
Sex Steroid Regulation of Oxidative Stress in Bone Cells: An In Vitro Study

 ,
,  ,
, 
 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Viability Assay
2.3. Scratch Wound Healing Assay
2.4. Transcriptional Activity Analysis
2.5. Trascriptomic Profile Analysis
2.6. Western Blotting Analysis
2.7. Total HDACs I/II Enzymatic Activity
2.8. H4 Global Acetylation Levels
2.9. Statistical Analysis
3. Results
3.1. Effects of Steroids on Counteracting t-BHP-Induced Oxidative Stress in Pre-Osteoblast MC3T3-E1 Cells
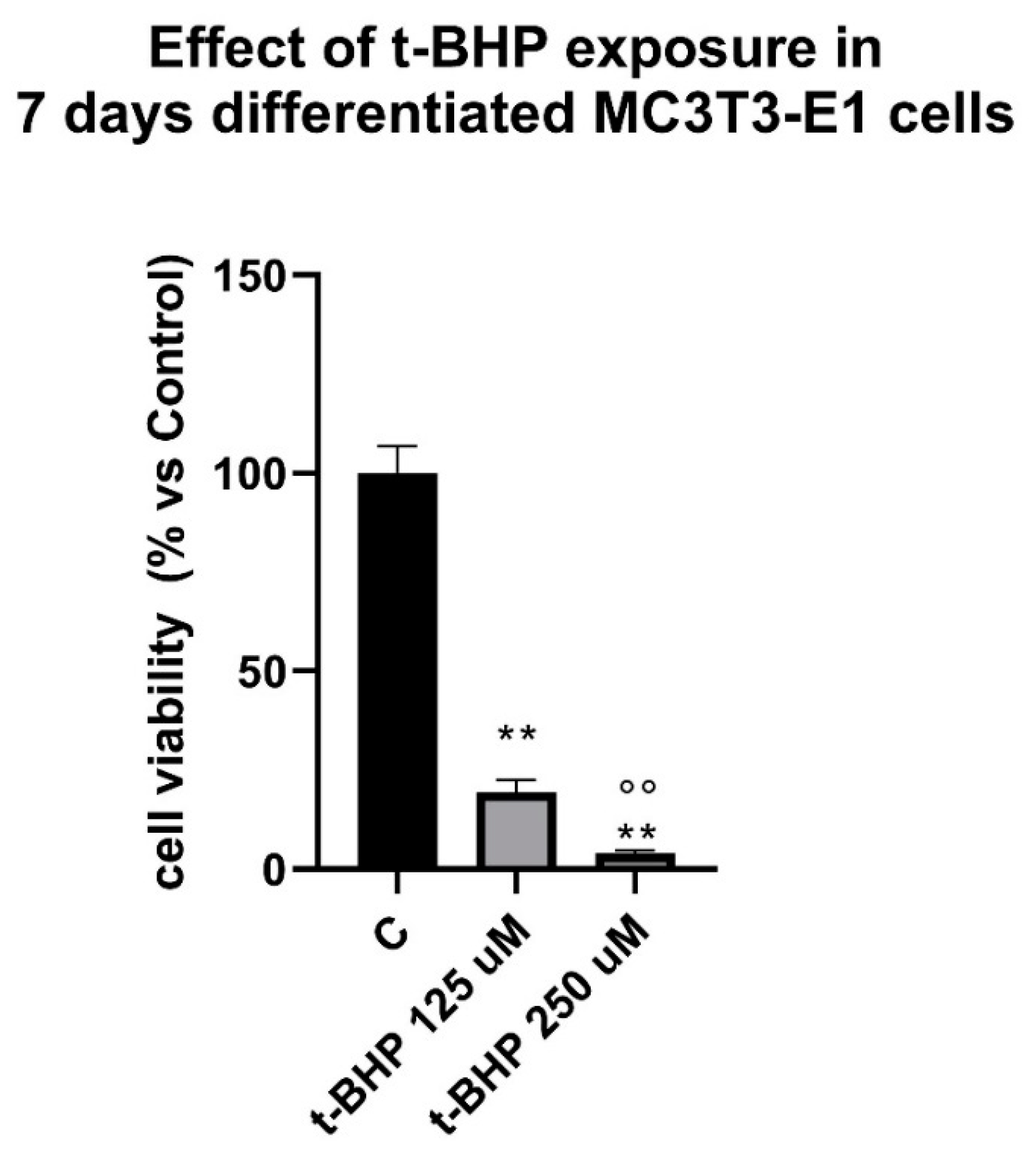
3.2. Effects of Steroids on Counteracting t-BHP-Induced Oxidative Stress in 7-Days Differentiated MC3T3-E1 Cells on Cells Viability
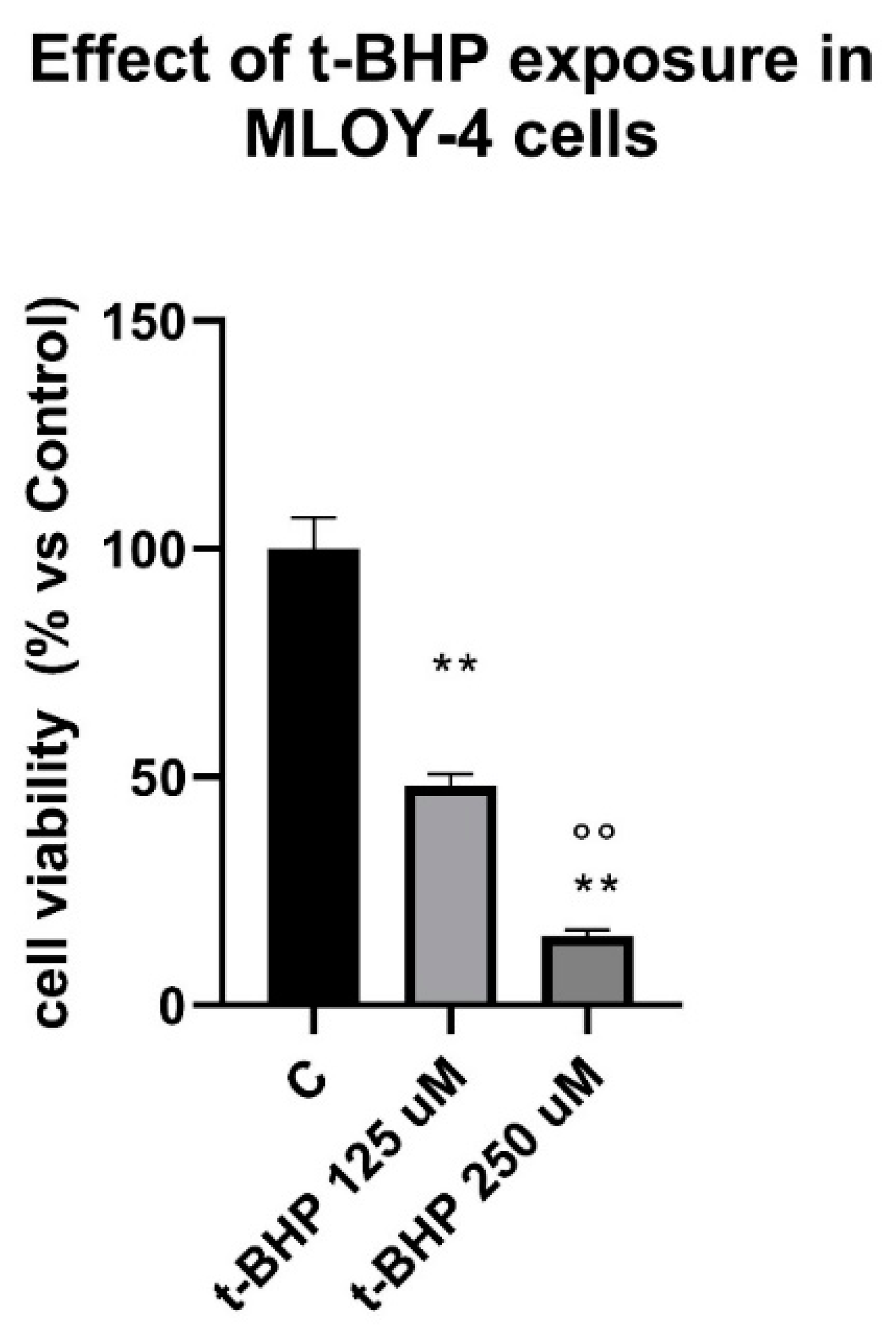
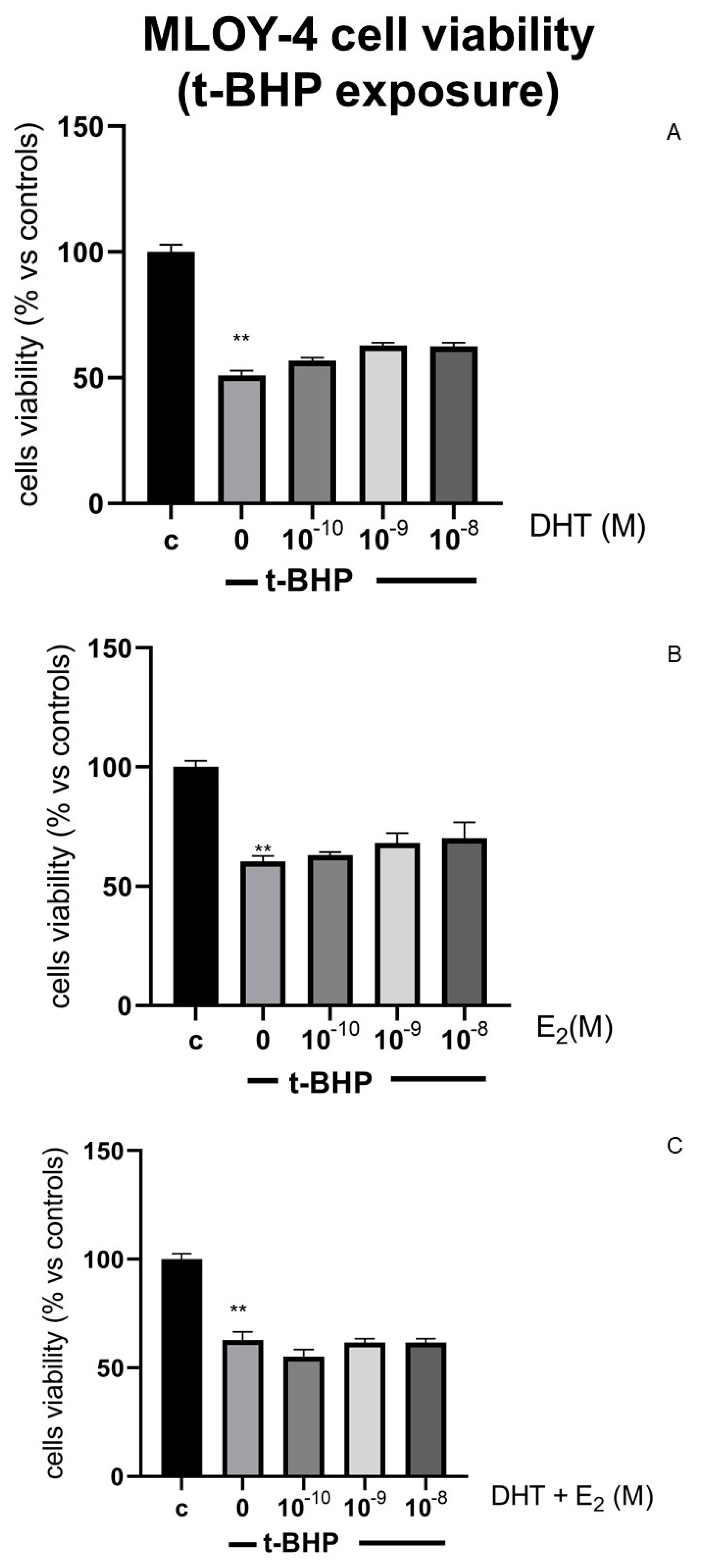
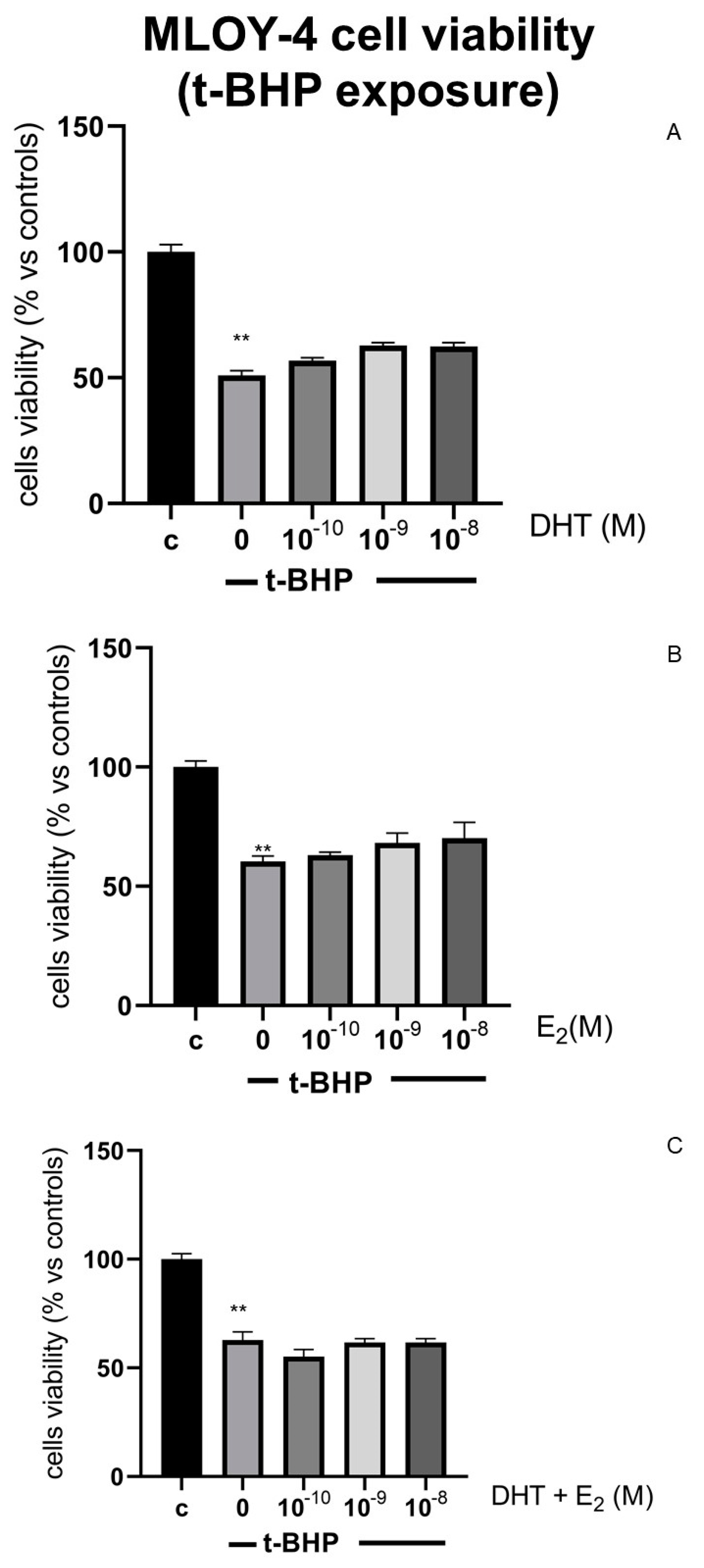
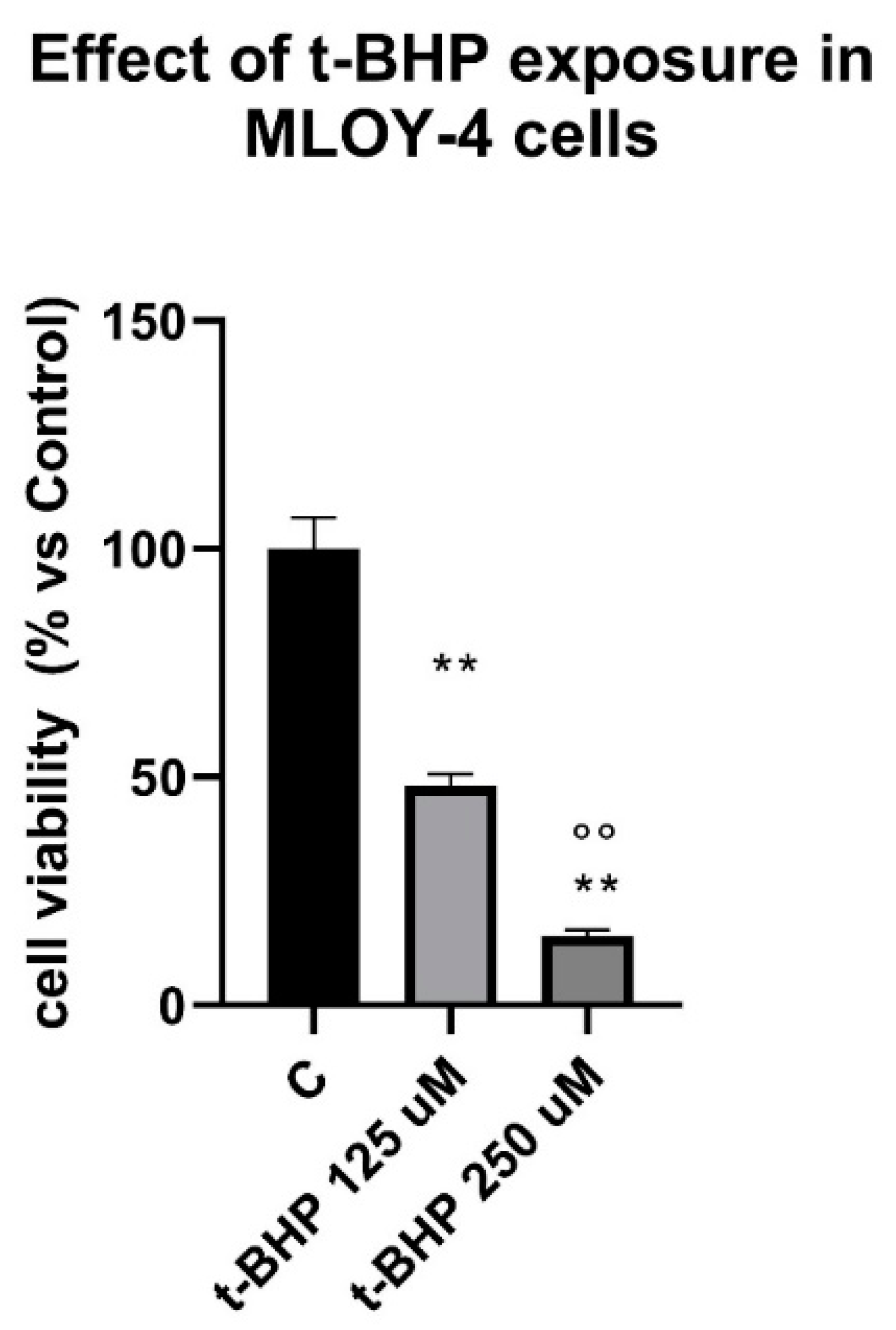
3.3. Effects of Steroids on Counteracting t-BHP-Induced Oxidative Stress in Osteocytes Like Cells, MLOY-4
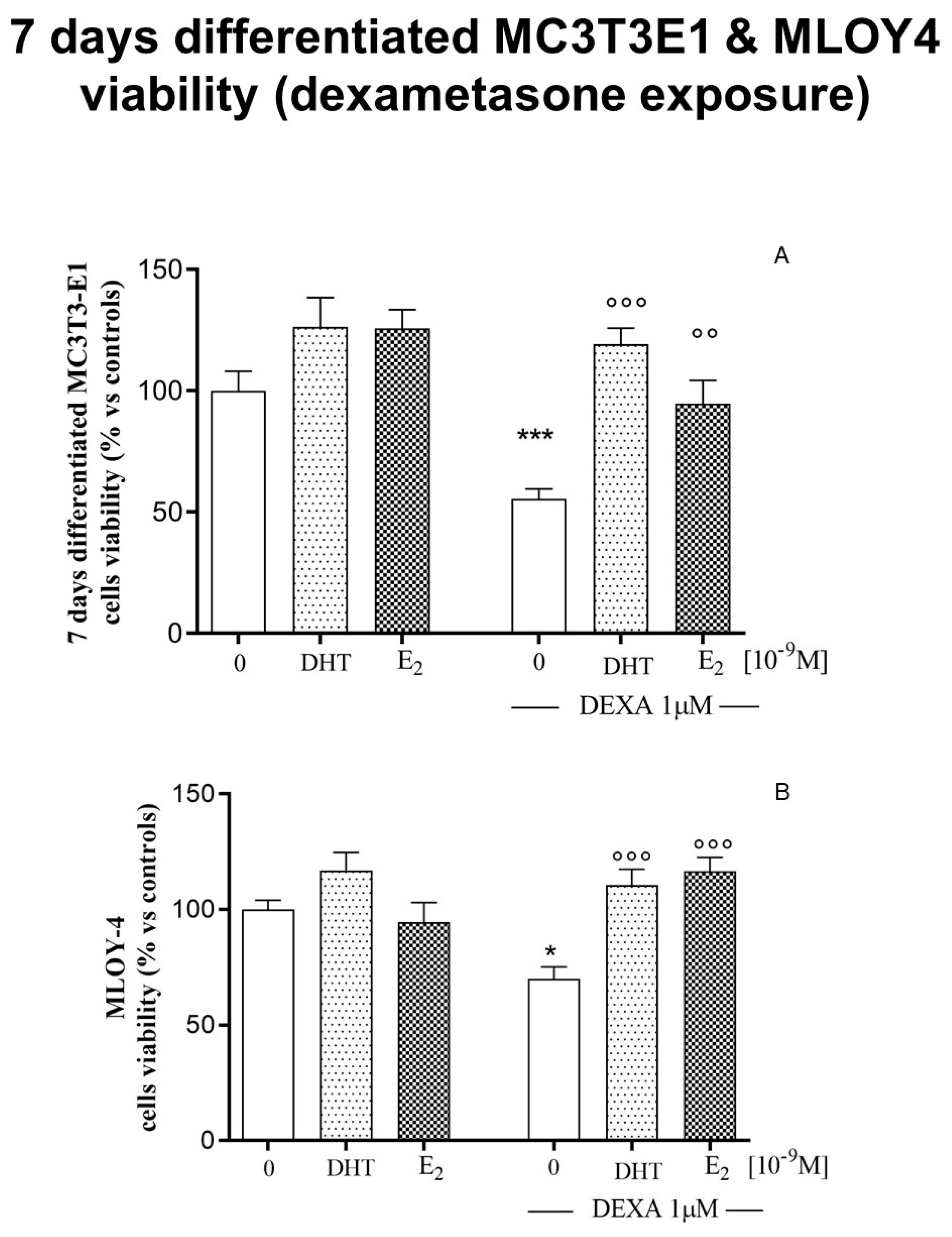
3.4. Effects of Steroids on Counteracting Dexamethasone-Induced Oxidative Stress in 7-Days Differentiated MC3T3-E1 Cells and in Osteocyte-Like Cells MLOY-4
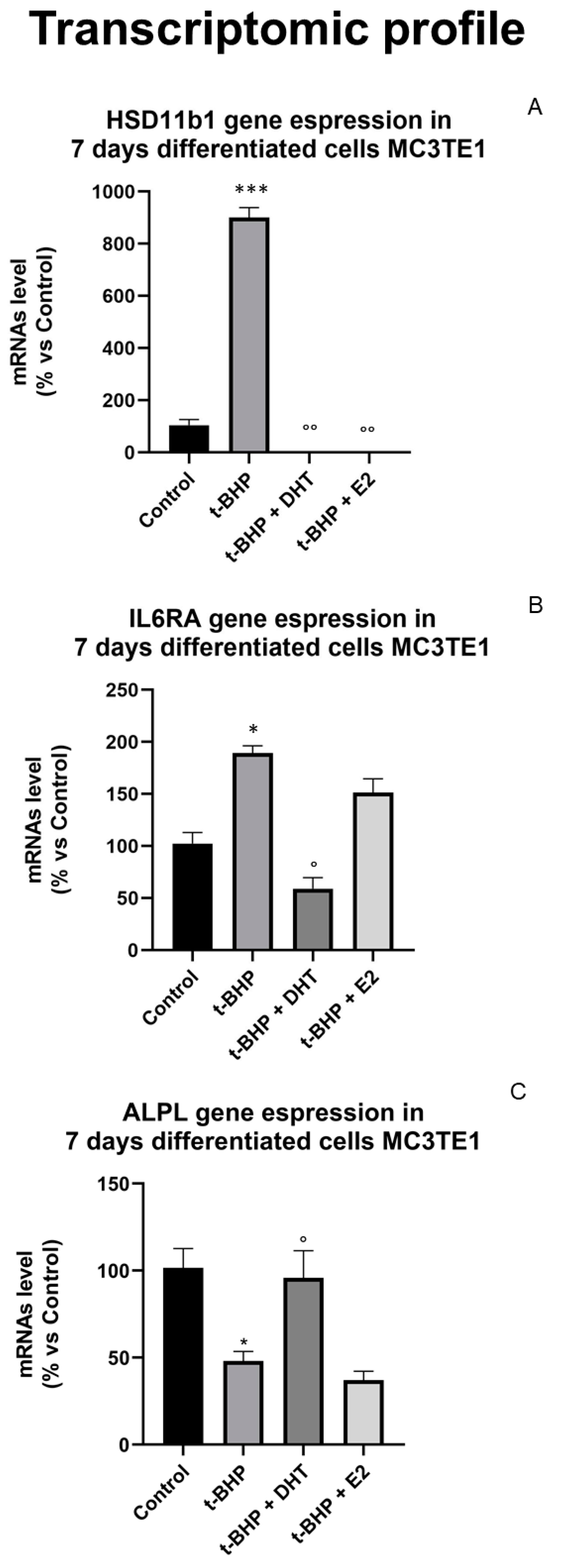
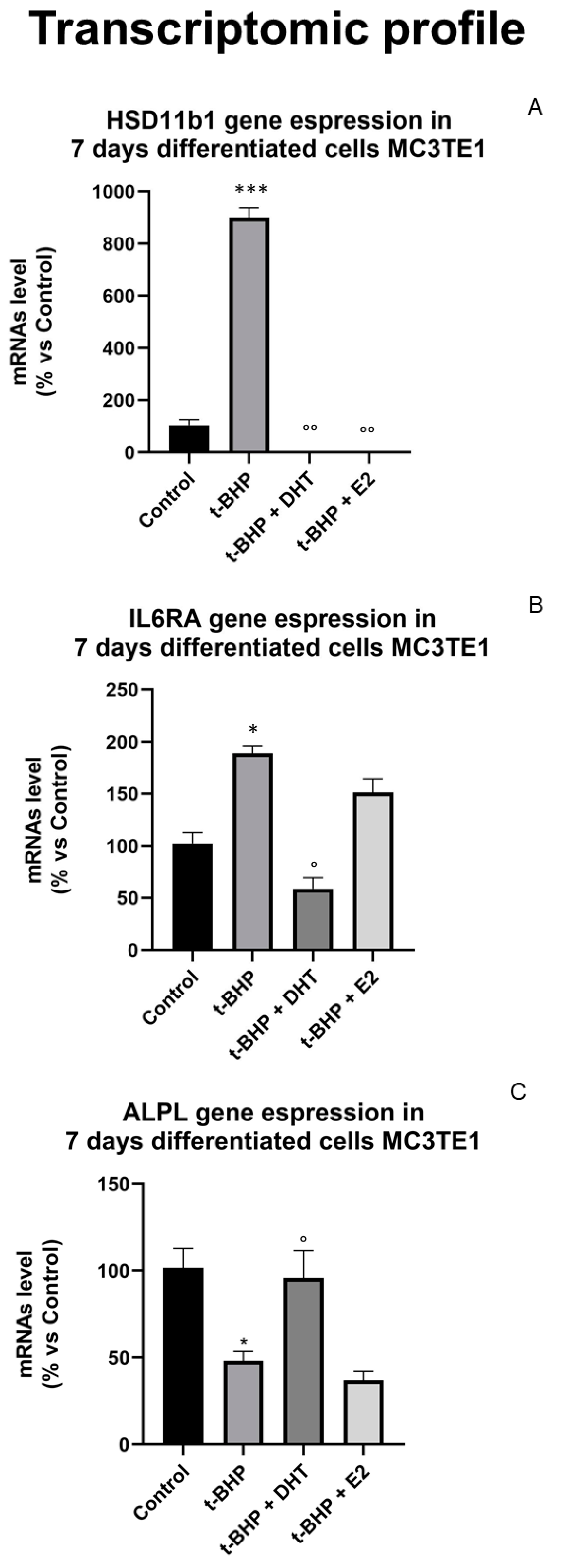
3.5. Transcriptomic Profile: Effect of Oxidative Stress and Steroids on Gene Expression
3.6. Epigenetic Mechanism: Effect of Oxidative Stress and Steroids on Histone Modification
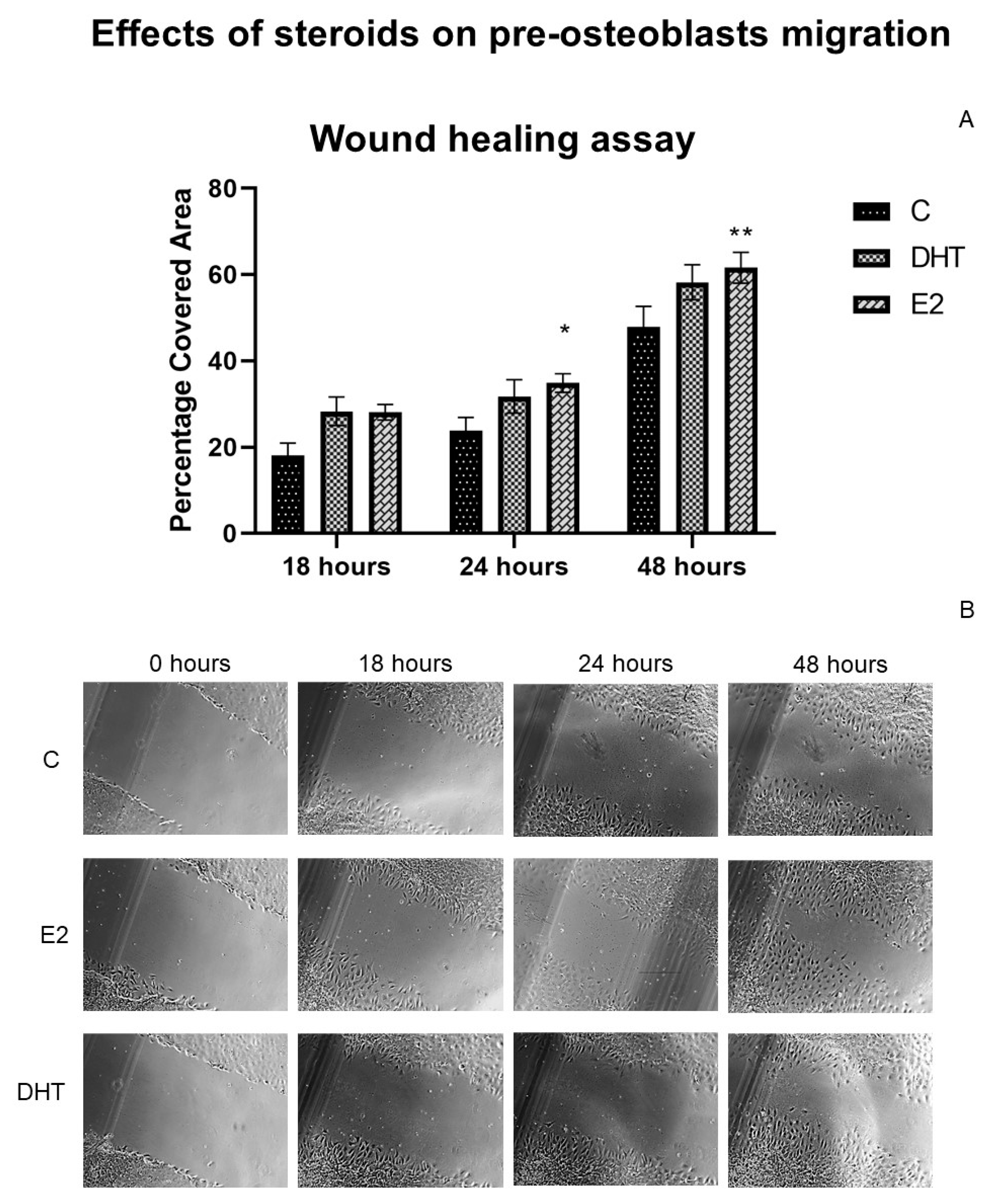
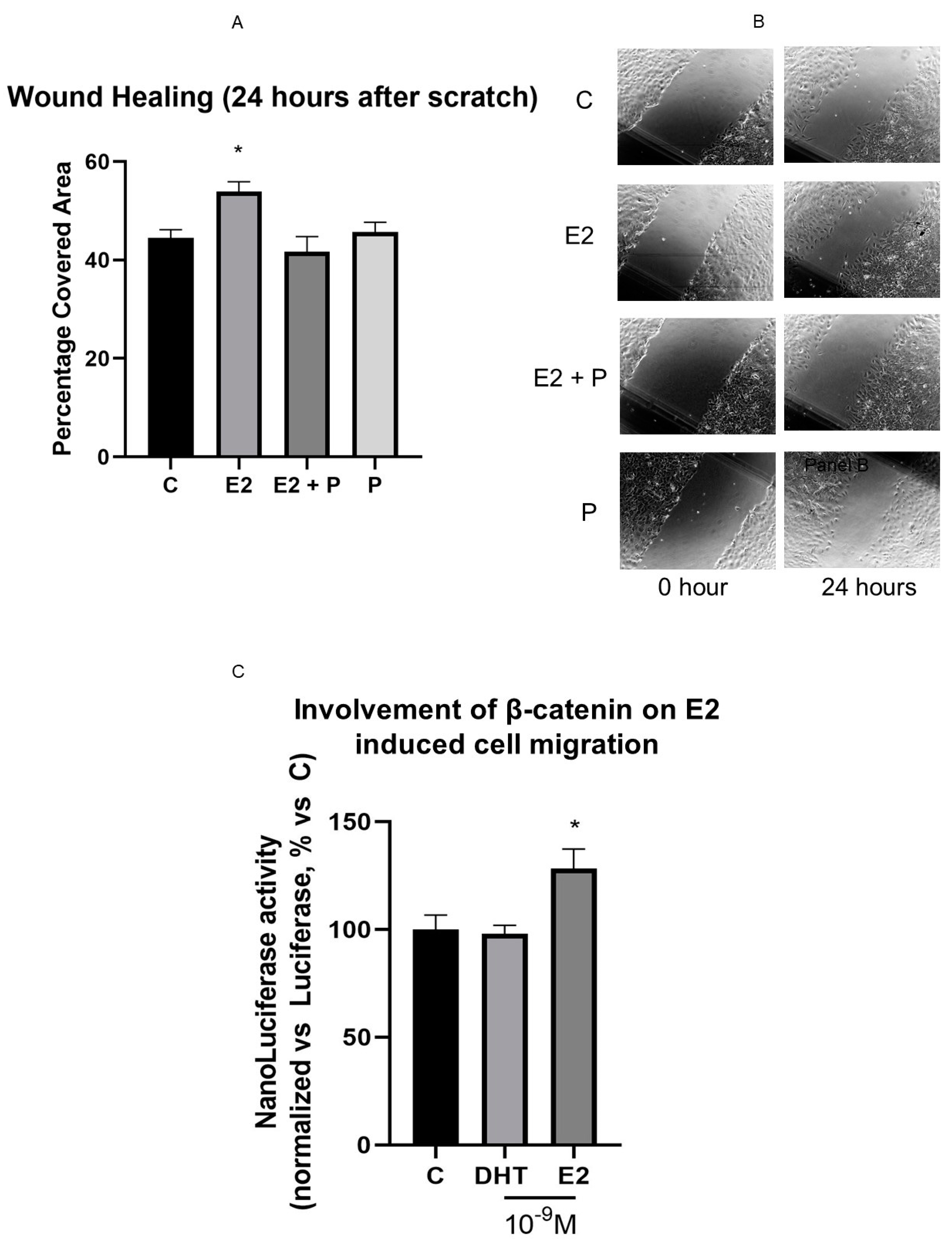
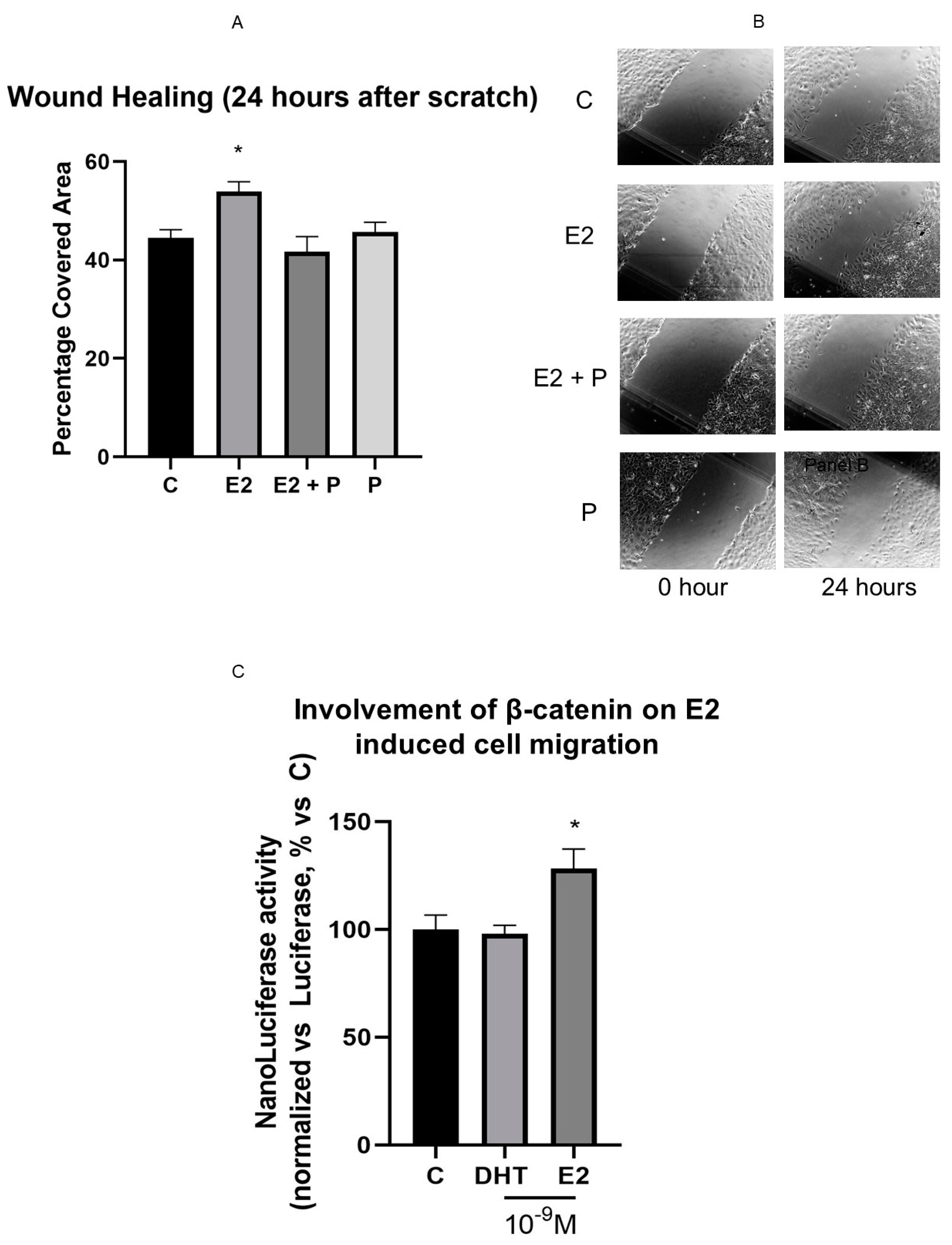
3.7. Effects of Hormonal Steroids on MC3T3E-1 Cell Migration
3.8. β-Catenin Is Involved in E2 Effect on Cell Motility
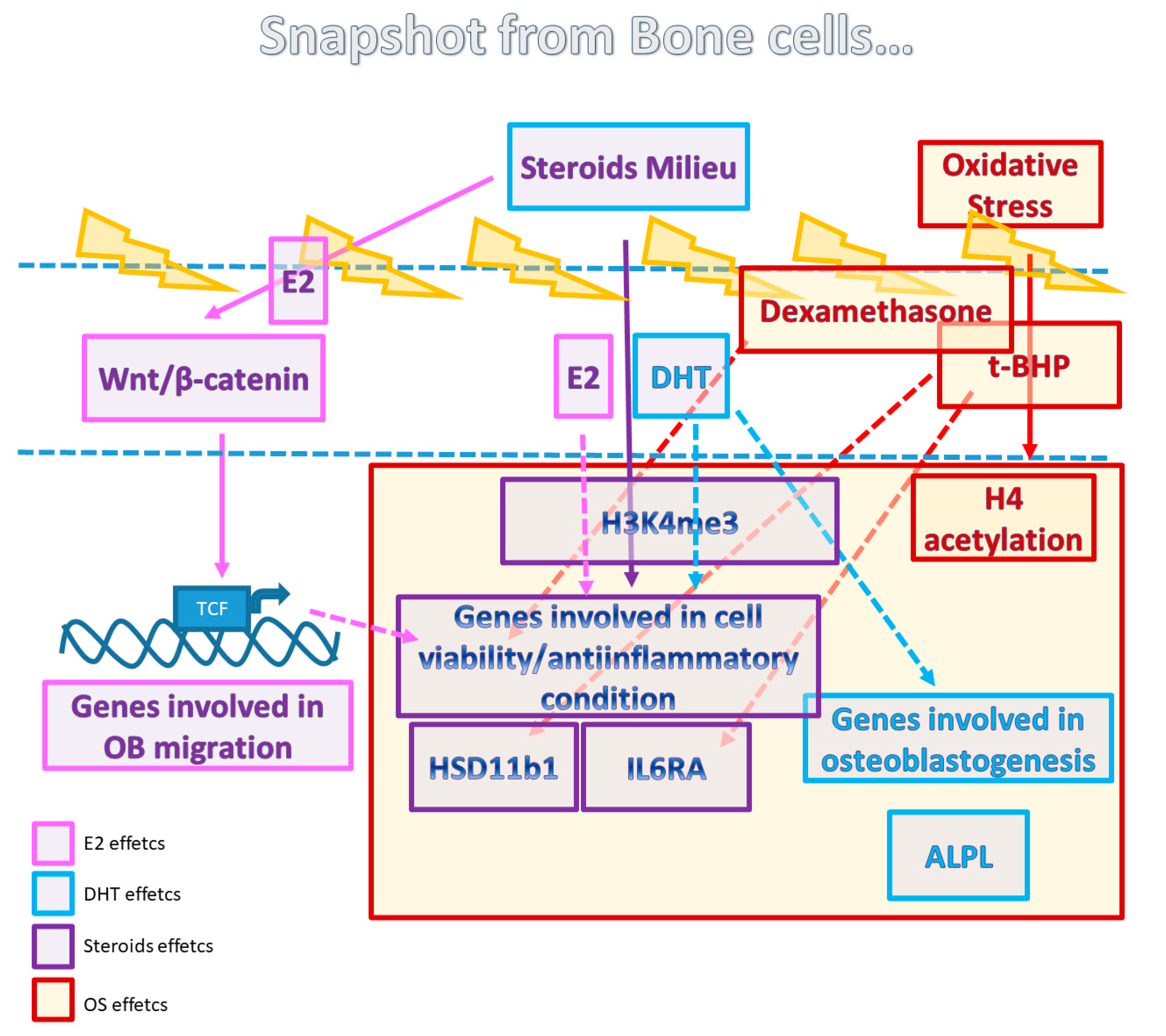
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Almeida, M.; Laurent, M.R.; Dubois, V.; Claessens, F.; O’Brien, C.A.; Bouillon, R.; Vanderschueren, D.; Manolagas, S.C. Estrogens and Androgens in Skeletal Physiology and Pathophysiology. Physiol. Rev. 2017, 97, 135–187. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.; O’Brien, C.A. Basic biology of skeletal aging: Role of stress response pathways. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2013, 68, 1197–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finkelstein, J.S.; Lee, H.; Leder, B.Z.; Burnett-Bowie, S.A.; Goldstein, D.W.; Hahn, C.W.; Hirsch, S.C.; Linker, A.; Perros, N.; Servais, A.B.; et al. Gonadal steroid-dependent effects on bone turnover and bone mineral density in men. J. Clin. Investig. 2016, 126, 1114–1125. [Google Scholar] [CrossRef]
