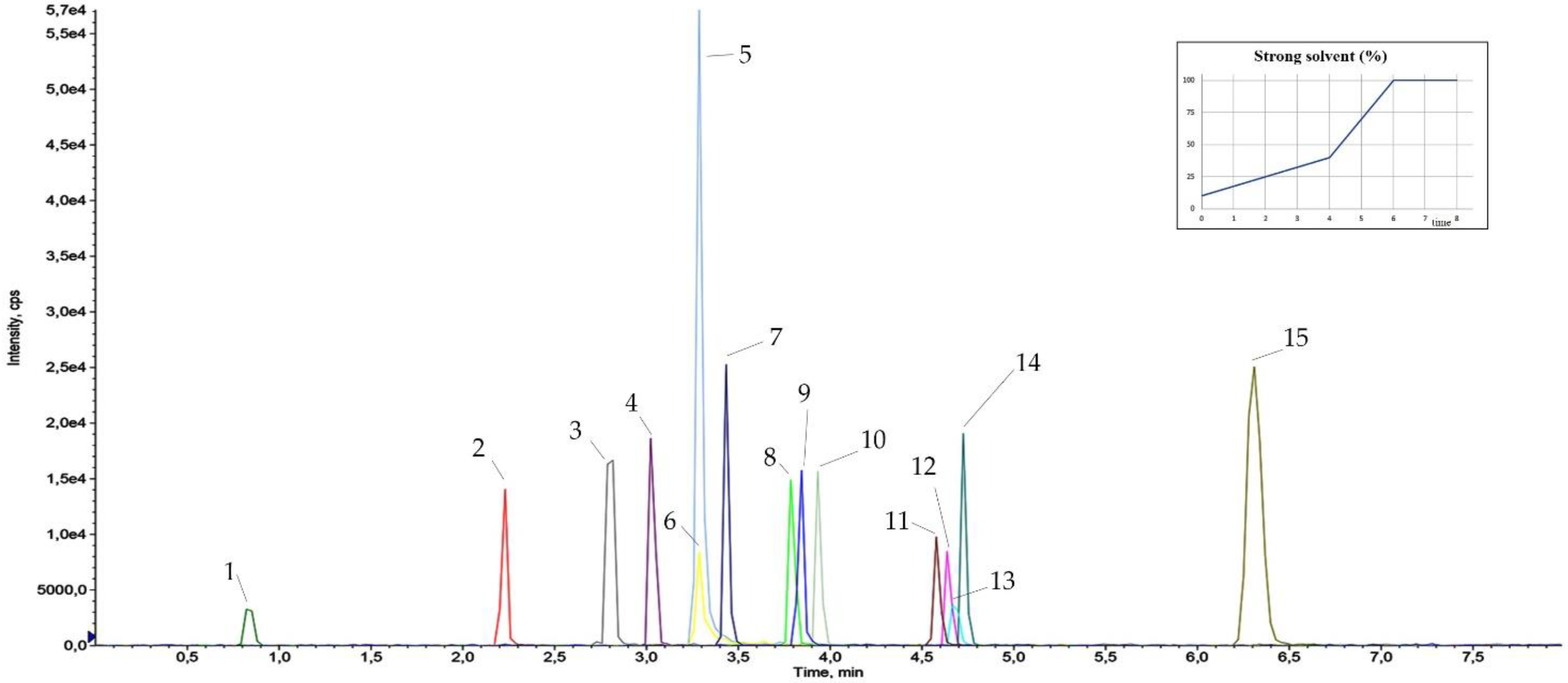
3.3. Dilute and Shoot and Chromatographic Separation
3.4. Results of Method Validation
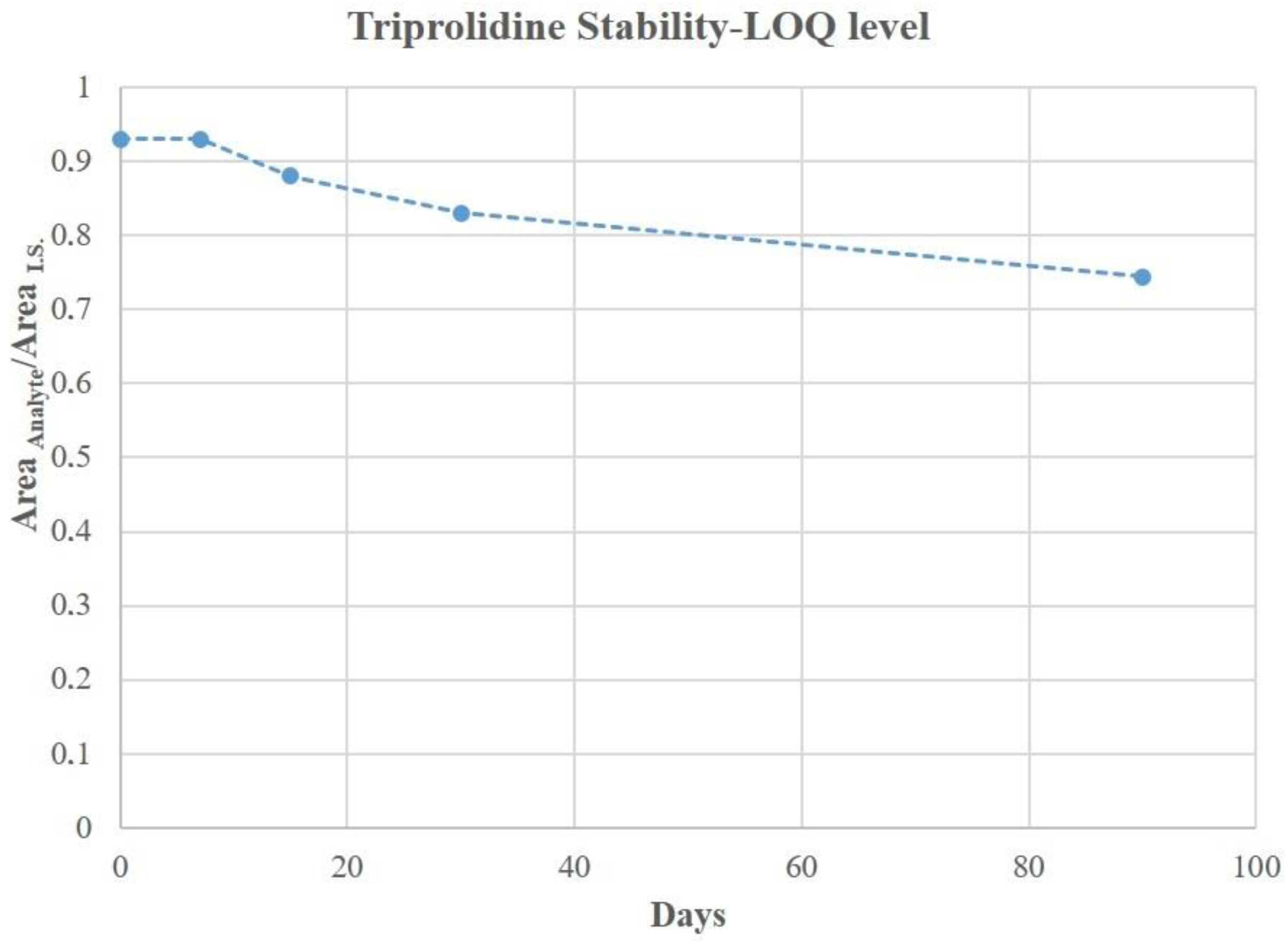
3.5. DBS Stability
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Belloni, A.; Morgan, D.; Paris, V. Pharmaceutical Expenditure and Policies: Past Trends and Future Challenges; OECD Health Working Papers 2016; OECD Publishing: Paris, France, 2016; Available online: https://www.oecd-ilibrary.org/social-issues-migration-health/pharmaceutical-expenditure-and-policies_5jm0q1f4cdq7-en (accessed on 1 January 2021).
- The Medicines Utilization Monitoring Centre. National Report on Medicines Use in Italy; Italian Medicines Agency: Rome, Italy, 2018. Available online: www.aifa.gov.it (accessed on 1 December 2020).
