
Alcohol-Induced Blood-Brain Barrier Impairment: An In Vitro Study


 ,
,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatments
2.2. MTT Assay
2.3. Western Blotting
2.4. Measurement of ROS
2.5. Immunofluorescent Labelling
2.6. Statistical Analysis
3. Results
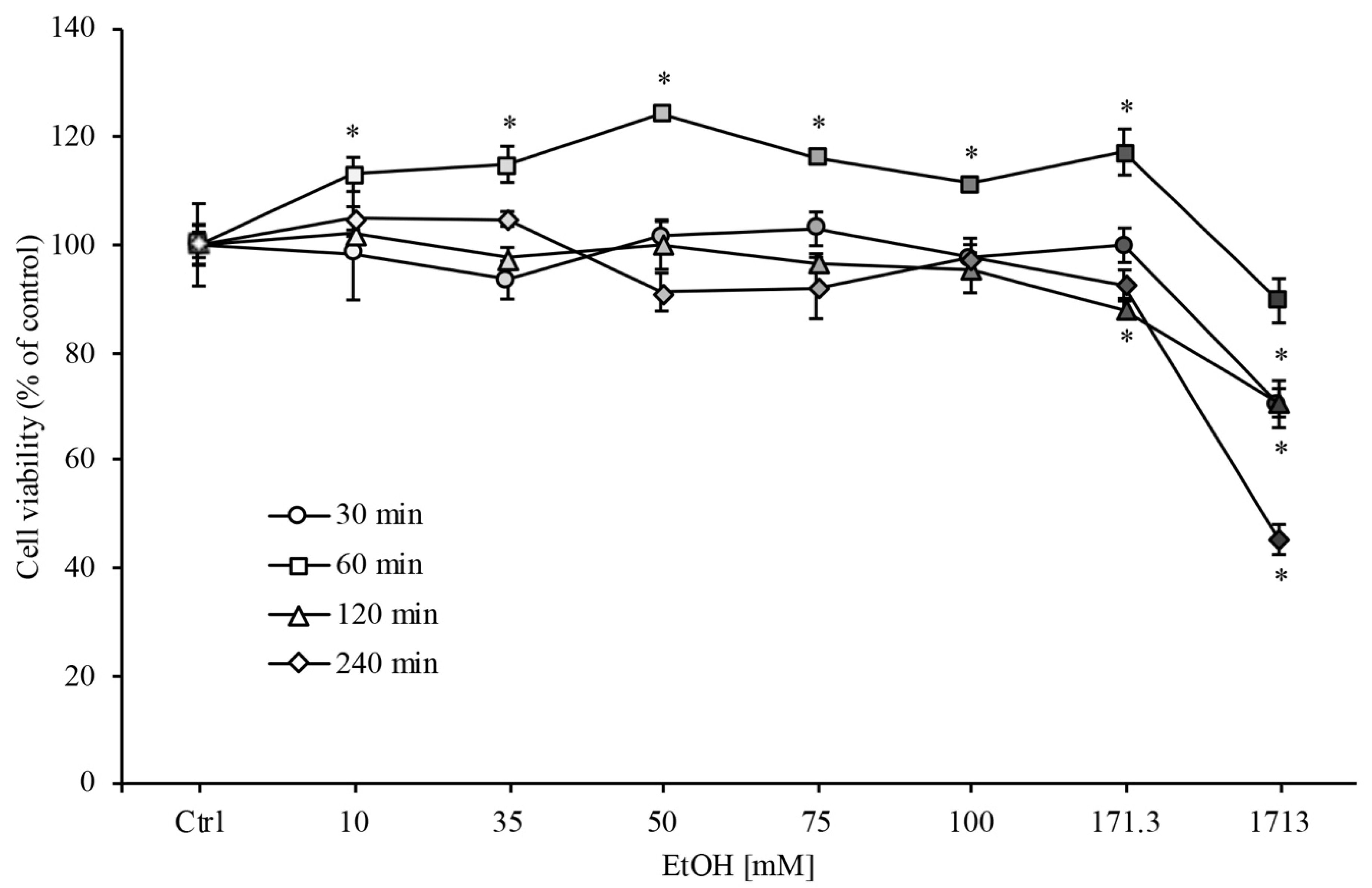
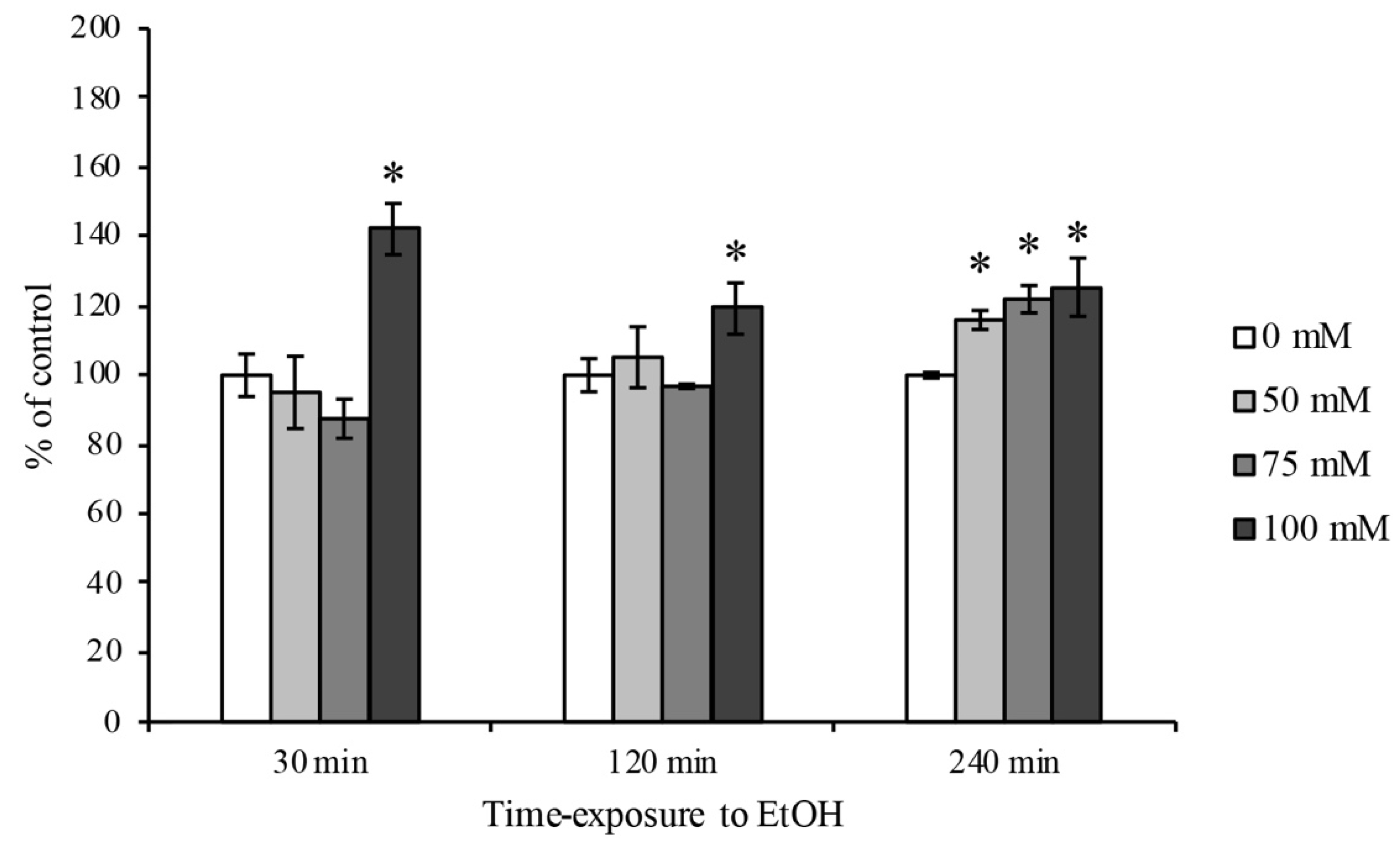
3.1. RBE4 Cell Viability
3.2. Evaluation of ROS Generation
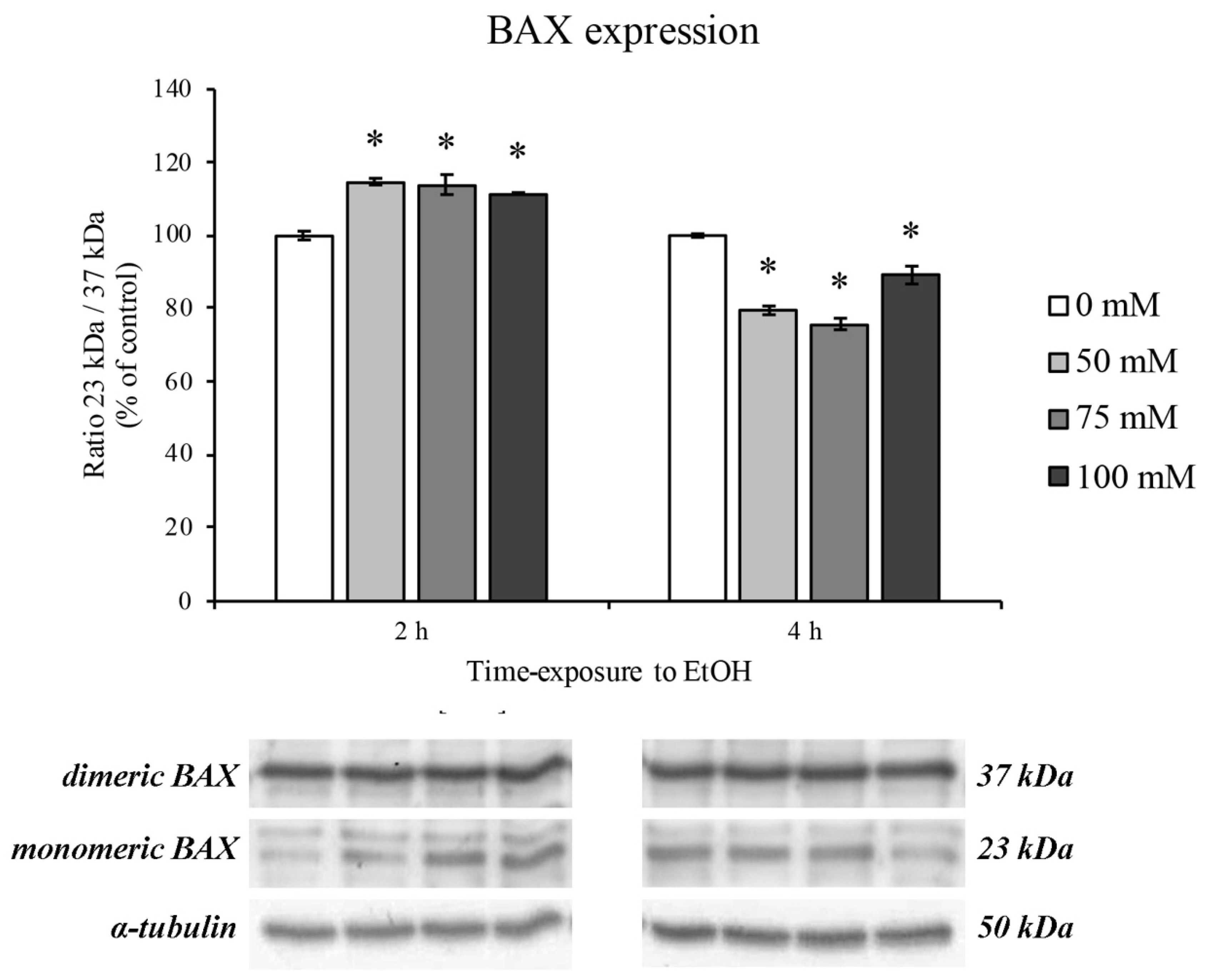
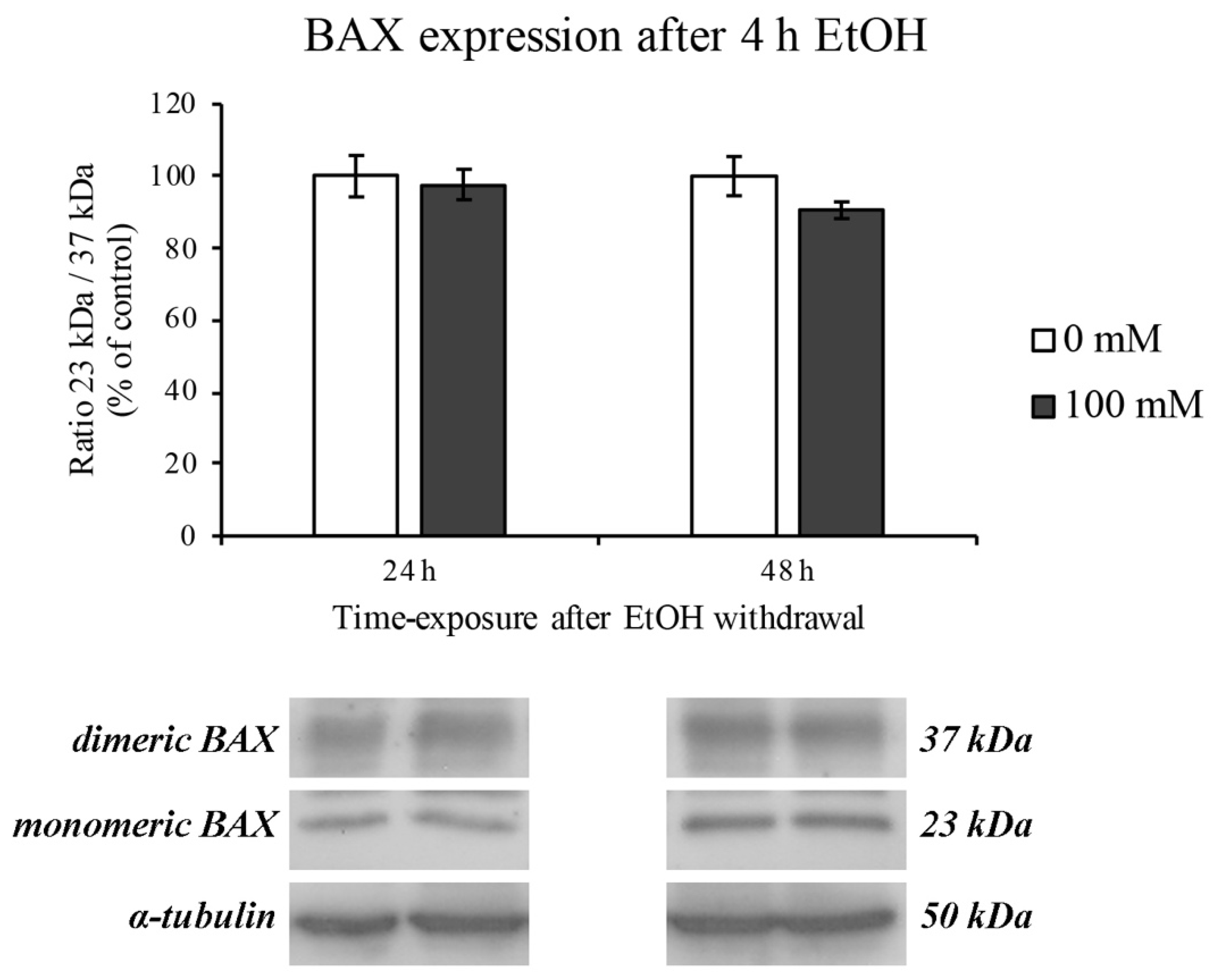
3.3. BAX Protein Expression Levels
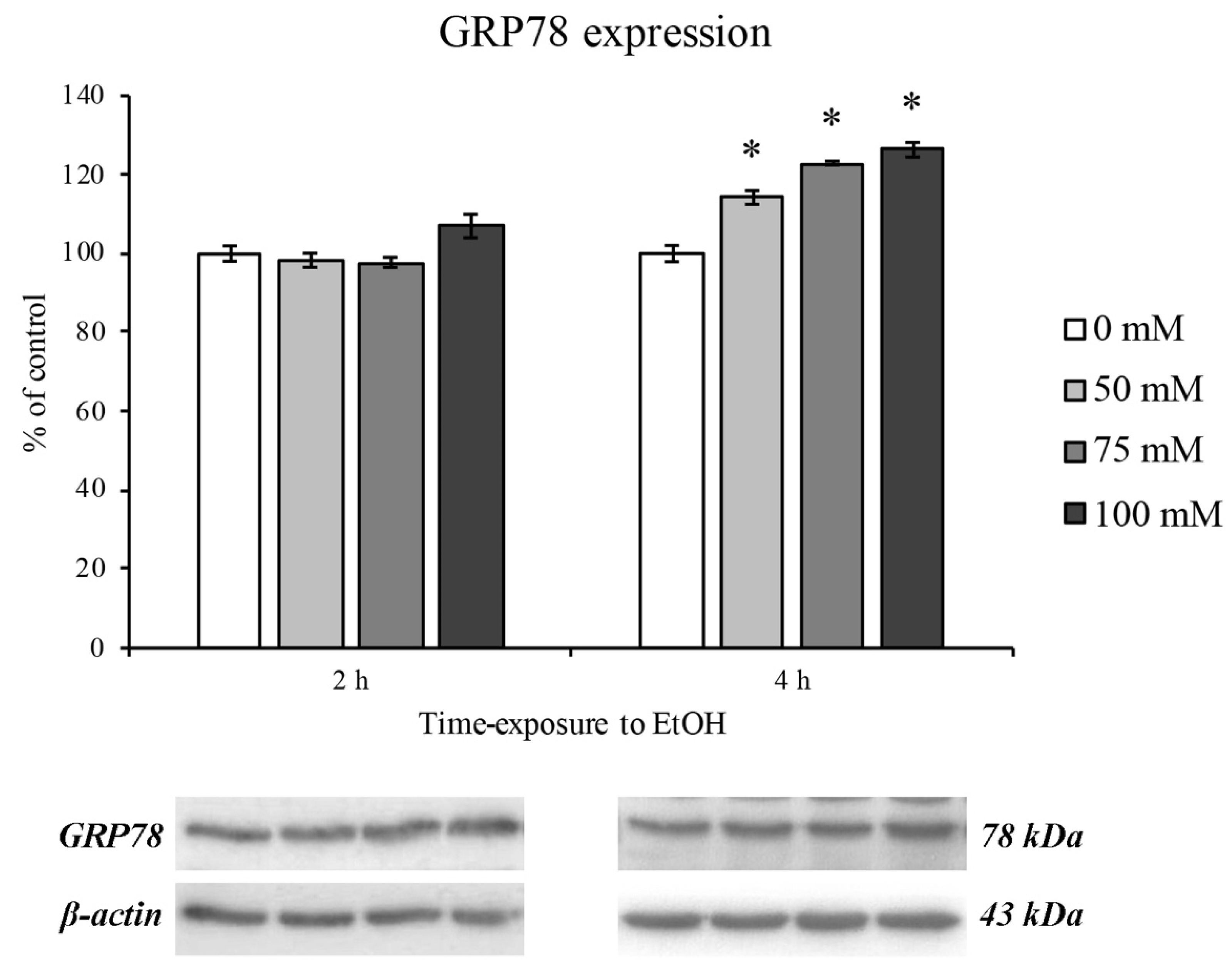
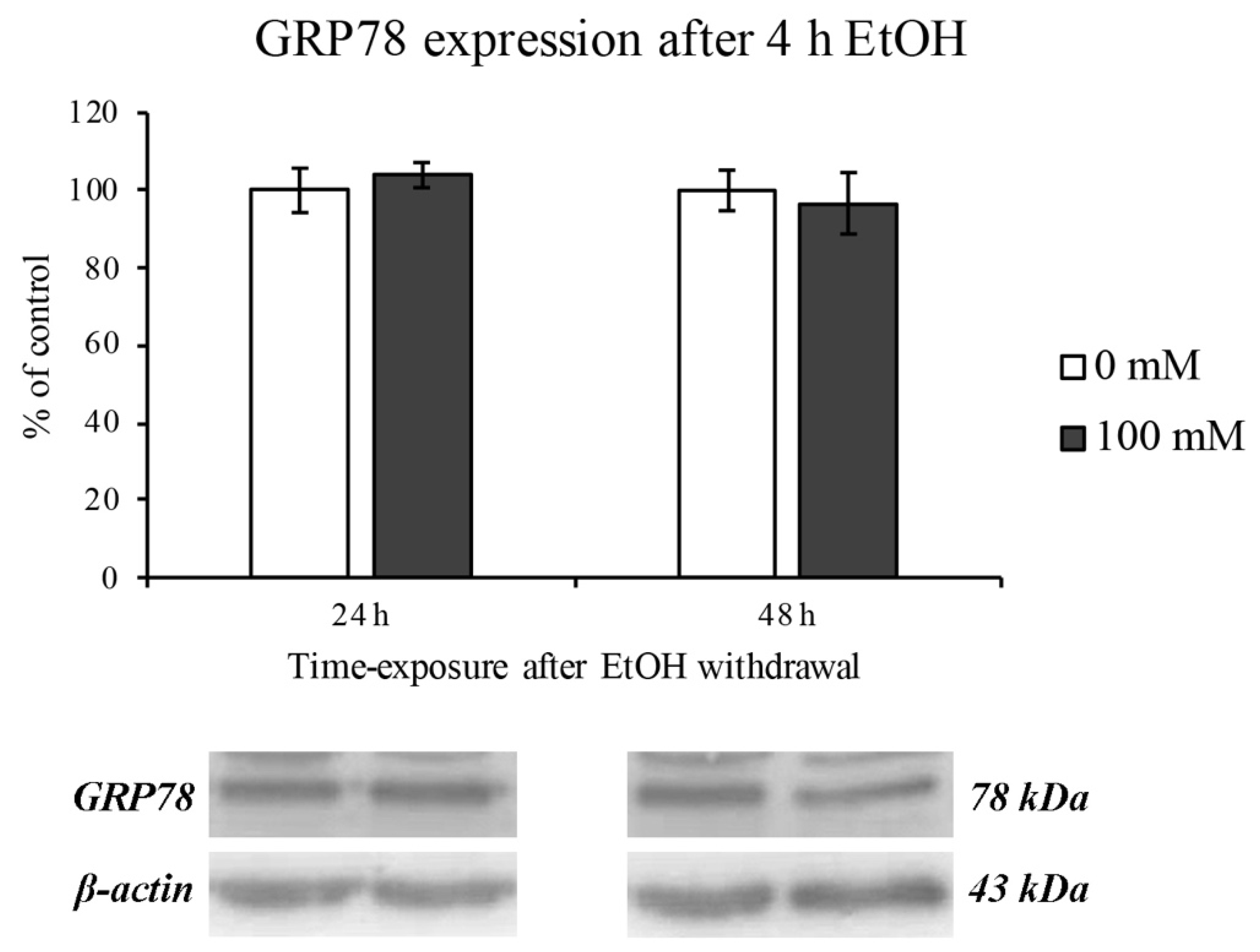
3.4. EtOH-Dependent ER Stress
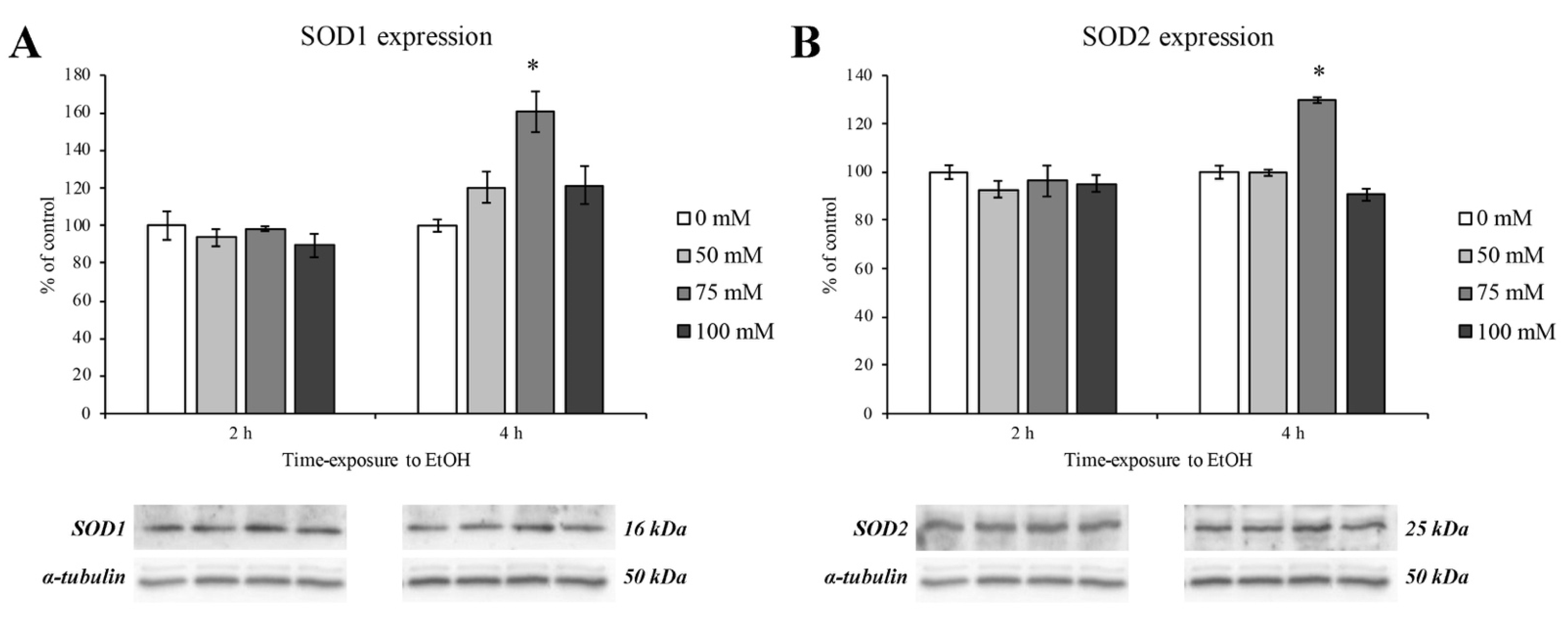
3.5. Expression of Antioxidant Enzymes
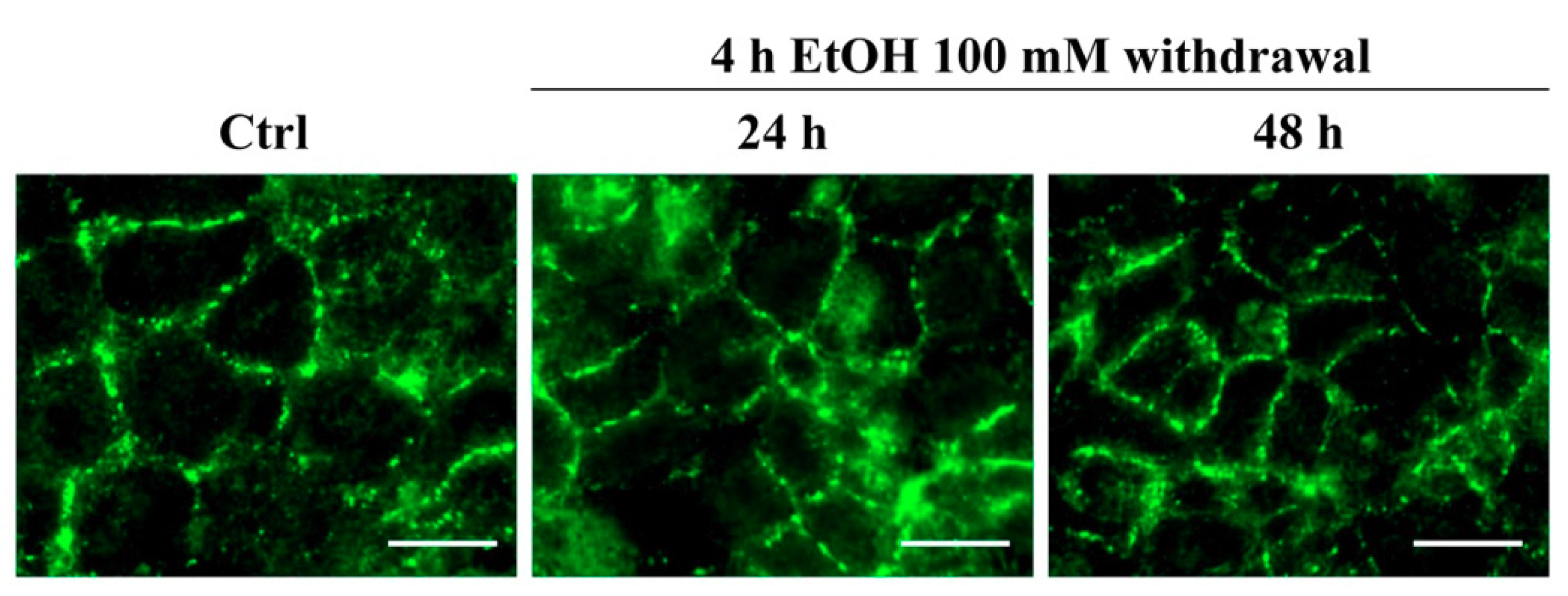
3.6. The Effect of EtOH on the Tight Junction Protein ZO-1
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- World Health Organization. Management of Substance Abuse Team. In Global Status Report on Alcohol and Health 2018; World Health Organization: Geneva, Switzerland, 2018; ISBN 978-92-4-156563-9. [Google Scholar]
- Osna, N.A.; Donohue, T.M., Jr.; Kharbanda, K.K. Alcoholic Liver Disease: Pathogenesis and Current Management. Alcohol Res. Curr. Rev. 2017, 38, 147–161. [Google Scholar]
- Larsson, S.C.; Burgess, S.; Mason, A.M.; Michaëlsson, K. Alcohol Consumption and Cardiovascular Disease: A Mendelian Randomization Study. Circ. Genom. Precis. Med. 2020, 13. [Google Scholar] [CrossRef]
- Bagnardi, V.; Rota, M.; Botteri, E.; Tramacere, I.; Islami, F.; Fedirko, V.; Scotti, L.; Jenab, M.; Turati, F.; Pasquali, E.; et al. Alcohol Consumption and Site-Specific Cancer Risk: A Comprehensive Dose–Response Meta-Analysis. Br. J. Cancer 2015, 112, 580–593. [Google Scholar] [CrossRef]
- Trevejo-Nunez, G.; de Wit, M. Alcohol Use As a Risk Factor in Infections and Healing. Alcohol Res. Curr. Rev. 2015, 37, 177–184. [Google Scholar]
- Bishehsari, F.; Desai, V.; Voigt, R.M.; Forsyth, C.B.; Keshavarzian, A. Alcohol and Gut-Derived Inflammation. Alcohol Res. Curr. Rev. 2017, 38, 163–171. [Google Scholar]
- Hanck, C.; Whitcomb, D.C. Alcoholic Pancreatitis. Gastroenterol. Clin. N. Am. 2004, 33, 751–765. [Google Scholar] [CrossRef] [PubMed]