- Huhtaniemi, I.T.; Tajar, A.; Lee, D.M.; O’Neill, T.W.; Finn, J.D.; Bartfai, G.; Boonen, S.; Casanueva, F.F.; Giwercman, A.; Han, T.S.; et al. Comparison of serum testosterone and estradiol measurements in 3174 European men using platform immunoassay and mass spectrometry; relevance for the diagnostics in aging men. Eur. J. Endocrinol. 2012, 166, 983–991. [Google Scholar] [CrossRef]
- Gennari, C.; Agnusdei, D.; Nardi, P.; Civitelli, R. Estrogen preserves a normal intestinal responsiveness to 1,25-dihydroxyvitamin D3 in oophorectomized women. J. Clin. Endocrinol. Metab. 1990, 71, 1288–1293. [Google Scholar] [CrossRef] [PubMed]
- Beato, M.; Klug, J. Steroid hormone receptors: An update. Hum. Reprod. Update 2000, 6, 225–236. [Google Scholar] [CrossRef] [Green Version]
- Nelson, H.D. Menopause. Lancet 2008, 371, 760–770. [Google Scholar] [CrossRef]
- Nakamura, T.; Imai, Y.; Matsumoto, T.; Sato, S.; Takeuchi, K.; Igarashi, K.; Harada, Y.; Azuma, Y.; Krust, A.; Yamamoto, Y.; et al. Estrogen prevents bone loss via estrogen receptor alpha and induction of Fas ligand in osteoclasts. Cell 2007, 130, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Youn, M.Y.; Inoue, K.; Takada, I.; Kouzmenko, A.; Kato, S. Nuclear receptors in bone physiology and diseases. Physiol. Rev. 2013, 93, 481–523. [Google Scholar] [CrossRef] [Green Version]
- Kawano, H.; Sato, T.; Yamada, T.; Matsumoto, T.; Sekine, K.; Watanabe, T.; Nakamura, T.; Fukuda, T.; Yoshimura, K.; Yoshizawa, T.; et al. Suppressive function of androgen receptor in bone resorption. Proc. Natl. Acad. Sci. USA 2003, 100, 9416–9421. [Google Scholar] [CrossRef] [Green Version]
- Ucer, S.; Iyer, S.; Kim, H.N.; Han, L.; Rutlen, C.; Allison, K.; Thostenson, J.D.; de Cabo, R.; Jilka, R.L.; O’Brien, C.; et al. The Effects of Aging and Sex Steroid Deficiency on the Murine Skeleton Are Independent and Mechanistically Distinct. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2017, 32, 560–574. [Google Scholar] [CrossRef] [PubMed]