- Dart, R.C.; Surratt, H.L.; Cicero, T.J.; Parrino, M.W.; Geoff Severtson, S.; Bucher-Bartelson, B.; Green, J.L. Trends in Opioid Analgesic Abuse and Mortality in the United States. N. Engl. J. Med. 2015, 372, 241–248. [Google Scholar] [CrossRef]
- Bisht, M.; Bist, S.S. Ototoxicity: The Hidden Menace. Indian J. Otolaryngol. Head Neck Surg. 2009, 63, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Cianfrone, G.; Pentangelo, D.; Cianfrone, F.; Mazzei, F.; Turchetta, R.; Orlando, M.P.; Altissimi, G. Pharmacological drugs inducing ototoxicity, vestibular symptoms and tinnitus: A reasoned and updated guide. Eur. Rev. Med. Pharmacolo. 2011, 15, 601–636. [Google Scholar]
- Kolla, B.P.; Lovely, J.K.; Mansukhani, M.P.; Morgenthaler, T.I. Zolpidem is independently associated with increased risk of inpatient falls. J. Hosp. Med. 2013, 8, 1–6. [Google Scholar] [CrossRef]
- Mets, M.A.J.; Volkerts, E.R.; Olivier, B.; Verster, J.C. Effect of hypnotic drugs on body balance and standing steadiness. Sleep Med. Rev. 2010, 14, 259–267. [Google Scholar] [CrossRef]
- Hegmann, K.T.; Weiss, M.S.; Bowden, K.; Branco, F.; DuBrueler, K.; Els, C.; Mandel, S.; McKinney, D.W.; Miguel, R.; Mueller, K.L.; et al. ACOEM Practice Guidelines: Opioids and Safety-Sensitive Work. J. Occup. Environ. Med. 2014, 56, e46–e53. [Google Scholar] [CrossRef]
- Bachs, L.C.; Engeland, A.; Mørland, J.G.; Skurtveit, S. The risk of motor vehicle accidents involving drivers with prescriptions for codeine or tramadol. Clin. Pharm. 2009, 85, 596–599. [Google Scholar] [CrossRef]
- Italian Workers’ Compensation Authority—INAIL Press Room—Accidents and Occupational Diseases, the Inail Open Data for the First Eight Months of 2019. Available online: https://www.inail.it/cs/internet/comunicazione/sala-stampa/comunicati-stampa/com-stampa-open-data-agosto-2019.html (accessed on 1 December 2020).
- Italian Workers’ Compensation Authority—INAIL Database—Occupational Diseases—Reported—Total Management (Industry and Services, Agriculture, State Account). Last update 2018. Available online: https://internetws.inail.it/BDSbi/saw.dll?Dashboard&NQUser=PUBLIC2&PortalPath=/shared/BDS%202.0%20-%20Prestazioni/_portal/MP_DN_TT_CM_ICD_TEM&Page=MP_DN_TT_CM_ICD_TEM_Settore (accessed on 1 November 2020).
- Kowalski-McGraw, M.; Green-McKenzie, J.; Pandalai, S.P.; Schulte, P.A. Characterizing the interrelationships of prescription opioid and benzodiazepine drugs with worker health and workplace hazards. Occup. Environ. Med. 2017, 59, 1114–1126. [Google Scholar] [CrossRef]
- Fitzgerald, R.L.; Griffin, T.L.; Yun, Y.-M.; Godfrey, R.A.; West, R.; Pesce, A.J.; Herold, D.A. Dilute and Shoot: Analysis of drugs of abuse using selected reaction monitoring for quantification and full scan product ion spectra for identification. J. Anal. Toxicol. 2012, 36, 106–111. [Google Scholar] [CrossRef]
- Alcántara-Durán, J.; Moreno-González, D.; Beneito-Cambra, M.; García-Reyes, J.F. Dilute-and-shoot coupled to nano-flow liquid chromatography high resolution mass spectrometry for the determination of drugs of abuse and sport drugs in human urine. Talanta 2018, 182, 218–224. [Google Scholar] [CrossRef]
- De Wilde, L.; Roels, K.; Polet, M.; Van Eenoo, P.; Deventer, K. Identification and confirmation of diuretics and masking agents in urine by turbulent flow online solid-phase extraction coupled with liquid chromatography-triple quadrupole mass spectrometry for doping control. J. Chromatogr. A 2018, 1579, 31–40. [Google Scholar] [CrossRef]
- Guthrie, R.; Susi, A. A simple phenylalanine method for detecting phenylketonuria in large populations of new-born infants. Pediatrics 1963, 32, 338–343. [Google Scholar] [PubMed]
