
Associations between Brain Reserve Proxies and Clinical Progression in Alzheimer’s Disease Dementia

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Participants
2.2. Clinical Assessment and Cognitive Reserve Proxies
2.3. Neuroimaging Data and Brain Reserve Proxies
2.3.1. MRI
2.3.2. FDG-PET
2.3.3. Florbetapir PET
2.4. Statistical Analysis
3. Results
3.1. Participant Characteristics
3.2. Clinical Characteristics and Reserve Proxies
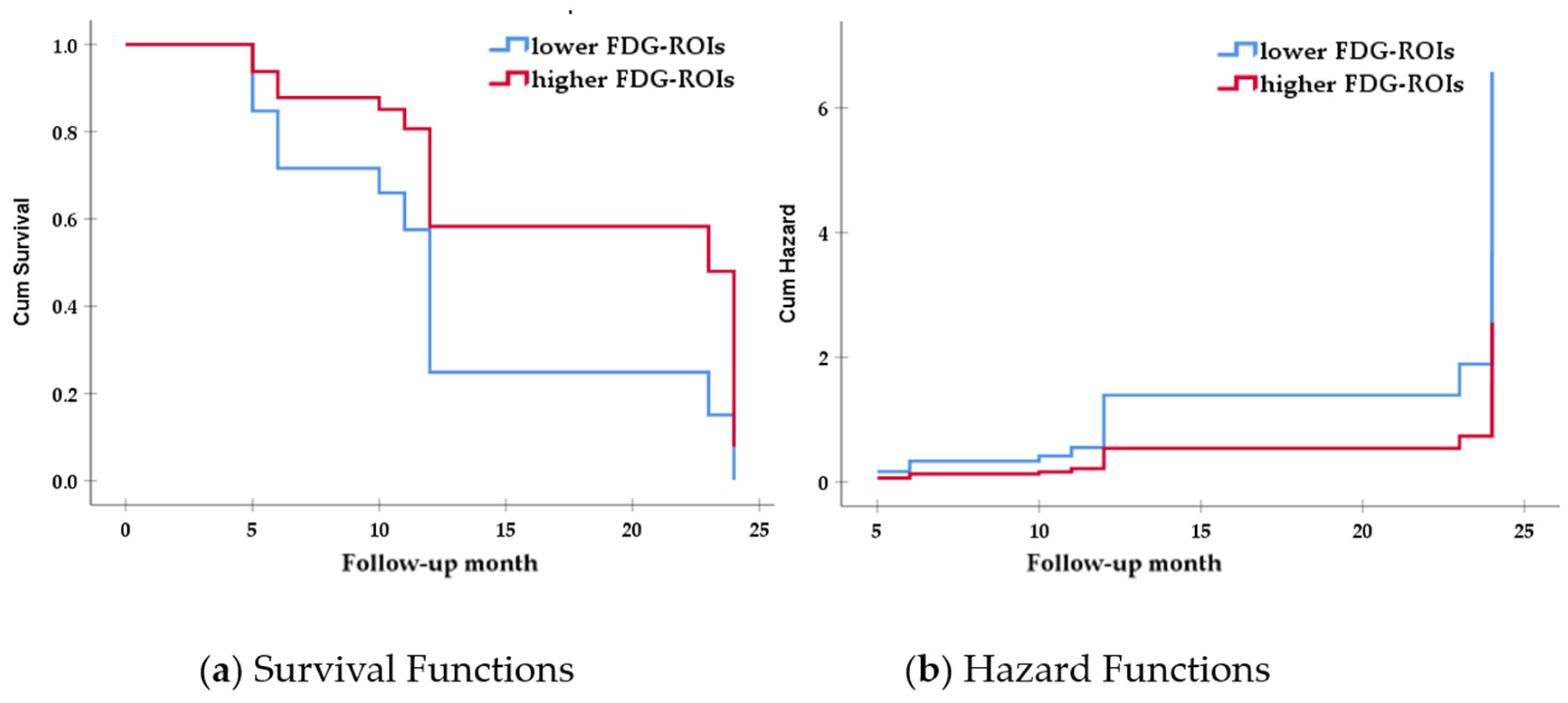
3.3. Associaations between Reserve Proxies and Clinical Progression
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rajan, K.B.; Weuve, J.; Barnes, L.L.; Wilson, R.S.; Evans, D.A. Prevalence and incidence of clinically diagnosed Alzheimer’s disease dementia from 1994 to 2012 in a population study. Alzheimers Dement. 2019, 15, 1–7. [Google Scholar] [CrossRef]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-5®); American Psychiatric Pub: Washington, DC, USA, 2013. [Google Scholar]
- Stern, Y. What is cognitive reserve? Theory and research application of the reserve concept. J. Int. Neuropsychol. Soc. 2002, 8, 448–460. [Google Scholar] [CrossRef]
- Katzman, R.; Terry, R.; DeTeresa, R.; Brown, T.; Davies, P.; Fuld, P.; Renbing, X.; Peck, A. Clinical, pathological, and neurochemical changes in dementia: A subgroup with preserved mental status and numerous neocortical plaques. Ann. Neurol. 1988, 23, 138–144. [Google Scholar] [CrossRef]
- Mortimer, J.A. Brain reserve and the clinical expression of Alzheimer’s disease. Geriatrics 1997, 52, S50–S53. [Google Scholar] [PubMed]
- Stern, Y. Cognitive reserve in ageing and Alzheimer’s disease. Lancet Neurol. 2012, 11, 1006–1012. [Google Scholar] [CrossRef] [Green Version]
- Lebel, C.; Beaulieu, C. Longitudinal development of human brain wiring continues from childhood into adulthood. J. Neurosci. 2011, 31, 10937–10947. [Google Scholar] [CrossRef] [PubMed]
- Wolf, H.; Julin, P.; Gertz, H.J.; Winblad, B.; Wahlund, L.O. Intracranial volume in mild cognitive impairment, Alzheimer’s disease and vascular dementia: Evidence for brain reserve? Int. J. Geriatr. Psychiatry 2004, 19, 995–1007. [Google Scholar] [CrossRef]
- Stern, Y. An approach to studying the neural correlates of reserve. Brain Imag. Behav. 2017, 11, 410–416. [Google Scholar] [CrossRef]
- Stern, Y.; Arenaza-Urquijo, E.M.; Bartres-Faz, D.; Belleville, S.; Cantilon, M.; Chetelat, G.; Ewers, M.; Franzmeier, N.; Kempermann, G.; Kremen, W.S.; et al. Whitepaper: Defining and investigating cognitive reserve, brain reserve, and brain maintenance. Alzheimers Dement. 2018, 16, 1305–1311. [Google Scholar] [CrossRef] [PubMed]
- Barulli, D.; Stern, Y. Efficiency, capacity, compensation, maintenance, plasticity: Emerging concepts in cognitive reserve. Trends Cogn. Sci. 2013, 17, 502–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamballais, S.; Zijlmans, J.L.; Vernooij, M.W.; Ikram, M.K.; Luik, A.I.; Ikram, M.A. The Risk of Dementia in Relation to Cognitive and Brain Reserve. J. Alzheimers Dis. 2020, 77, 607–618. [Google Scholar] [CrossRef]