- Casati, L.; Sendra, R.; Poletti, A.; Negri-Cesi, P.; Celotti, F. Androgen receptor activation by polychlorinated biphenyls: Epigenetic effects mediated by the histone demethylase Jarid1b. Epigenetics 2013, 8, 1061–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leader, J.E.; Wang, C.; Fu, M.; Pestell, R.G. Epigenetic regulation of nuclear steroid receptors. Biochem. Pharmacol. 2006, 72, 1589–1596. [Google Scholar] [CrossRef] [PubMed]
- Manolagas, S.C.; O’Brien, C.A.; Almeida, M. The role of estrogen and androgen receptors in bone health and disease. Nat. Rev. Endocrinol. 2013, 9, 699–712. [Google Scholar] [CrossRef]
- Almeida, M.; Iyer, S.; Martin-Millan, M.; Bartell, S.M.; Han, L.; Ambrogini, E.; Onal, M.; Xiong, J.; Weinstein, R.S.; Jilka, R.L.; et al. Estrogen receptor-α signaling in osteoblast progenitors stimulates cortical bone accrual. J. Clin. Investig. 2013, 123, 394–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manolagas, S.C.; Kousteni, S.; Jilka, R.L. Sex steroids and bone. Recent Prog. Horm. Res. 2002, 57, 385–409. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Carboni, G.A.; Guemes, M.; Bailey, S.; Anaya, E.; Corselli, M.; Peault, B.; Krum, S.A. GATA4 regulates estrogen receptor-alpha-mediated osteoblast transcription. Mol. Endocrinol. 2011, 25, 1126–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monroe, D.G.; Secreto, F.J.; Hawse, J.R.; Subramaniam, M.; Khosla, S.; Spelsberg, T.C. Estrogen receptor isoform-specific regulation of the retinoblastoma-binding protein 1 (RBBP1) gene—Roles of AF1 and enhancer elements. J. Biol. Chem. 2006, 281, 28596–28604. [Google Scholar] [CrossRef] [Green Version]
- Weitzmann, M.N.; Pacifici, R. Estrogen deficiency and bone loss: An inflammatory tale. J. Clin. Investig. 2006, 116, 1186–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baron, R.; Kneissel, M. WNT signaling in bone homeostasis and disease: From human mutations to treatments. Nat. Med. 2013, 19, 179–192. [Google Scholar] [CrossRef]
- Albers, J.; Keller, J.; Baranowsky, A.; Beil, F.T.; Catala-Lehnen, P.; Schulze, J.; Amling, M.; Schinke, T. Canonical Wnt signaling inhibits osteoclastogenesis independent of osteoprotegerin. J. Cell Biol. 2013, 200, 537–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Stern, P.H. Sex-specific effects of estrogen and androgen on gene expression in human monocyte-derived osteoclasts. J. Cell. Biochem. 2011, 112, 3714–3721. [Google Scholar] [CrossRef]