- Kvaskoff, D.; Heath, A.K.; Simila, H.A.; Ko, P.; English, D.R.; Eyles, D.W. Minimizing matrix effects for the accurate quantification of 25-Hydroxyvitamin D metabolites in dried blood spots by LC-MS/MS. Clin. Chem. 2016, 62, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Yakkundi, S.; Mulla, H.; Pandya, H.; Turner, M.A.; McElnay, J. Quantitative analysis of methyl and propyl parabens in neonatal DBS using LC–MS/MS. Bioanalysis 2016, 8, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- den Burger, J.C.G.; Wilhelm, A.J.; Chahbouni, A.C.; Vos, R.M.; Sinjewel, A.; Swart, E.L. Haematocrit corrected analysis of creatinine in dried blood spots through potassium measurement. Anal. Bioanal. Chem. 2015, 407, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Knapen, L.M.; de Beer, Y.; Brüggemann, R.J.M.; Stolk, L.M.; de Vries, F.; Tjan-Heijnen, V.C.G.; van Erp, N.P.; Croes, S. Development and validation of an analytical method using UPLC–MS/MS to quantify everolimus in dried blood spots in the oncology setting. J. Pharm. Biomed. Anal. 2018, 149, 106–113. [Google Scholar] [CrossRef]
- Gandhi, A.; Beekman, C.; Parker, R.; Fang, L.; Babiskin, A.; Matta, M.K. Novel and rapid LC–MS/MS method for quantitative analysis of methylphenidate in dried blood spots. Bioanalysis 2018, 10, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Gallay, J.; Prodhom, S.; Mercier, T.; Bardinet, C.; Spaggiari, D.; Pothin, E.; Buclin, T.; Genton, B.; Decosterd, L.A. LC–MS/MS method for the simultaneous analysis of seven antimalarials and two active metabolites in dried blood spots for applications in field trials: Analytical and clinical validation. J. Pharm. Biomed. Anal. 2018, 154, 263–277. [Google Scholar] [CrossRef]
- Tretzel, L.; Thomas, A.; Piper, T.; Hedeland, M.; Geyer, H.; Schänzer, W.; Thevis, M. Fully automated determination of nicotine and its major metabolites in whole blood by means of a DBS online-SPE LC-HR-MS/MS approach for sports drug testing. J. Pharm. Biomed. 2016, 123, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Luginbühl, M.; Schröck, A.; König, S.; Schürch, S.; Weinmann, W. Determination of fatty acid ethyl esters in dried blood spots by LC–MS/MS as markers for ethanol intake: Application in a drinking study. Anal. Bioanal. Chem. 2016, 408, 3503–3509. [Google Scholar] [CrossRef] [PubMed]
- Rosting, C.; Gjelstad, A.; Grønhaug Halvorsen, T. Expanding the knowledge on dried blood spots and LC-MS-based protein analysis: Two different sampling materials and six protein targets. Anal. Bioanal. Chem. 2017, 409, 3383–3392. [Google Scholar] [CrossRef]
- Sharma, A.; Jaiswal, S.; Shukla, M.; Lal, J. Dried blood spots: Concepts, present status, and future perspectives in bioanalysis. Drug Test. Anal. 2014, 6, 399–414. [Google Scholar] [CrossRef]
- Venzon Antunes, M.; Feiffer Charão, M.; Linden, R. Dried blood spots analysis with mass spectrometry: Potentials and pitfalls in therapeutic drug monitoring. Clin. Biochem. 2016, 49, 1035–1046. [Google Scholar] [CrossRef]
- Murphy, J.E.; Peltier, T.; Anderson, D.; Ward, E.S. A comparison of venous versus capillary measurements of drug concentration. Ther. Drug Monit. 1990, 12, 264–267. [Google Scholar] [CrossRef]
- Ritzmo, C.B.S.; Albertioni, F.; Cosic, K.R.; Söderhäll, S.; Eksborg, S. Therapeutic Drug Monitoring of Methotrexate on the Pediatric Oncology Ward: Can Blood Sampling From Central Venous Accesses Substitute for Capillary Finger Punctures? Ther. Drug Monit. 2007, 29, 447–451. [Google Scholar] [CrossRef]