- van Loenhoud, A.C.; Groot, C.; Vogel, J.W.; van der Flier, W.M.; Ossenkoppele, R. Is intracranial volume a suitable proxy for brain reserve? Alzheimers Res. Ther. 2018, 10, 91. [Google Scholar] [CrossRef]
- Le Carret, N.; Lafont, S.; Letenneur, L.; Dartigues, J.-F.; Mayo, W.; Fabrigoule, C. The Effect of Education on Cognitive Performances and Its Implication for the Constitution of the Cognitive Reserve. Develop. Neuropsychol. 2003, 23, 317–337. [Google Scholar] [CrossRef]
- Dekhtyar, S.; Wang, H.X.; Fratiglioni, L.; Herlitz, A. Childhood school performance, education and occupational complexity: A life-course study of dementia in the Kungsholmen Project. Int. J. Epidemiol. 2016, 45, 1207–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.X.; MacDonald, S.W.; Dekhtyar, S.; Fratiglioni, L. Association of lifelong exposure to cognitive reserve-enhancing factors with dementia risk: A community-based cohort study. PLoS Med. 2017, 14, e1002251. [Google Scholar] [CrossRef]
- Devita, M.; Bordignon, A.; Trevisan, C.; Sergi, G.; Girardi, A.; Mapelli, D.; Manzato, E.; Coin, A. Longitudinal investigation of the role of cognitive reserve in the evolution of dementia in outpatients prescribed AChEI. J. Clin. Exp. Neuropsychol. 2020, 42, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Devita, M.; Mondini, S.; Bordignon, A.; Sergi, G.; Girardi, A.; Manzato, E.; Mapelli, D.; Coin, A. The importance of cognitive reserve in comprehensive geriatric assessment for dementia. Aging Clin. Exp. Res. 2020, 32, 1179–1181. [Google Scholar] [CrossRef]
- Jia, F.; Li, Y.; Li, M.; Cao, F. Subjective Cognitive Decline, Cognitive Reserve Indicators, and the Incidence of Dementia. J. Am. Med. Dir. Assoc. 2020, 22, 1449–1455. [Google Scholar] [CrossRef]
- Jia, F.; Liu, F.; Li, X.; Shi, X.; Liu, Y.; Cao, F. Cognitive reserve, modifiable-risk-factor profile and incidence of dementia: Results from a longitudinal study of CFAS Wales. Aging Ment. Health 2020, 1–7. [Google Scholar] [CrossRef]
- Scherrer, J.F.; Morley, J.E. Lowering dementia risk and slowing progression of disease: The role of cognitive reserve and cognitive training. Br. J. Psychiatry 2020, 218, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Yang, R.; Dintica, C.; Qi, X.; Song, R.; Bennett, D.A.; Xu, W. Association of lifespan cognitive reserve indicator with the risk of mild cognitive impairment and its progression to dementia. Alzheimers Dement. 2020, 16, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, G.; Zhukovsky, P.; Voineskos, A.; Grady, C. A profile of brain reserve in adults at genetic risk of Alzheimer’s disease. Alzheimers Dement. 2021, 13, e12208. [Google Scholar] [CrossRef]
- Serra, L.; Petrosini, L.; Salaris, A.; Pica, L.; Bruschini, M.; Di Domenico, C.; Caltagirone, C.; Marra, C.; Bozzali, M. Testing for the Myth of Cognitive Reserve: Are the Static and Dynamic Cognitive Reserve Indexes a Representation of Different Reserve Warehouses? J. Alzheimers Dis. 2019, 72, 111–126. [Google Scholar] [CrossRef] [Green Version]