- Feldman, D.E.; McPherson, K.L.; Biesecker, C.L.; Wiers, C.E.; Manza, P.; Volkow, N.D.; Wang, G.-J. Neuroimaging of Inflammation in Alcohol Use Disorder: A Review. Sci. China Inf. Sci. 2020, 63, 170102. [Google Scholar] [CrossRef]
- Li, Q.; Liu, D.; Pan, F.; Ho, C.S.H.; Ho, R.C.M. Ethanol Exposure Induces Microglia Activation and Neuroinflammation through TLR4 Activation and SENP6 Modulation in the Adolescent Rat Hippocampus. Neural Plast. 2019, 2019, 1–12. [Google Scholar] [CrossRef]
- Erickson, E.K.; Grantham, E.K.; Warden, A.S.; Harris, R.A. Neuroimmune Signaling in Alcohol Use Disorder. Pharmacol. Biochem. Behav. 2019, 177, 34–60. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, A.K.; Löfving, S.; Callaghan, R.C.; Allebeck, P. Alcohol Use Disorders and Risk of Parkinson’s Disease: Findings from a Swedish National Cohort Study 1972–2008. BMC Neurol. 2013, 13, 190. [Google Scholar] [CrossRef] [PubMed]
- Christoffersen, S. Death from Seizures Induced by Chronic Alcohol Abuse—Does It Exist? Seizure 2007, 16, 379–383. [Google Scholar] [CrossRef]
- Crews, F.T.; Collins, M.A.; Dlugos, C.; Littleton, J.; Wilkins, L.; Neafsey, E.J.; Pentney, R.; Snell, L.D.; Tabakoff, B.; Zou, J.; et al. Alcohol-Induced Neurodegeneration: When, Where and Why? Alcohol. Clin. Exp. Res. 2004, 28, 350–364. [Google Scholar] [CrossRef] [PubMed]
- Diamond, I.; Messing, R.O. Neurologic Effects of Alcoholism. West. J. Med. 1994, 161, 279–287. [Google Scholar]
- Feigin, V.L.; Nichols, E.; Alam, T.; Bannick, M.S.; Beghi, E.; Blake, N.; Culpepper, W.J.; Dorsey, E.R.; Elbaz, A.; Ellenbogen, R.G.; et al. Global, Regional, and National Burden of Neurological Disorders, 1990–2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar] [CrossRef]
- O’Callaghan, J.P.; Sriram, K. Glial Fibrillary Acidic Protein and Related Glial Proteins as Biomarkers of Neurotoxicity. Expert Opin. Drug Saf. 2005, 4, 433–442. [Google Scholar] [CrossRef]
- Leyns, C.E.G.; Holtzman, D.M. Glial Contributions to Neurodegeneration in Tauopathies. Mol. Neurodegener. 2017, 12, 50. [Google Scholar] [CrossRef]