- Palm, C.; Björk, O.; Björkholm, M.; Eksborg, S. Quantification of doxorubicin in plasma-a comparative study of capillary and venous blood sampling. Anti Cancer Drugs 2001, 12, 859–864. [Google Scholar] [CrossRef]
- Sadones, N.; Archer, J.R.H.; Ingels, A.-S.M.E.; Dargan, P.I.; Wood, D.M.; Wood, M.; Neels, H.; Lambert, W.E.; Stove, C.P. Do capillary dried blood spot concentrations of gamma-hydroxybutyric acid mirror those in venous blood? A comparative study. Drug Test. Anal. 2015, 7, 336–340. [Google Scholar] [CrossRef]
- Rittau, A.M.; McLachlan, A.J. Investigating paracetamol pharmacokinetics using venous and capillary blood and saliva sampling. J. Pharm. Pharmacol. 2012, 64, 705–711. [Google Scholar] [CrossRef]
- Jantos, R.; Schumacher, M.; Skopp, G. Comparison of opioid analysis in whole blood and dried blood spots. In Proceedings of the 49th Annual Meeting of The International Association of Forensic Toxicologists (TIAFT), San Francisco, CA, USA, 25–30 September 2011. [Google Scholar]
- Goggin, M.; Lundberg, R.; Janis, G. Measuring circulating levels of 13 opiate and opioid analgesics from dried blood spot samples. In Proceedings of the American Association of Pharmaceutical Scientists (AAPS), Chicago, IL, USA, 14–17 October 2012. [Google Scholar]
- Jantos, R.; Skopp, G. Comparison of drug analysis in whole blood and dried blood spots. Toxichem. Krimtech. 2011, 78, 268–275. [Google Scholar]
- Youhnovski, N.; Bergeron, A.; Furtado, M.; Garofolo, F. Pre-cut dried blood spot (PCDBS): An alternative to dried blood spot (DBS) technique to overcome hematocrit impact. Rapid Commun. Mass Spectrom. 2011, 25, 2951–2958. [Google Scholar] [CrossRef]
- Li, F.; Zulkoski, J.; Fast, D.; Michael, S. Perforated dried blood spots: A novel format for accurate microsampling. Bioanalysis 2011, 3, 2321–2333. [Google Scholar] [CrossRef]
- The European Monitoring Centre for Drugs and Drug Addiction (EMCDDA). Drug Profiles (Database). Available online: https://www.emcdda.europa.eu/publications/drug-profiles_en (accessed on 1 March 2021).
- Hiemke, C.; Baumann, P.; Bergemann, N.; Conca, A.; Dietmaier, O.; Egberts, K.; Fric, M.; Gerlach, M.; Greiner, C.; Gründer, G.; et al. AGNP Consensus guidelines for therapeutic drug monitoring in psychiatry: Update 2011. Pharmacopsychiatry 2011, 44, 195–235. [Google Scholar] [CrossRef]
- Ishizaki, T.; Oyama, Y.; Suganuma, T.; Sasaki, T.; Nakaya, H.; Shibuya, T.; Sato, T. A dose ranging study of atenolol in hypertension: Fall in blood pressure and plasma renin activity, beta-blockade and steady-state pharmacokinetics. Br. J. Clin. Pharmac. 1983, 16, 17–25. [Google Scholar] [CrossRef]
- Blyden, G.T.; Greenblatt, D.J.; Scavone, J.M.; Shader, R.I. Pharmacokinetics of diphenhydramine and a demethylated metabolite following intravenous and oral administration. J. Clin. Pharmacol. 1986, 26, 529–533. [Google Scholar] [CrossRef]
- Capiau, S.; Veenhof, H.; Koster, R.A.; Bergqvist, Y.; Boettcher, M.; Halmingh, O.; Keevil, B.G.; Koch, B.C.P.; Linden, R.; Pistos, C.; et al. Official international association for therapeutic drug monitoring and clinical toxicology guideline: Development and validation of Dried Blood Spot–based methods for therapeutic drug monitoring. Ther. Drug Monit. 2019, 41, 410–430. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Guideline on Bioanalytical Method Validation; European Medicines Agency: London, UK, 2011. [Google Scholar]
- Food and Drug Administration, US Department of Health and Human Services. Guidance for Industry, Bioanalytical Method Validation; Food and Drug Administration: Rockville, MD, USA, 2018.
