
Towards A Novel Multi-Epitopes Chimeric Vaccine for Simulating Strong Immune Responses and Protection against Morganella morganii


 ,
,  ,
,  ,
, 
Abstract
1. Introduction
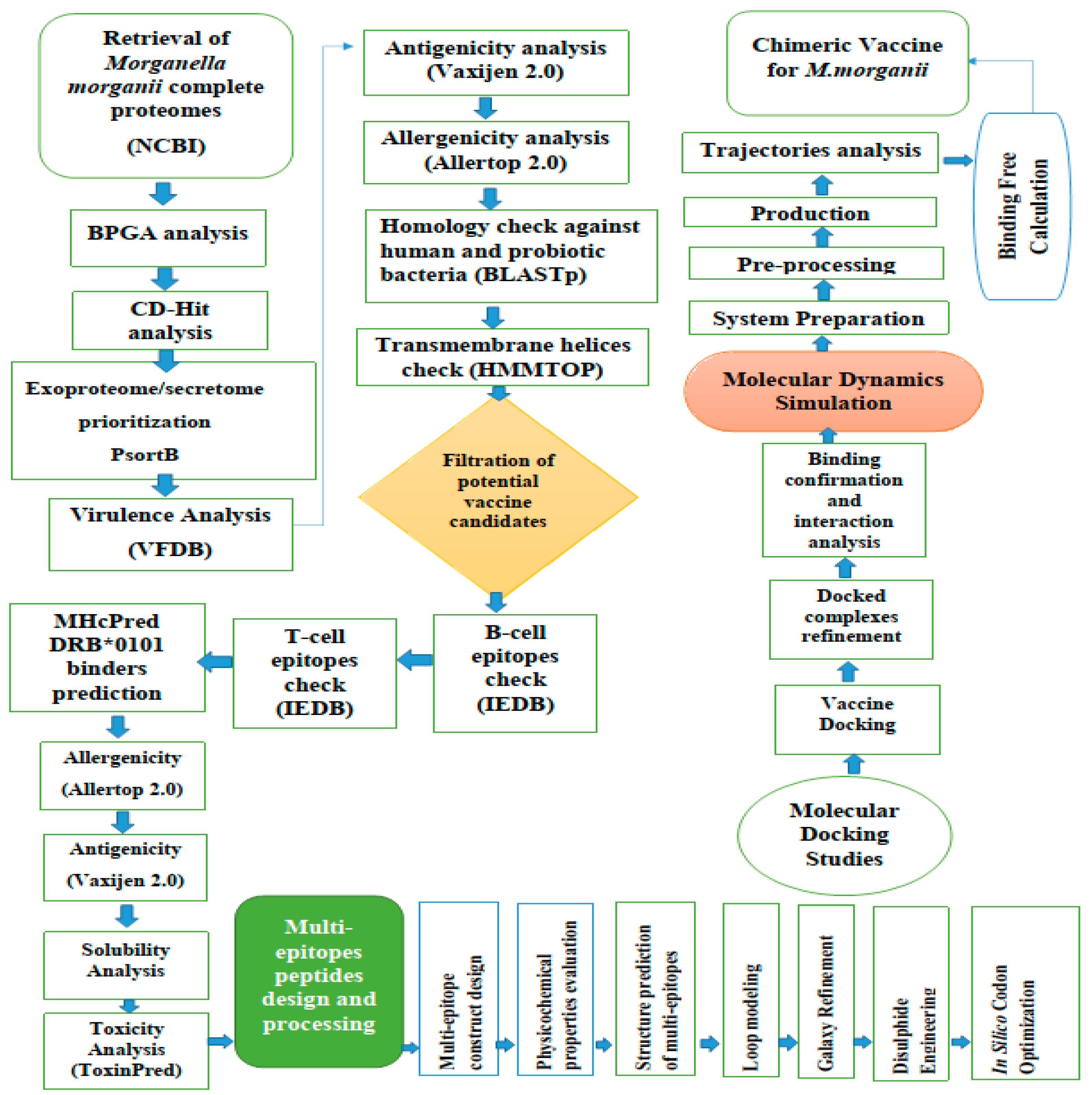