- Sidoryk-Wegrzynowicz, M.; Strużyńska, L. Dysfunctional Glia: Contributors to Neurodegenerative Disorders. Neural Regen. Res. 2021, 16, 218. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood–Brain Barrier Breakdown in Alzheimer Disease and Other Neurodegenerative Disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Carvey, P.M.; Hendey, B.; Monahan, A.J. The Blood-Brain Barrier in Neurodegenerative Disease: A Rhetorical Perspective. J. Neurochem. 2009, 111, 291–314. [Google Scholar] [CrossRef] [PubMed]
- Noe, C.R.; Noe-Letschnig, M.; Handschuh, P.; Noe, C.A.; Lanzenberger, R. Dysfunction of the Blood-Brain Barrier—A Key Step in Neurodegeneration and Dementia. Front. Aging Neurosci. 2020, 12, 185. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Prat, A. The Blood–Brain Barrier. Cold Spring Harb Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Zhou, L.; Agalliu, D.; Cahoy, J.D.; Kaushal, A.; Barres, B.A. The Mouse Blood-Brain Barrier Transcriptome: A New Resource for Understanding the Development and Function of Brain Endothelial Cells. PLoS ONE 2010, 5, e13741. [Google Scholar] [CrossRef]
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes Regulate the Blood–Brain Barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, J.I.; Katayama, T.; Prat, A. Glial Influence on the Blood Brain Barrier: Glial Influence on the Blood Brain Barrier. Glia 2013, 61, 1939–1958. [Google Scholar] [CrossRef] [PubMed]
- Greene, C.; Campbell, M. Tight Junction Modulation of the Blood Brain Barrier: CNS Delivery of Small Molecules. Tissue Barriers 2016, 4, e1138017. [Google Scholar] [CrossRef]
- Stamatovic, S.; Keep, R.; Andjelkovic, A. Brain Endothelial Cell-Cell Junctions: How to “Open” the Blood Brain Barrier. Curr. Neuropharmacol. 2008, 6, 179–192. [Google Scholar] [CrossRef]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-Brain Barrier Breakdown in the Aging Human Hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef]
- Weiss, N.; Miller, F.; Cazaubon, S.; Couraud, P.-O. The Blood-Brain Barrier in Brain Homeostasis and Neurological Diseases. Biochim. Biophys. Acta (BBA) Biomembr. 2009, 1788, 842–857. [Google Scholar] [CrossRef]