- Petersen, R.C.; Aisen, P.S.; Beckett, L.A.; Donohue, M.C.; Gamst, A.C.; Harvey, D.J.; Jack, C.R., Jr.; Jagust, W.J.; Shaw, L.M.; Toga, A.W.; et al. Alzheimer’s Disease Neuroimaging Initiative (ADNI): Clinical characterization. Neurology 2010, 74, 201–209. [Google Scholar] [CrossRef] [Green Version]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939–944. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, R.I.; Kurosaki, T.T.; Harrah, C.H., Jr.; Chance, J.M.; Filos, S. Measurement of functional activities in older adults in the community. J. Gerontol. 1982, 37, 323–329. [Google Scholar] [CrossRef]
- Saykin, A.J.; Shen, L.; Foroud, T.M.; Potkin, S.G.; Swaminathan, S.; Kim, S.; Risacher, S.L.; Nho, K.; Huentelman, M.J.; Craig, D.W.; et al. Alzheimer’s Disease Neuroimaging. Alzheimer’s Disease Neuroimaging Initiative biomarkers as quantitative phenotypes: Genetics core aims, progress, and plans. Alzheimers Dement. 2010, 6, 265–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, H.E.; O’Connell, A. Dementia: The estimation of premorbid intelligence levels using the New Adult Reading Test. Cortex 1978, 14, 234–244. [Google Scholar] [CrossRef]
- Grober, E.; Sliwinski, M. Development and validation of a model for estimating premorbid verbal intelligence in the elderly. J. Clin. Exp. Neuropsychol. 1991, 13, 933–949. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bernstein, M.A.; Fox, N.C.; Thompson, P.; Alexander, G.; Harvey, D.; Borowski, B.; Britson, P.J.J.L.W.; Ward, C.; Dale, A.M.; et al. The Alzheimer’s Disease Neuroimaging Initiative (ADNI): MRI methods. J. Magn. Reson. Imag. 2008, 27, 685–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischl, B.; Dale, A.M. Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proc. Natl. Acad. Sci. USA 2000, 97, 11050–11055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischl, B.; Sereno, M.I.; Tootell, R.B.; Dale, A.M. High-resolution intersubject averaging and a coordinate system for the cortical surface. Hum. Brain Mapp 1999, 8, 272–284. [Google Scholar] [CrossRef]
- Courchesne, E.; Chisum, H.J.; Townsend, J.; Cowles, A.; Covington, J.; Egaas, B.; Harwood, M.; Hinds, S.; Press, G.A. Normal brain development and aging: Quantitative analysis at in vivo MR imaging in healthy volunteers. Radiology 2000, 216, 672–682. [Google Scholar] [CrossRef]
- Lenroot, R.K.; Giedd, J.N. Brain development in children and adolescents: Insights from anatomical magnetic resonance imaging. Neurosci. Biobehav. Rev. 2006, 30, 718–729. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Landau, S.M.; Mintun, M.A.; Joshi, A.D.; Koeppe, R.A.; Petersen, R.C.; Aisen, P.S.; Weiner, M.W.; Jagust, W.J. Alzheimer’s Disease Neuroimaging Initiative. Amyloid deposition, hypometabolism, and longitudinal cognitive decline. Ann. Neurol. 2012, 72, 578–586. [Google Scholar] [CrossRef]