2. Materials and Methods
2.1. M. Morganii Predicted Proteomes Retrieval
2.2. Subtraction of Core Proteins
2.3. The Prioritization Phase of the Vaccine Targets
2.3.1. Epitope Prediction Phase
2.3.2. Multi-Epitope Vaccine Design and Processing
2.3.3. Physiochemical Properties of the Final Vaccine Construct
2.4. Structure Modelling of the Vaccine
2.5. Disulfide Engineering and Codon Optimization
2.6. Molecular Docking and Refinement
2.7. Molecular Dynamics Simulations
2.8. Binding Free Energies Calculation
2.9. C-immune Simulations
3. Results
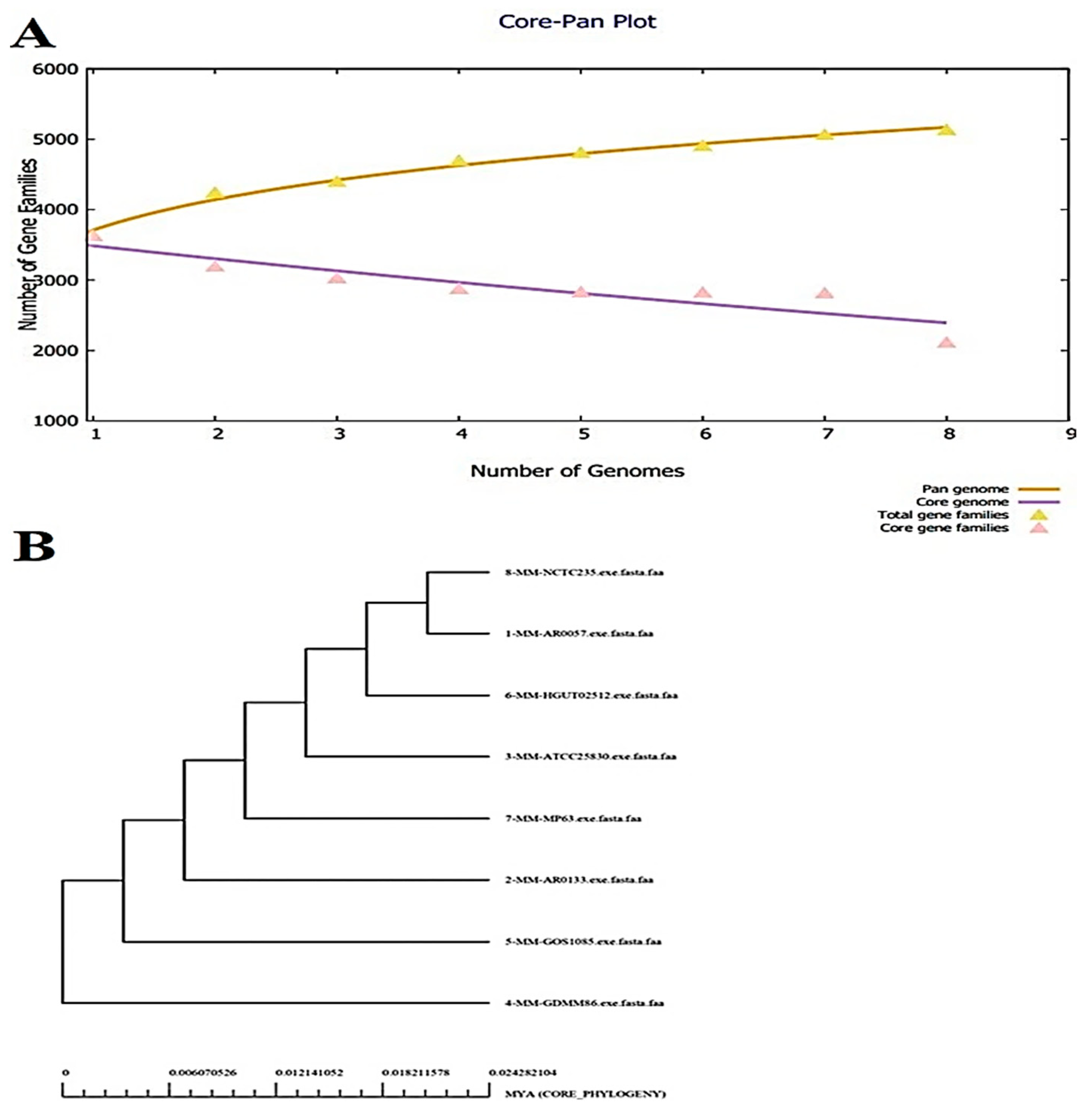
3.1. Bacterial Pan-genome Analysis (BPGA)