- Zlokovic, B.V. The Blood-Brain Barrier in Health and Chronic Neurodegenerative Disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef]
- Manzo-Avalos, S.; Saavedra-Molina, A. Cellular and Mitochondrial Effects of Alcohol Consumption. Int. J. Environ. Res. Public Health 2010, 7, 4281–4304. [Google Scholar] [CrossRef]
- Dguzeh, U.; Haddad, N.; Smith, K.; Johnson, J.; Doye, A.; Gwathmey, J.; Haddad, G. Alcoholism: A Multi-Systemic Cellular Insult to Organs. Int. J. Environ. Res. Public Health 2018, 15, 1083. [Google Scholar] [CrossRef]
- Lochhead, J.J.; McCaffrey, G.; Quigley, C.E.; Finch, J.; DeMarco, K.M.; Nametz, N.; Davis, T.P. Oxidative Stress Increases Blood–Brain Barrier Permeability and Induces Alterations in Occludin during Hypoxia–Reoxygenation. J. Cereb. Blood Flow Metab. 2010, 30, 1625–1636. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Li, Y.; Zhang, H.; An, N.; Wei, Y.; Wang, L.; Tian, C.; Yuan, M.; Sun, Y.; Xing, Y.; et al. Oxidative Stress-Mediated Blood-Brain Barrier (BBB) Disruption in Neurological Diseases. Oxid. Med. Cell. Longev. 2020, 2020, 1–27. [Google Scholar] [CrossRef]
- Branca, J.J.V.; Maresca, M.; Morucci, G.; Mello, T.; Becatti, M.; Pazzagli, L.; Colzi, I.; Gonnelli, C.; Carrino, D.; Paternostro, F.; et al. Effects of Cadmium on ZO-1 Tight Junction Integrity of the Blood Brain Barrier. Int. J. Mol. Sci. 2019, 20, 6010. [Google Scholar] [CrossRef] [PubMed]
- Branca, J.J.V.; Maresca, M.; Morucci, G.; Becatti, M.; Paternostro, F.; Gulisano, M.; Ghelardini, C.; Salvemini, D.; Di Cesare Mannelli, L.; Pacini, A. Oxaliplatin-Induced Blood Brain Barrier Loosening: A New Point of View on Chemotherapy-Induced Neurotoxicity. Oncotarget 2018, 9, 23426–23438. [Google Scholar] [CrossRef] [PubMed]
- Cherpitel, C.J.; Bond, J.; Ye, Y.; Borges, G.; MacDonald, S.; Stockwell, T.; Giesbrecht, N.; Cremonte, M. Alcohol-Related Injury in the ER: A Cross-National Meta-Analysis from the Emergency Room Collaborative Alcohol Analysis Project (ERCAAP). J. Stud. Alcohol. 