- De Kesel, P.M.M.; Capiau, S.; Lambert, W.E.; Stove, C.P. Current strategies for coping with the hematocrit problem in dried blood spot analysis. Bioanalysis 2014, 6, 1871–1874. [Google Scholar] [CrossRef]
- Denniff, P.; Spooner, N. The effect of hematocrit on assay bias when using DBS samples for the quantitative bioanalysis of drugs. Bioanalysis 2010, 2, 1385–1395. [Google Scholar] [CrossRef]
- Chieh Chao, T.; Trybala, A.; Starov, V.; Das, D.B. Influence of haematocrit level on the kinetics of blood spreading on thin porous medium during dried blood spot sampling. Colloids Surf. A Phys. Eng. Asp. 2014, 451, 38–47. [Google Scholar] [CrossRef]
- Wagner, M.; Tonoli, D.; Varesio, E.; Hopfgartner, G. The use of mass spectrometry to analyze dried blood spots. Mass Spectrom. Rev. 2016, 35, 361–438. [Google Scholar] [CrossRef] [PubMed]
- Velghe, S.; Delahaye, L.; Stove, C.P. Is the hematocrit still an issue in quantitative dried blood spot analysis? J. Pharm. Biomed. Anal. 2019, 163, 188–196. [Google Scholar] [CrossRef]
- De Kesel, P.M.M.; Sadones, N.; Capiau, S.; Lambert, W.E.; Stove, C.P. Hemato-critical issues in quantitative analysis of dried blood spots: Challenges and solutions. Bioanalysis 2013, 5, 2023–2041. [Google Scholar] [CrossRef] [PubMed]
- Deventer, K.; Pozo, O.J.; Verstraete, A.G.; Van Eenoo, P. Dilute-and-shoot-liquid chromatography-mass spectrometry for urine analysis in doping control and analytical toxicology. Trends Anal. Chem. 2014, 55, 1–13. [Google Scholar] [CrossRef]
- Viette, V.; Fathi, M.; Rudaz, S.; Hochstrasser, D.; Veuthey, J.-L. Current role of liquid chromatography coupled to mass spectrometry in clinical toxicology screening methods. Clin. Chem. Lab. Med. 2011, 49, 1091–1103. [Google Scholar] [CrossRef]
- Tölgyesi, A.; Virender, K.S. Determination of acrylamide in gingerbread and other food samples by HILIC-MS/MS: A dilute-and-shoot method. J. Chrom. B 2020, 1136, 121933. [Google Scholar] [CrossRef]
- Kaplan, M.; Olgun, E.O.; Karaoglu, O. Determination of Grayanotoxins in Honey by Liquid Chromatography Tandem Mass Spectrometry Using Dilute-and-Shoot Sample Preparation Approach. J. Agric. Food Chem. 2014, 62, 5485–5491. [Google Scholar] [CrossRef] [PubMed]
- Esposito, S.; Bracacel, E.; Nibbio, M.; Speziale, R.; Orsatti, L.; Veneziano, M.; Monteagudo, E.; Bonelli, F. Use of ‘dilute-and-shoot’ liquid chromatography-high resolution mass spectrometry in preclinical research: Application to a DMPK study of perhexiline in mouse plasma. J. Pharm. Biomed. Anal. 2016, 118, 70–80. [Google Scholar] [CrossRef]
- Kwok, W.H.; Choi, T.L.S.; Kwok, K.Y.; Chan, G.H.M.; Wong, J.K.Y.; Wan, T.S.M. Doping control analysis of 46 polar drugs in horse plasma and urine using a ‘dilute-and-shoot’ ultra high performance liquid chromatography-high resolution mass spectrometry approach. J. Chrom. A 2016, 1451, 41–49. [Google Scholar] [CrossRef]
- Bukowski, M.R.; Voeller, K.; Jahns, L. Simple and sensitive dilute-and-shoot analysis of carotenoids in human plasma. J. Chrom. B 2018, 1095, 32–38. [Google Scholar] [CrossRef]