3.2. CD-Hit Analysis
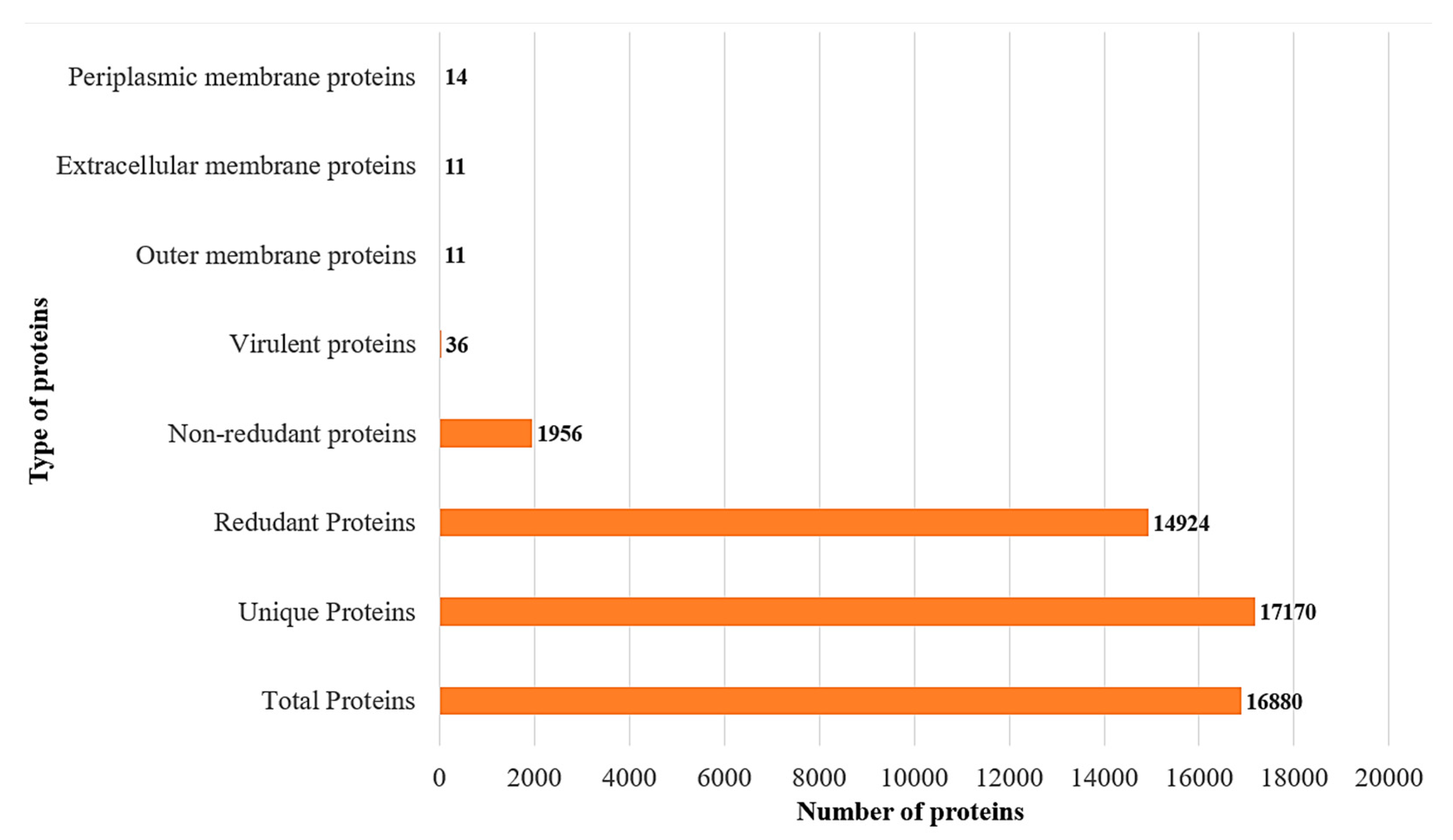
3.3. Subcellular Localization and Virulence Analysis
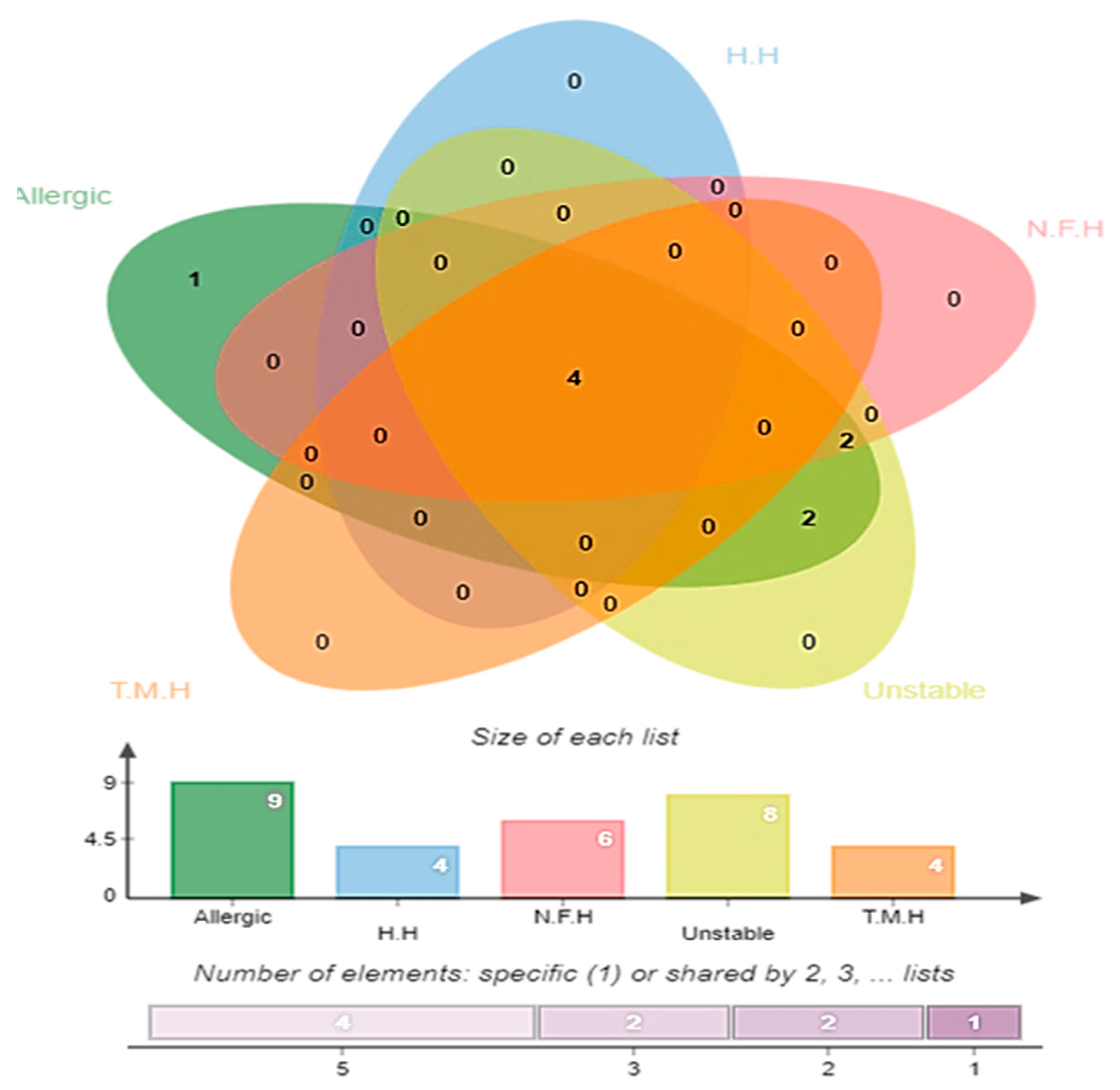
3.4. Antigenicity, Allergenicity, Human and Normal Microbiota