2003, 64, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Deutch, S.R.; Christian, C.; Hoyer, S.; Christensen, E.F.; Dragsholt, C.; Hansen, A.C.; Kristensen, I.B.; Hougaard, K. Drug and Alcohol Use among Patients Admitted to a Danish Trauma Centre: A Prospective Study from a Regional Trauma Centre in Scandinavia. Eur. J. Emerg. Med. 2004, 11, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Haorah, J.; Heilman, D.; Knipe, B.; Chrastil, J.; Leibhart, J.; Ghorpade, A.; Miller, D.W.; Persidsky, Y. Ethanol-Induced Activation of Myosin Light Chain Kinase Leads to Dysfunction of Tight Junctions and Blood-Brain Barrier Compromise. Alcohol. Clin. Exp. Res. 2005, 29, 999–1009. [Google Scholar] [CrossRef]
- Saeed, R.W.; Varma, S.; Peng, T.; Tracey, K.J.; Sherry, B.; Metz, C.N. Ethanol Blocks Leukocyte Recruitment and Endothelial Cell Activation In Vivo and In Vitro. J. Immunol. 2004, 173, 6376–6383. [Google Scholar] [CrossRef]
- Garner, T.P.; Reyna, D.E.; Priyadarshi, A.; Chen, H.-C.; Li, S.; Wu, Y.; Ganesan, Y.T.; Malashkevich, V.N.; Cheng, E.H.; Gavathiotis, E. An Autoinhibited Dimeric Form of BAX Regulates the BAX Activation Pathway. Mol. Cell 2016, 63, 485–497. [Google Scholar] [CrossRef]
- Zeeshan, H.; Lee, G.; Kim, H.-R.; Chae, H.-J. Endoplasmic Reticulum Stress and Associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef]
- Wilson, D.F.; Matschinsky, F.M. Ethanol Metabolism: The Good, the Bad, and the Ugly. Med. Hypotheses 2020, 140, 109638. [Google Scholar] [CrossRef]
- Haorah, J.; Knipe, B.; Persidsky, Y. Stabilization of Superoxide Dismutase by AcetylLcarnitine in Human Brain Endothelium during Alcohol Exposure: Novel Protective Approach. Free Radic. Biol. Med. 2011, 51, 1601–1609. [Google Scholar] [CrossRef] [PubMed]
- Huber, J.D.; Egleton, R.D.; Davis, T.P. Molecular Physiology and Pathophysiology of Tight Junctions in the Blood–Brain Barrier. Trends Neurosci. 2001, 24, 719–725. [Google Scholar] [CrossRef]
- Vorbrodt, A.W.; Dobrogowska, D.H. Molecular Anatomy of Intercellular Junctions in Brain Endothelial and Epithelial Barriers: Electron Microscopist’s View. Brain Res. Rev. 2003, 42, 221–242. [Google Scholar] [CrossRef]