- Knopman, D.S.; Lundt, E.S.; Therneau, T.M.; Vemuri, P.; Lowe, V.J.; Kantarci, K.; Gunter, J.L.; Senjem, M.L.; Mielke, M.M.; Machulda, M.M.; et al. Entorhinal cortex tau, amyloid-beta, cortical thickness and memory performance in non-demented subjects. Brain 2019, 142, 1148–1160. [Google Scholar] [CrossRef]
- Das, S.R.; Xie, L.; Wisse, L.E.M.; Ittyerah, R.; Tustison, N.J.; Dickerson, B.C.; Yushkevich, P.A.; Wolk, D.A. Alzheimer’s Disease Neuroimaging Initiative. Longitudinal and cross-sectional structural magnetic resonance imaging correlates of AV-1451 uptake. Neurobiol. Aging 2018, 66, 49–58. [Google Scholar] [CrossRef] [Green Version]
- Jueptner, M.; Weiller, C. Review: Does measurement of regional cerebral blood flow reflect synaptic activity? Implications for PET and fMRI. Neuroimage 1995, 2, 148–156. [Google Scholar] [CrossRef]
- Sokoloff, L. Relationships among local functional activity, energy metabolism, and blood flow in the central nervous system. Fed Proc. 1981, 40, 2311–2316. [Google Scholar]
- Kim, M.J.; Lee, K.M.; Son, Y.D.; Jeon, H.A.; Kim, Y.B.; Cho, Z.H. Increased basal forebrain metabolism in mild cognitive impairment: An evidence for brain reserve in incipient dementia. J. Alzheimers Dis. 2012, 32, 927–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverman, D.H.; Small, G.W.; Chang, C.Y.; Lu, C.S.; Kung De Aburto, M.A.; Chen, W.; Czernin, J.; Rapoport, S.I.; Pietrini, P.; Alexander, G.E.; et al. Positron emission tomography in evaluation of dementia: Regional brain metabolism and long-term outcome. JAMA 2001, 286, 2120–2127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toro, R.; Perron, M.; Pike, B.; Richer, L.; Veillette, S.; Pausova, Z.; Paus, T. Brain size and folding of the human cerebral cortex. Cereb. Cortex 2008, 18, 2352–2357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida-Meza, P.; Steptoe, A.; Cadar, D. Markers of cognitive reserve and dementia incidence in the English Longitudinal Study of Ageing. Br. J. Psychiatry 2020, 1–9. [Google Scholar]
- Soldan, A.; Pettigrew, C.; Cai, Q.; Wang, J.; Wang, M.C.; Moghekar, A.; Miller, M.I.; Albert, M.; Team, B.R. Cognitive reserve and long-term change in cognition in aging and preclinical Alzheimer’s disease. Neurobiol. Aging 2017, 60, 164–172. [Google Scholar] [CrossRef]
- Rouillard, M.; Audiffren, M.; Albinet, C.; Ali Bahri, M.; Garraux, G.; Collette, F. Contribution of four lifelong factors of cognitive reserve on late cognition in normal aging and Parkinson’s disease. J. Clin. Exp. Neuropsychol. 2017, 39, 142–162. [Google Scholar] [CrossRef]
- Lane, A.P.; Windsor, T.D.; Andel, R.; Luszcz, M.A. Is Occupational Complexity Associated with Cognitive Performance or Decline? Results from the Australian Longitudinal Study of Ageing. Gerontology 2017, 63, 550–559. [Google Scholar] [CrossRef]

{kind=link}
| Progressive Group (n = 87) | Stable Group (n = 33) | F | χ2 | p Value | |
|---|---|---|---|---|---|
| Age (SD), y | 74.59 (8.07) | 73.57 (8.01) | 0.38 | 0.54 | |
| a Education (SD), y | 15.55 (2.57) | 16.21 (2.75) | 1.52 | 0.22 | |
| Female, n (%) | 34 (39.1) | 16 (48.5) | 0.87 | 0.35 | |
| FU, month | 11.68 (5.88) | 10.73 (5.69) | 0.64 | 0.43 | |