- Sinnesael, M.; Jardi, F.; Deboel, L.; Laurent, M.R.; Dubois, V.; Zajac, J.D.; Davey, R.A.; Carmeliet, G.; Claessens, F.; Vanderschueren, D. The androgen receptor has no direct antiresorptive actions in mouse osteoclasts. Mol. Cell. Endocrinol. 2015, 411, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Manolagas, S.C. From estrogen-centric to aging and oxidative stress: A revised perspective of the pathogenesis of osteoporosis. Endocr. Rev. 2010, 31, 266–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lean, J.M.; Davies, J.T.; Fuller, K.; Jagger, C.J.; Kirstein, B.; Partington, G.A.; Urry, Z.L.; Chambers, T.J. A crucial role for thiol antioxidants in estrogen-deficiency bone loss. J. Clin. Investig. 2003, 112, 915–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, M.; Han, L.; Martin-Millan, M.; Plotkin, L.I.; Stewart, S.A.; Roberson, P.K.; Kousteni, S.; O’Brien, C.A.; Bellido, T.; Parfitt, A.M.; et al. Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J. Biol. Chem. 2007, 282, 27285–27297. [Google Scholar] [CrossRef] [Green Version]
- Guillaumet-Adkins, A.; Yanez, Y.; Peris-Diaz, M.D.; Calabria, I.; Palanca-Ballester, C.; Sandoval, J. Epigenetics and Oxidative Stress in Aging. Oxidative Med. Cell. Longev. 2017, 2017, 9175806. [Google Scholar] [CrossRef]
- Husain, A.; Jeffries, M.A. Epigenetics and Bone Remodeling. Curr. Osteoporos. Rep. 2017, 15, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Willbanks, A.; Leary, M.; Greenshields, M.; Tyminski, C.; Heerboth, S.; Lapinska, K.; Haskins, K.; Sarkar, S. The Evolution of Epigenetics: From Prokaryotes to Humans and Its Biological Consequences. Genet. Epigenetics 2016, 8, 25–36. [Google Scholar] [CrossRef] [Green Version]
- Casati, L.; Sendra, R.; Sibilia, V.; Celotti, F. Endocrine disrupters: The new players able to affect the epigenome. Front. Cell Dev. Biol. 2015, 3, 37. [Google Scholar] [CrossRef] [PubMed]
- Sudo, H.; Kodama, H.A.; Amagai, Y.; Yamamoto, S.; Kasai, S. In vitro differentiation and calcification in a new clonal osteogenic cell line derived from newborn mouse calvaria. J. Cell Biol. 1983, 96, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Windle, J.J.; Koop, B.A.; Mundy, G.R.; Bonewald, L.F. Establishment of an osteocyte-like cell line, MLO-Y4. J. Bone Miner. Res. 1997, 12, 2014–2023. [Google Scholar] [CrossRef]