- Papalimperi, A.; Athanaselis, S.; Mina, A.; Papoutsis, I.; Spiliopoulou, C.; Papadodima, S. Incidence of Fatalities of Road Traffic Accidents Associated with Alcohol Consumption and the Use of Psychoactive Drugs: A 7-Year Survey (2011–2017). Exp. Ther. Med. 2019, 18, 2299–2306. [Google Scholar] [CrossRef]
- Conner, K.R.; Bagge, C.L.; Goldston, D.B.; Ilgen, M.A. Alcohol and Suicidal Behavior. Am. J. Prev. Med. 2014, 47, S204–S208. [Google Scholar] [CrossRef]
- Pompili, M.; Serafini, G.; Innamorati, M.; Dominici, G.; Ferracuti, S.; Kotzalidis, G.D.; Serra, G.; Girardi, P.; Janiri, L.; Tatarelli, R.; et al. Suicidal Behavior and Alcohol Abuse. Int. J. Environ. Res. Public Health 2010, 7, 1392–1431. [Google Scholar] [CrossRef]
- Sprunger, J.G.; Eckhardt, C.I.; Parrott, D.J. Anger, Problematic Alcohol Use, and Intimate Partner Violence Victimisation and Perpetration: Anger and Alcohol Use in Intimate Partner Violence. Crim. Behav. Ment. Health 2015, 25, 273–286. [Google Scholar] [CrossRef]
- Peng, B.; Yang, Q.; Joshi, R.B.; Liu, Y.; Akbar, M.; Song, B.-J.; Zhou, S.; Wang, X. Role of Alcohol Drinking in Alzheimer’s Disease, Parkinson’s Disease, and Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2020, 21, 2316. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Araiz, A.; Porcu, F.; Pérez-Hernández, M.; García-Gutiérrez, M.S.; Aracil-Fernández, M.A.; Gutierrez-López, M.D.; Guerri, C.; Manzanares, J.; O’Shea, E.; Colado, M.I. Disruption of Blood-Brain Barrier Integrity in Postmortem Alcoholic Brain: Preclinical Evidence of TLR4 Involvement from a Binge-like Drinking Model: TLR4 Involvement from a Binge-like Drinking Model. Addict. Biol. 2017, 22, 1103–1116. [Google Scholar] [CrossRef]
- Pan, W.; Barron, M.; Hsuchou, H.; Tu, H.; Kastin, A.J. Increased Leptin Permeation across the Blood–Brain Barrier after Chronic Alcohol Ingestion. Neuropsychopharmacology 2008, 33, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Haorah, J.; Knipe, B.; Leibhart, J.; Ghorpade, A.; Persidsky, Y. Alcohol-induced Oxidative Stress in Brain Endothelial Cells Causes Blood-brain Barrier Dysfunction. J. Leukoc. Biol. 2005, 78, 1223–1232. [Google Scholar] [CrossRef]