- Bell, D.S.; Shollenberger, D.; Cramer, H. Evaluation of Retention and Selectivity Using Biphenyl Stationary Phases. LCGC N. Am. 2017, 35, 360–365. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Structure | Ear Disorders | Nervous System Disorders | tr (min) | MRM Transition * (m/z) | Declustering Potential (V) | Collision Energy (V) | Internal Standard |
|---|---|---|---|---|---|---|---|---|
| Atenolol |  | Sleep disturbances, lightheadedness, lethargy, drowsiness, dizziness, vertigo | 0.83 | 267.1/116.1 | 92 | 28 | Atenolol d7 | |
| 267.1/73.9 | 33 | |||||||
| Tramadol |  | Drowsiness, dizziness, blurred vision | 2.23 | 264.0/58.1 | 43 | 43 | Tramadol d6 | |
| 264.0/246.1 | 17 | |||||||
| Venlafaxine |  | Tinnitus, hyperacusis, otitis media, labyrinthitis | Somnolence, dizziness, tremor, insomnia, blurred vision, vertigo | 2.80 | 278.1/58.0 | 53 | 43 | |
| 278.1/260.2 | 18 | |||||||
| Zolpidem |  | Vertigo, tinnitus, labyrinthitis, otitis externa | Somnolence, attention disorder, hypoesthesia, balance disorder, ataxia, dizziness, blurred vision | 3.03 | 308.0/235.2 | 82 | 49 | |
| 308.0/236.1 | 39 | |||||||
| Triprolidine |  | Dizziness, drowsiness, blurred vision | 3.28 | 279.1/208.1 | 40 | 22 | Diphenhydramine d3 | |
| 279.1/192.2 | 63 | |||||||
| Bromazepam |  | Drowsiness, ataxia, dizziness | 3.30 | 315.9/182.1 | 82 | 47 | ||
| 315.9/209.1 | 37 | |||||||
| Trazodone |  | Hypoacusis, tinnitus, vertigo | Dizziness, light-headedness, drowsiness, balance disorder, tremors | 3.43 | 372.1/176.1 | 82 | 35 | |
| 372.1/78.0 | 88 | |||||||
| Fentanyl |  | Somnolence, dizziness, confusion, lethargy, tremor, hallucination, insomnia | 3.79 | 337.2/188.1 | 48 | 33 | ||
| 337.2/105.0 | 55 | |||||||
| Diphenhydramine |  | Tinnitus, acute labyrinthitis | Sedation, somnolence, sleepiness, convulsions, tremor, unsteadiness, vertigo, drowsiness, dizziness, attention disorder | 3.84 | 256.1/167.1 | 35 | 18 | |
| 256.1/165.1 | 55 | |||||||
| Escitalopram |  | Tinnitus, otitis externa | Somnolence, insomnia, dizziness, lethargy, tremor, amnesia, ataxia | 3.93 | 325.2/262.1 | 66 | 29 | |
| 325.2/234.1 | 39 | |||||||
| Cetirizine |  | Deafness earache, tinnitus | Somnolence, dizziness, tremor, confusion, leg cramps, paralysis, syncope, vertigo | 4.58 | 389.1/166.1 | 48 | 59 | Cetirizine d8 |
| 389.1/165.1 | 90 | |||||||
| Cyproheptadine |  | Tinnitus, acute labyrinthitis | Drowsiness, sedation, coordination disorder, loss of coordination, vertigo | 4.64 | 288.0/96.1 | 94 | 36 | Cyproheptadine d3 |
| 288.0/215.1 | 72 | |||||||
| Alprazolam |  | Tinnitus | Ataxia, cognitive dysfunction, blurred vision, attention disorder, confusion, tremor, drowsiness, dizziness | 4.67 | 308.9/205.1 | 85 | 55 | |
| 308.9/274.0 | 36 | |||||||
| Delorazepam |  | Somnolence, insomnia, dizziness | 4.73 | 305.1/140.0 | 92 | 43 | ||
| 305.1/242.1 | 39 | |||||||
| Ebastine |  | Somnolence, insomnia, dizziness, weakness | 6.29 | 470.1/167.1 | 65 | 41 | Ebastine d5 | |
| 470.1/152.1 | 94 |
| Analytes | Absolute Recoveries (R %) | Intra-Day Precision (RSD %) | Inter-Day Precision (RSD %) | Trueness (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Low (n = 6) | Medium (n = 6) | High (n = 6) | Low (n = 6) | Medium (n = 6) | High (n = 6) | Low (n = 6) | Medium (n = 6) | High (n = 6) | Low (n = 6) | Medium (n = 6) | High (n = 6) | |