- Casati, L.; Pagani, F.; Limonta, P.; Vanetti, C.; Stancari, G.; Sibilia, V. Beneficial effects of delta-tocotrienol against oxidative stress in osteoblastic cells: Studies on the mechanisms of action. Eur. J. Nutr. 2020, 59, 1975–1987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dieci, E.; Casati, L.; Pagani, F.; Celotti, F.; Sibilia, V. Acylated and unacylated ghrelin protect MC3T3-E1 cells against tert-butyl hydroperoxide-induced oxidative injury: Pharmacological characterization of ghrelin receptor and possible epigenetic involvement. Amino Acids 2014, 46, 1715–1725. [Google Scholar] [CrossRef] [PubMed]
- Haidara, K.; Marion, M.; Gascon-Barré, M.; Denizeau, F.; Averill-Bates, D.A. Implication of caspases and subcellular compartments in tert-butylhydroperoxide induced apoptosis. Toxicol. Appl. Pharmacol. 2008, 229, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Kim, S.E.; Lee, D.Y.; Choi, D. Serum estradiol level according to dose and formulation of oral estrogens in postmenopausal women. Sci. Rep. 2021, 11, 3585. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Han, L.; Cong, W. Alpinumisoflavone rescues glucocorticoid-induced apoptosis of osteocytes via suppressing Nox2-dependent ROS generation. Pharmacol. Rep. 2018, 70, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Wiren, K.M.; Toombs, A.R.; Semirale, A.A.; Zhang, X. Osteoblast and osteocyte apoptosis associated with androgen action in bone: Requirement of increased Bax/Bcl-2 ratio. Bone 2006, 38, 637–651. [Google Scholar] [CrossRef]
- Michael, H.; Härkönen, P.L.; Väänänen, H.K.; Hentunen, T.A. Estrogen and testosterone use different cellular pathways to inhibit osteoclastogenesis and bone resorption. J. Bone Miner. Res. 2005, 20, 2224–2232. [Google Scholar] [CrossRef] [PubMed]
- Casati, L.; Pagani, F.; Fibiani, M.; Lo Scalzo, R.; Sibilia, V. Potential of delphinidin-3-rutinoside extracted from Solanum melongena L. as promoter of osteoblastic MC3T3-E1 function and antagonist of oxidative damage. Eur. J. Nutr. 2019, 58, 1019–1032. [Google Scholar] [CrossRef] [Green Version]