- Yu, H.; Wang, C.; Wang, X.; Wang, H.; Zhang, C.; You, J.; Wang, P.; Feng, C.; Xu, G.; Zhao, R.; et al. Long-term Exposure to Ethanol Downregulates Tight Junction Proteins through the Protein Kinase Cα Signaling Pathway in Human Cerebral Microvascular Endothelial Cells. Exp. Ther. Med. 2017, 14, 4789–4796. [Google Scholar] [CrossRef] [PubMed]
- Harrison, N.L.; Skelly, M.J.; Grosserode, E.K.; Lowes, D.C.; Zeric, T.; Phister, S.; Salling, M.C. Effects of Acute Alcohol on Excitability in the CNS. Neuropharmacology 2017, 122, 36–45. [Google Scholar] [CrossRef]
- Mureşan, C.; Eremia, I. Ethanol Stimulates the Formation of Free Oxygen Radicals in the Brain of Newborn Rats. Rom. J. Morphol. Embryol. 1997, 43, 113–117. [Google Scholar]
- Branca, J.J.V.; Fiorillo, C.; Carrino, D.; Paternostro, F.; Taddei, N.; Gulisano, M.; Pacini, A.; Becatti, M. Cadmium-Induced Oxidative Stress: Focus on the Central Nervous System. Antioxidants 2020, 9, 492. [Google Scholar] [CrossRef] [PubMed]
- Pelicano, H.; Lu, W.; Zhou, Y.; Zhang, W.; Chen, Z.; Hu, Y.; Huang, P. Mitochondrial Dysfunction and Reactive Oxygen Species Imbalance Promote Breast Cancer Cell Motility through a CXCL14-Mediated Mechanism. Cancer Res. 2009, 69, 2375–2383. [Google Scholar] [CrossRef]
- Kirkland, R.A.; Windelborn, J.A.; Kasprzak, J.M.; Franklin, J.L. A Bax-Induced Pro-Oxidant State Is Critical for Cytochrome c Release during Programmed Neuronal Death. J. Neurosci. 2002, 22, 6480–6490. [Google Scholar] [CrossRef]
- Celardo, I.; Costa, A.C.; Lehmann, S.; Jones, C.; Wood, N.; Mencacci, N.E.; Mallucci, G.R.; Loh, S.H.Y.; Martins, L.M. Mitofusin-Mediated ER Stress Triggers Neurodegeneration in Pink1/Parkin Models of Parkinson’s Disease. Cell Death Dis. 2016, 7, e2271. [Google Scholar] [CrossRef]
- Li, J.; Zhang, D.; Brundel, B.J.J.M.; Wiersma, M. Imbalance of ER and Mitochondria Interactions: Prelude to Cardiac Ageing and Disease? Cells 2019, 8, 1617. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S. The ER Chaperone and Signaling Regulator GRP78/BiP as a Monitor of Endoplasmic Reticulum Stress. Methods 2005, 35, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Oslowski, C.M. (Ed.) Stress Responses; Methods in Molecular Biology; Springer: New York, NY, USA, 2015; Volume 1292, ISBN 978-1-4939-2521-6. [Google Scholar]