Homology, and Transmembrane Helices and Stability Analysis
3.5. B-Cell and T-Cell Epitopes Prediction
3.5.1. B-Cell Epitope Prediction
3.5.2. B-Cell-Derived T-Cell Epitope Prediction
3.6. Epitope Prioritization Phase
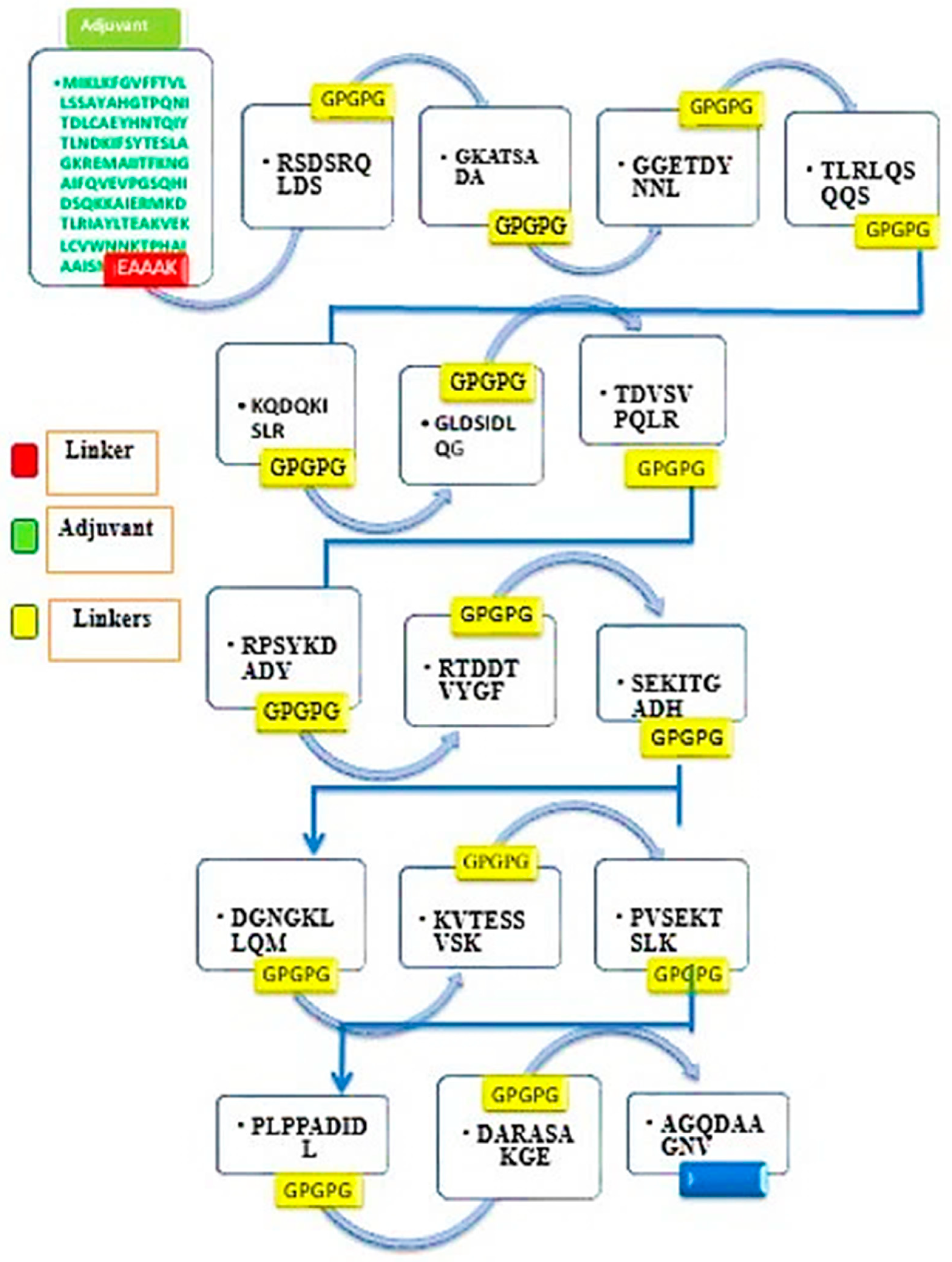
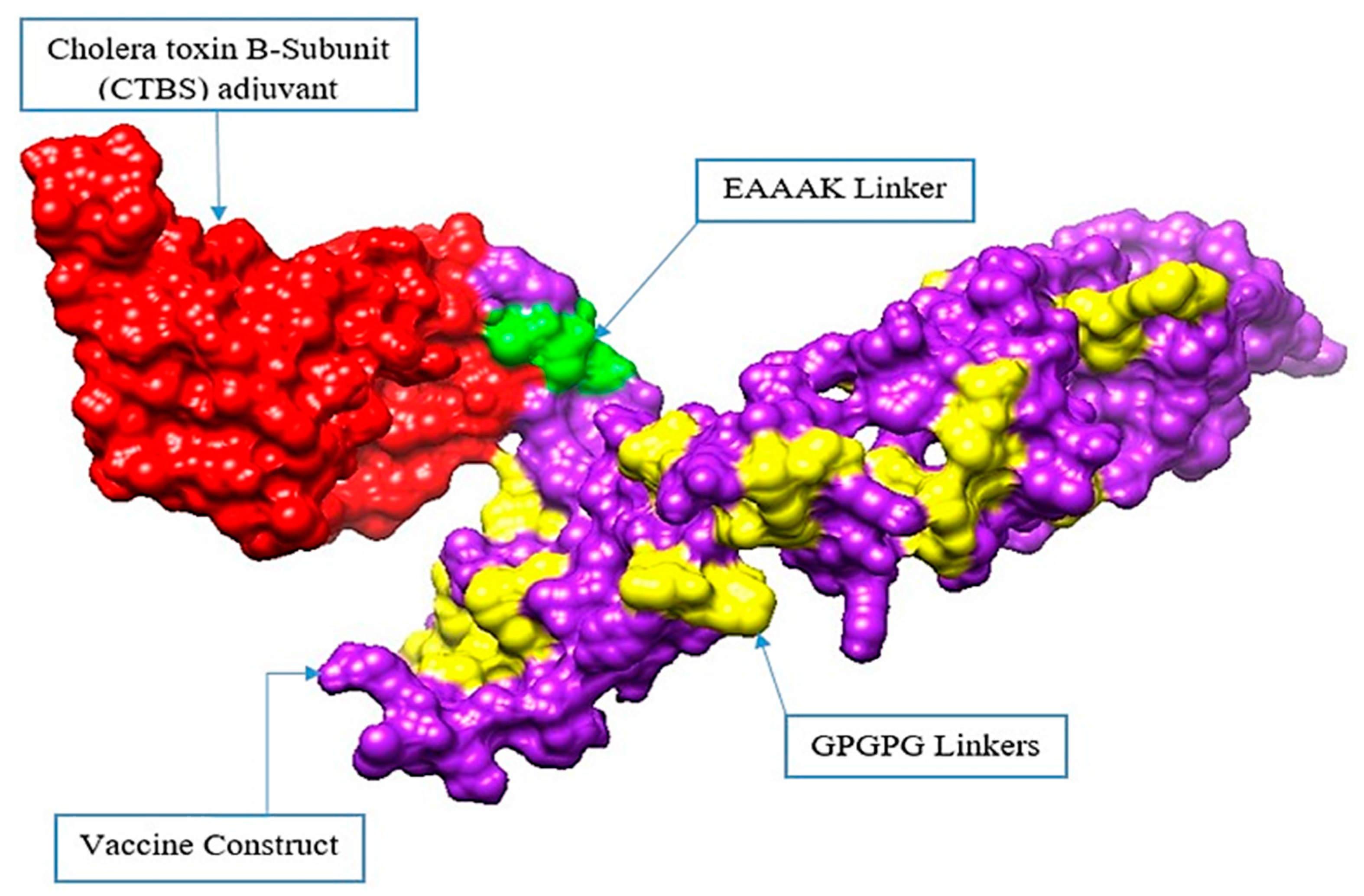
3.7. Multi-Epitope Vaccine Construction
3.8. Structure Prediction, Loop Modelling, and Refinement
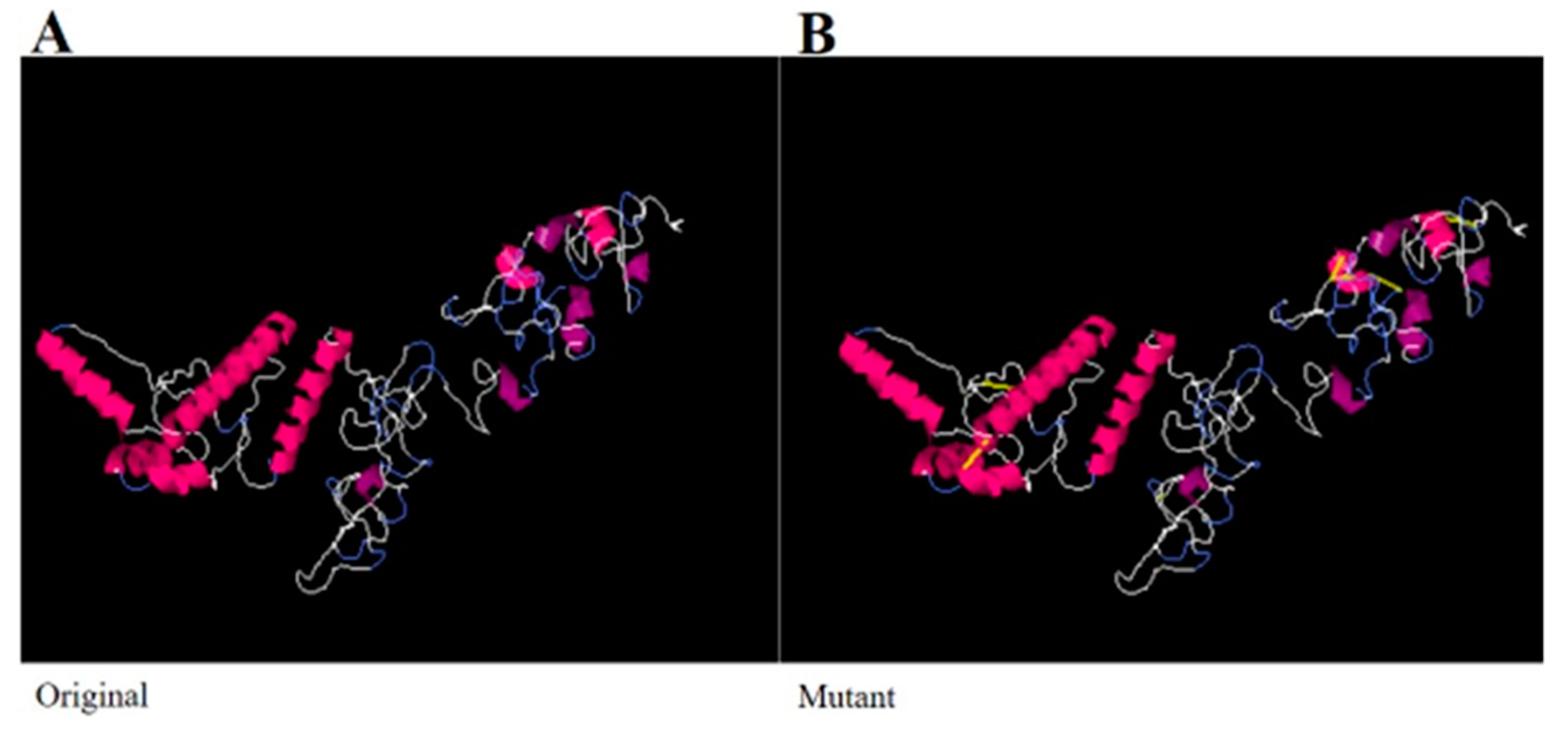