- Casati, L.; Pagani, F.; Maggi, R.; Ferrucci, F.; Sibilia, V. Food for Bone: Evidence for a Role for Delta-Tocotrienol in the Physiological Control of Osteoblast Migration. Int. J. Mol. Sci. 2020, 21, 4661. [Google Scholar] [CrossRef]
- Planz, V.; Wang, J.; Windbergs, M. Establishment of a cell-based wound healing assay for bio-relevant testing of wound therapeutics. J. Pharmacol. Toxicol. Methods 2018, 89, 19–25. [Google Scholar] [CrossRef]
- Casati, L.; Sendra, R.; Colciago, A.; Negri-Cesi, P.; Berdasco, M.; Esteller, M.; Celotti, F. Polychlorinated biphenyls affect histone modification pattern in early development of rats: A role for androgen receptor-dependent modulation? Epigenomics 2012, 4, 101–112. [Google Scholar] [CrossRef]
- Gao, Z.; Xu, Z.; Hung, M.S.; Lin, Y.C.; Wang, T.; Gong, M.; Zhi, X.; Jablons, D.M.; You, L. Procaine and procainamide inhibit the Wnt canonical pathway by promoter demethylation of WIF-1 in lung cancer cells. Oncol. Rep. 2009, 22, 1479–1484. [Google Scholar]
- Herencia, C.; Diaz-Tocados, J.M.; Jurado, L.; Montes de Oca, A.; Rodriguez-Ortiz, M.E.; Martin-Alonso, C.; Martinez-Moreno, J.M.; Vergara, N.; Rodriguez, M.; Almaden, Y.; et al. Procaine Inhibits Osteo/Odontogenesis through Wnt/beta-Catenin Inactivation. PLoS ONE 2016, 11, e0156788. [Google Scholar] [CrossRef]
- Hadjidakis, D.J.; Androulakis, I.I. Bone remodeling. Ann. N. Y. Acad. Sci. 2006, 1092, 385–396. [Google Scholar] [CrossRef]
- Hendrickx, G.; Boudin, E.; Van Hul, W. A look behind the scenes: The risk and pathogenesis of primary osteoporosis. Nat. Rev. Rheumatol. 2015, 11, 462–474. [Google Scholar] [CrossRef] [PubMed]
- Chapman, K.; Holmes, M.; Seckl, J. 11β-hydroxysteroid dehydrogenases: Intracellular gate-keepers of tissue glucocorticoid action. Physiol. Rev. 2013, 93, 1139–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, K.; Hardy, R.; Ahasan, M.M.; Eijken, M.; van Leeuwen, J.P.; Filer, A.; Thomas, A.M.; Raza, K.; Buckley, C.D.; Stewart, P.M.; et al. Synergistic induction of local glucocorticoid generation by inflammatory cytokines and glucocorticoids: Implications for inflammation associated bone loss. Ann. Rheum. Dis. 2010, 69, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Amarasekara, D.S.; Kim, S.; Rho, J. Regulation of Osteoblast Differentiation by Cytokine Networks. Int. J. Mol. Sci. 2021, 22, 2851. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Hadoke, P.W.; Wu, J.; Vesey, A.T.; Lerman, D.A.; Dweck, M.R.; Newby, D.E.; Smith, L.B.; MacRae, V.E. Ablation of the androgen receptor from vascular smooth muscle cells demonstrates a role for testosterone in vascular calcification. Sci. Rep. 2016, 6, 24807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filaire, E.; Toumi, H. Reactive oxygen species and exercise on bone metabolism: Friend or enemy? Jt. Bone Spine 2012, 79, 341–346. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Jamora, C.; DasGupta, R.; Kocieniewski, P.; Fuchs, E. Links between signal transduction, transcription and adhesion in epithelial bud development. Nature 2003, 422, 317–322. [Google Scholar] [CrossRef] [Green Version]
- Novak, A.; Dedhar, S. Signaling through beta-catenin and Lef/Tcf. Cell. Mol. Life Sci. 1999, 56, 523–537. [Google Scholar] [CrossRef]
- Feng, Q.; Zheng, S.; Zheng, J. The emerging role of microRNAs in bone remodeling and its therapeutic implications for osteoporosis. Biosci. Rep. 2018, 38, 104350. [Google Scholar] [CrossRef] [Green Version]
- Raut, N.; Wicks, S.M.; Lawal, T.O.; Mahady, G.B. Epigenetic regulation of bone remodeling by natural compounds. Pharmacol. Res. 2019, 147, 104350. [Google Scholar] [CrossRef] [PubMed]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef]
- De Boer, J.; Licht, R.; Bongers, M.; van der Klundert, T.; Arends, R.; van Blitterswijk, C. Inhibition of histone acetylation as a tool in bone tissue engineering. Tissue Eng. 2006, 12, 2927–2937. [Google Scholar] [CrossRef]
- Cho, H.H.; Park, H.T.; Kim, Y.J.; Bae, Y.C.; Suh, K.T.; Jung, J.S. Induction of osteogenic differentiation of human mesenchymal stem cells by histone deacetylase inhibitors. J. Cell. Biochem. 2005, 96, 533–542. [Google Scholar] [CrossRef]
- Vrtacnik, P.; Zupan, J.; Mlakar, V.; Kranjc, T.; Marc, J.; Kern, B.; Ostanek, B. Epigenetic enzymes influenced by oxidative stress and hypoxia mimetic in osteoblasts are differentially expressed in patients with osteoporosis and osteoarthritis. Sci. Rep. 2018, 8, 16215. [Google Scholar] [CrossRef] [PubMed]