- Murphy, M.P. Mitochondrial Dysfunction Indirectly Elevates ROS Production by the Endoplasmic Reticulum. Cell Metab. 2013, 18, 145–146. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dilution | Manifacturer | Host | |
|---|---|---|---|
| GRP78 | 1:500 | ThermoFisher Sientific, Milan, Italy | rabbit |
| ZO-1 | 1:500 | ThermoFisher Sientific, Milan, Italy | rabbit |
| SOD1 SOD2 | 1:5000 | GeneTex, Prodotti Gianni, Milan, Italy | rabbit |
| BAX | 1:200 | Santa Cruz Biotechnology, Santa Cruz, CA, USA | rabbit |
| β-actin | 1:10,000 | Santa Cruz Biotechnology, Santa Cruz, CA, USA | mouse |
| α-tubulin | 1:10,000 | Santa Cruz Biotechnology, Santa Cruz, CA, USA | mouse |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carrino, D.; Branca, J.J.V.; Becatti, M.; Paternostro, F.; Morucci, G.; Gulisano, M.; Di Cesare Mannelli, L.; Pacini, A. Alcohol-Induced Blood-Brain Barrier Impairment: An In Vitro Study. Int. J. Environ. Res. Public Health 2021, 18, 2683. https://doi.org/10.3390/ijerph18052683
Carrino D, Branca JJV, Becatti M, Paternostro F, Morucci G, Gulisano M, Di Cesare Mannelli L, Pacini A. Alcohol-Induced Blood-Brain Barrier Impairment: An In Vitro Study. International Journal of Environmental Research and Public Health. 2021; 18(5):2683. https://doi.org/10.3390/ijerph18052683
Chicago/Turabian StyleCarrino, Donatello, Jacopo Junio Valerio Branca, Matteo Becatti, Ferdinando Paternostro, Gabriele Morucci, Massimo Gulisano, Lorenzo Di Cesare Mannelli, and Alessandra Pacini. 2021. "Alcohol-Induced Blood-Brain Barrier Impairment: An In Vitro Study" International Journal of Environmental Research and Public Health 18, no. 5: 2683. https://doi.org/10.3390/ijerph18052683
APA StyleCarrino, D., Branca, J. J. V., Becatti, M., Paternostro, F., Morucci, G., Gulisano, M., Di Cesare Mannelli, L., & Pacini, A. (2021). Alcohol-Induced Blood-Brain Barrier Impairment: An In Vitro Study. International Journal of Environmental Research and Public Health, 18(5), 2683. https://doi.org/10.3390/ijerph18052683