| Atenolol | 81 | 85 | 84 | 6.1 | 7.2 | 9.1 | 14.2 | 13.5 | 14.9 | 103 | 91 | 94 |
| Tramadol | 95 | 91 | 97 | 9.8 | 5.7 | 10.6 | 9.8 | 6.6 | 6.6 | 93 | 92 | 103 |
| Venlafaxine | 75 | 76 | 73 | 8.3 | 10.0 | 8.8 | 10.1 | 9.6 | 13.6 | 78 | 90 | 98 |
| Zolpidem | 93 | 89 | 91 | 9.1 | 9.0 | 7.9 | 14.4 | 13.0 | 13.0 | 84 | 98 | 101 |
| Triprolidine | 107 | 101 | 100 | 9.0 | 10.2 | 6.2 | 10.9 | 6.8 | 6.8 | 102 | 93 | 100 |
| Bromazepam | 76 | 71 | 77 | 9.1 | 8.6 | 7.3 | 14.4 | 14.0 | 14.2 | 86 | 105 | 102 |
| Trazodone | 93 | 91 | 94 | 4.6 | 7.6 | 9.3 | 8.5 | 6.9 | 6.9 | 103 | 96 | 103 |
| Fentanyl | 89 | 95 | 90 | 8.1 | 9.9 | 6.7 | 6.3 | 3.8 | 3.8 | 98 | 101 | 101 |
| Diphenhydramine | 91 | 91 | 87 | 6.8 | 8.5 | 10.1 | 14.7 | 13.2 | 13.2 | 103 | 88 | 98 |
| Escitalopram | 90 | 84 | 88 | 7.7 | 10.1 | 9.6 | 14.6 | 14.8 | 13.8 | 105 | 97 | 98 |
| Cetirizine | 75 | 82 | 76 | 9.5 | 9.7 | 4.3 | 7.1 | 4.3 | 5.2 | 99 | 101 | 102 |
| Cyproheptadine | 85 | 88 | 89 | 6.0 | 6.7 | 8.3 | 9.5 | 8.8 | 7.8 | 105 | 105 | 103 |
| Alprazolam | 94 | 99 | 97 | 9.7 | 5.3 | 9.3 | 7.3 | 6.7 | 6.7 | 93 | 100 | 102 |
| Delorazepam | 99 | 103 | 106 | 7.6 | 2.5 | 3.4 | 8.1 | 6.5 | 7.2 | 92 | 90 | 101 |
| Ebastine | 99 | 105 | 100 | 7.7 | 7.2 | 7.8 | 8.4 | 6.9 | 7.3 | 101 | 102 | 98 |
| Analytes | LODs (pg µL−1) | LOQs (pg µL−1) | Matrix Effect (%) |
|---|---|---|---|
| Atenolol | 0.8 | 2.4 | 14 |
| Tramadol | 0.1 | 0.3 | 13 |
| Venlafaxine | 0.7 | 2.1 | 9 |
| Zolpidem | 0.1 | 0.3 | 3 |
| Triprolidine | 0.4 | 1.2 | 13 |
| Bromazepam | 4.9 | 15 | 16 |
| Trazodone | 0.1 | 0.3 | 8 |
| Fentanyl | 0.5 | 1.5 | 20 |
| Diphenhydramine | 0.2 | 0.6 | 19 |
| Escitalopram | 0.9 | 2.7 | 23 |
| Cetirizine | 0.2 | 0.6 | 13 |
| Cyproheptadine | 0.1 | 0.3 | 29 |
| Alprazolam | 0.5 | 1.5 | 8 |
| Delorazepam | 0.6 | 1.8 | 8 |
| Ebastine | 1.4 | 4.2 | 16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mainero Rocca, L.; L’Episcopo, N.; Gordiani, A.; Vitali, M.; Staderini, A. A ‘Dilute and Shoot’ Liquid Chromatography-Mass Spectrometry Method for Multiclass Drug Analysis in Pre-Cut Dried Blood Spots. Int. J. Environ. Res. Public Health 2021, 18, 3068. https://doi.org/10.3390/ijerph18063068
Mainero Rocca L, L’Episcopo N, Gordiani A, Vitali M, Staderini A. A ‘Dilute and Shoot’ Liquid Chromatography-Mass Spectrometry Method for Multiclass Drug Analysis in Pre-Cut Dried Blood Spots. International Journal of Environmental Research and Public Health. 2021; 18(6):3068. https://doi.org/10.3390/ijerph18063068
Chicago/Turabian StyleMainero Rocca, Lucia, Nunziata L’Episcopo, Andrea Gordiani, Matteo Vitali, and Alessandro Staderini. 2021. "A ‘Dilute and Shoot’ Liquid Chromatography-Mass Spectrometry Method for Multiclass Drug Analysis in Pre-Cut Dried Blood Spots" International Journal of Environmental Research and Public Health 18, no. 6: 3068. https://doi.org/10.3390/ijerph18063068
APA StyleMainero Rocca, L., L’Episcopo, N., Gordiani, A., Vitali, M., & Staderini, A. (2021). A ‘Dilute and Shoot’ Liquid Chromatography-Mass Spectrometry Method for Multiclass Drug Analysis in Pre-Cut Dried Blood Spots. International Journal of Environmental Research and Public Health, 18(6), 3068. https://doi.org/10.3390/ijerph18063068