- Gambacurta, A.; Merlini, G.; Ruggiero, C.; Diedenhofen, G.; Battista, N.; Bari, M.; Balsamo, M.; Piccirillo, S.; Valentini, G.; Mascetti, G.; et al. Human osteogenic differentiation in Space: Proteomic and epigenetic clues to better understand osteoporosis. Sci. Rep. 2019, 9, 8343. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Acronym | t-BHP Effect | DHT + t-BHP Effect | E2 + t-BHP Effect |
|---|---|---|---|---|
| Alkaline Phosphatase | ALPL |  |  = C = C | |
| Calcitonin Receptor | CALCR | |  | |
| Chloride Voltage Gated Channel 7 | CLCN7 | | | |
| Hydroxysteroid 11-β Dehydrogenase 1 | HSD11B1 |  |  = C = C | = C |
| Interleukin-6 | IL6 | | | |
| Interleukin-6 receptor subunit alpha | IL6-RA | | = C | |
| Methylenetetrahydrofolate Reductase | MTHFR | | | |
| Nuclear Factor Of Activated T Cells 1 | NFATC1 | | | |
| Runt-related transcription factor 2 | RUNX-2 | | | |
| Sex hormone-binding globulin precursor | SHBG | | = C | = C |
| Osteoprotegerin | TNFSR11B1 | | | |
| Vascular endothelial growth factor | VEGFA | | | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sibilia, V.; Bottai, D.; Maggi, R.; Pagani, F.; Chiaramonte, R.; Giannandrea, D.; Citro, V.; Platonova, N.; Casati, L. Sex Steroid Regulation of Oxidative Stress in Bone Cells: An In Vitro Study. Int. J. Environ. Res. Public Health 2021, 18, 12168. https://doi.org/10.3390/ijerph182212168
Sibilia V, Bottai D, Maggi R, Pagani F, Chiaramonte R, Giannandrea D, Citro V, Platonova N, Casati L. Sex Steroid Regulation of Oxidative Stress in Bone Cells: An In Vitro Study. International Journal of Environmental Research and Public Health. 2021; 18(22):12168. https://doi.org/10.3390/ijerph182212168
Chicago/Turabian StyleSibilia, Valeria, Daniele Bottai, Roberto Maggi, Francesca Pagani, Raffaella Chiaramonte, Domenica Giannandrea, Valentina Citro, Natalia Platonova, and Lavinia Casati. 2021. "Sex Steroid Regulation of Oxidative Stress in Bone Cells: An In Vitro Study" International Journal of Environmental Research and Public Health 18, no. 22: 12168. https://doi.org/10.3390/ijerph182212168
APA StyleSibilia, V., Bottai, D., Maggi, R., Pagani, F., Chiaramonte, R., Giannandrea, D., Citro, V., Platonova, N., & Casati, L. (2021). Sex Steroid Regulation of Oxidative Stress in Bone Cells: An In Vitro Study. International Journal of Environmental Research and Public Health, 18(22), 12168. https://doi.org/10.3390/ijerph182212168