| CDR, 0.5, n (%) | 37 (42.5) | 16 (48.5) | 0.67 | 0.72 | |
| 1.0 | 49 (56.3) | 17 (51.5) | |||
| 2.0 | 1 (1.1) | 0 (0.0) | |||
| CDR SB | 4.51 (1.67) | 4.35 (1.64) | 0.23 | 0.63 | |
| APOE ε+, n (%) | 59 (67.8) | 23 (69.7) | 0.04 | 0.84 | |
| Aβ+, n (%) | 77 (88.5) | 27 (81.8) | 0.93 | 0.34 | |
| MMSE | 22.78 (2.01) | 23.67 (2.13) | 4.49 | 0.04 | |
| FAQ | 13.84 (7.07) | 10.91 (6.66) | 4.22 | 0.04 | |
| a ANARTERR | 17.78 (9.13) | 13.94 (9.54) | 4.14 | 0.04 | |
| b FDG-ROIs | 5.28 (0.68) | 5.61 (0.88) | 4.78 | 0.03 | |
| HC | 5847.26 (852.97) | 6008.33 (1079.91) | 0.55 | 0.46 | |
| EC | 2895.81 (658.73) | 3006.46 (678.28) | 0.49 | 0.48 | |
| b ICV | 1,502,750.00 (172,421.14) | 1,527,000.00 (181,148.63) | 0.42 | 0.52 |
| B | SE B | Wald | p Value | HR | 95% CI | ||
|---|---|---|---|---|---|---|---|
| Lower | Upper | ||||||
| Total group | |||||||
| Age | 0.018 | 0.015 | 1.485 | 0.223 | 1.018 | 0.989 | 1.049 |
| Gender | −0.098 | 0.243 | 0.162 | 0.688 | 0.907 | 0.563 | 1.460 |
| MMSE | 0.014 | 0.061 | 0.055 | 0.815 | 1.027 | 0.993 | 1.062 |
| FAQ | 0.027 | 0.017 | 2.481 | 0.115 | 1.027 | 0.993 | 1.062 |
| FDG-ROIs 0: lower * 1: higher | −0.577 | 0.271 | 4.532 | 0.033 | 0.562 | 0.330 | 0.955 |
| Larger ICV group only | |||||||
| Age | 0.004 | 0.029 | 0.021 | 0.884 | 1.004 | 0.949 | 1.062 |
| Gender | 0.057 | 0.554 | 0.011 | 0.918 | 1.059 | 0.357 | 3.138 |
| MMSE | −0.062 | 0.084 | 0.546 | 0.460 | 0.940 | 0.797 | 1.108 |
| FAQ | 0.035 | 0.026 | 1.807 | 0.179 | 1.036 | 0.984 | 1.091 |
| FDG-ROIs 0: lower * 1: higher | −0.947 | 0.430 | 4.838 | 0.028 | 0.388 | 0.167 | 0.902 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoon, H.-J.; Kim, S.-G.; Kim, S.H.; Woo, J.I.; Seo, E.H.; For the Alzheimer’s Disease Neuroimaing Initiative. Associations between Brain Reserve Proxies and Clinical Progression in Alzheimer’s Disease Dementia. Int. J. Environ. Res. Public Health 2021, 18, 12159. https://doi.org/10.3390/ijerph182212159
Yoon H-J, Kim S-G, Kim SH, Woo JI, Seo EH, For the Alzheimer’s Disease Neuroimaing Initiative. Associations between Brain Reserve Proxies and Clinical Progression in Alzheimer’s Disease Dementia. International Journal of Environmental Research and Public Health. 2021; 18(22):12159. https://doi.org/10.3390/ijerph182212159
Chicago/Turabian StyleYoon, Hyung-Jun, Seung-Gon Kim, Sang Hoon Kim, Jong Inn Woo, Eun Hyun Seo, and For the Alzheimer’s Disease Neuroimaing Initiative. 2021. "Associations between Brain Reserve Proxies and Clinical Progression in Alzheimer’s Disease Dementia" International Journal of Environmental Research and Public Health 18, no. 22: 12159. https://doi.org/10.3390/ijerph182212159
APA StyleYoon, H.-J., Kim, S.-G., Kim, S. H., Woo, J. I., Seo, E. H., & For the Alzheimer’s Disease Neuroimaing Initiative. (2021). Associations between Brain Reserve Proxies and Clinical Progression in Alzheimer’s Disease Dementia. International Journal of Environmental Research and Public Health, 18(22), 12159. https://doi.org/10.3390/ijerph182212159