3.9. Disulfide Engineering and Codon Optimization
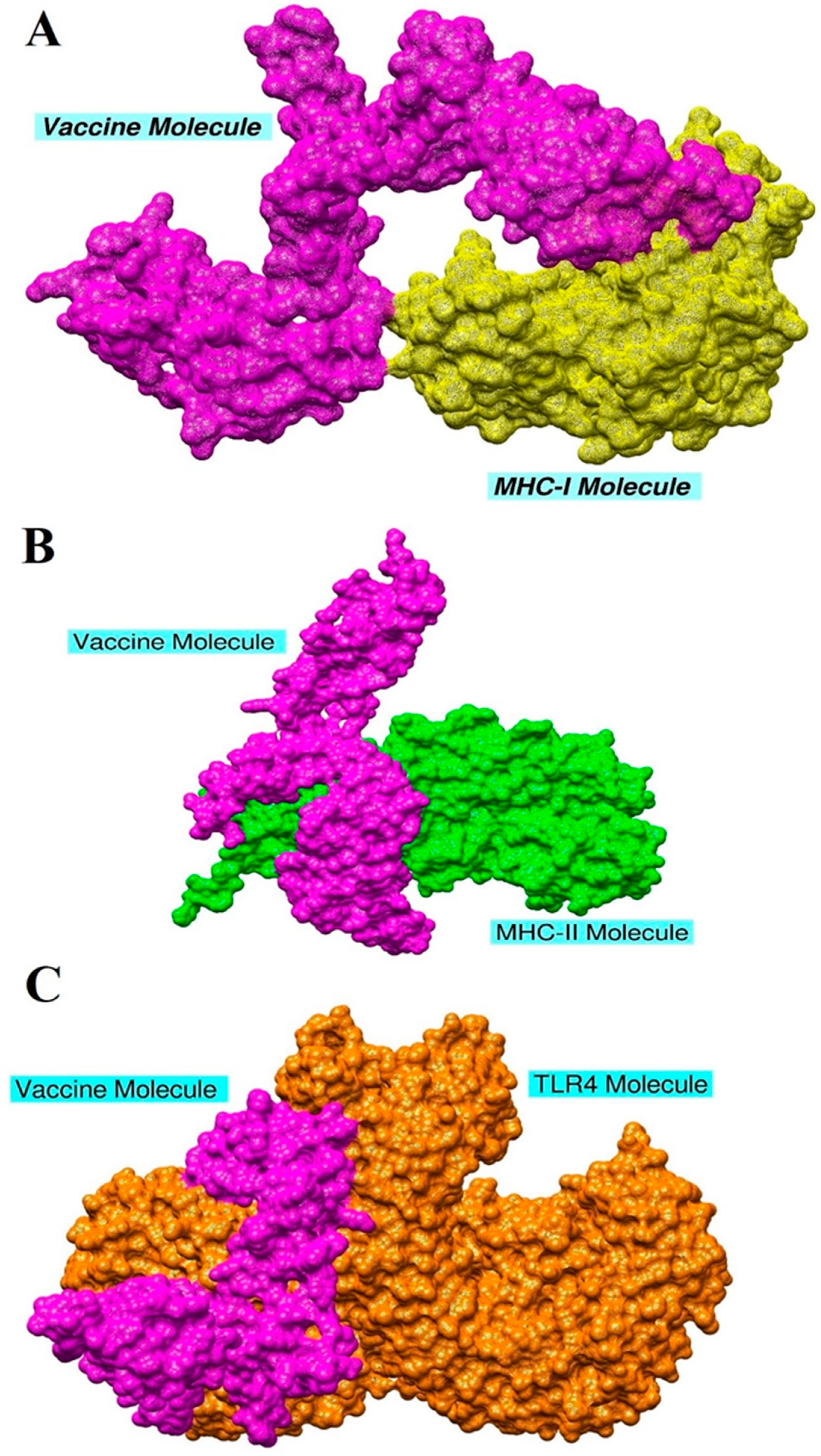
3.10. Molecular Docking and Refinement
3.11. Chemical Interactions of the Vaccine with MHC-I, MHC-II, and TLR-4
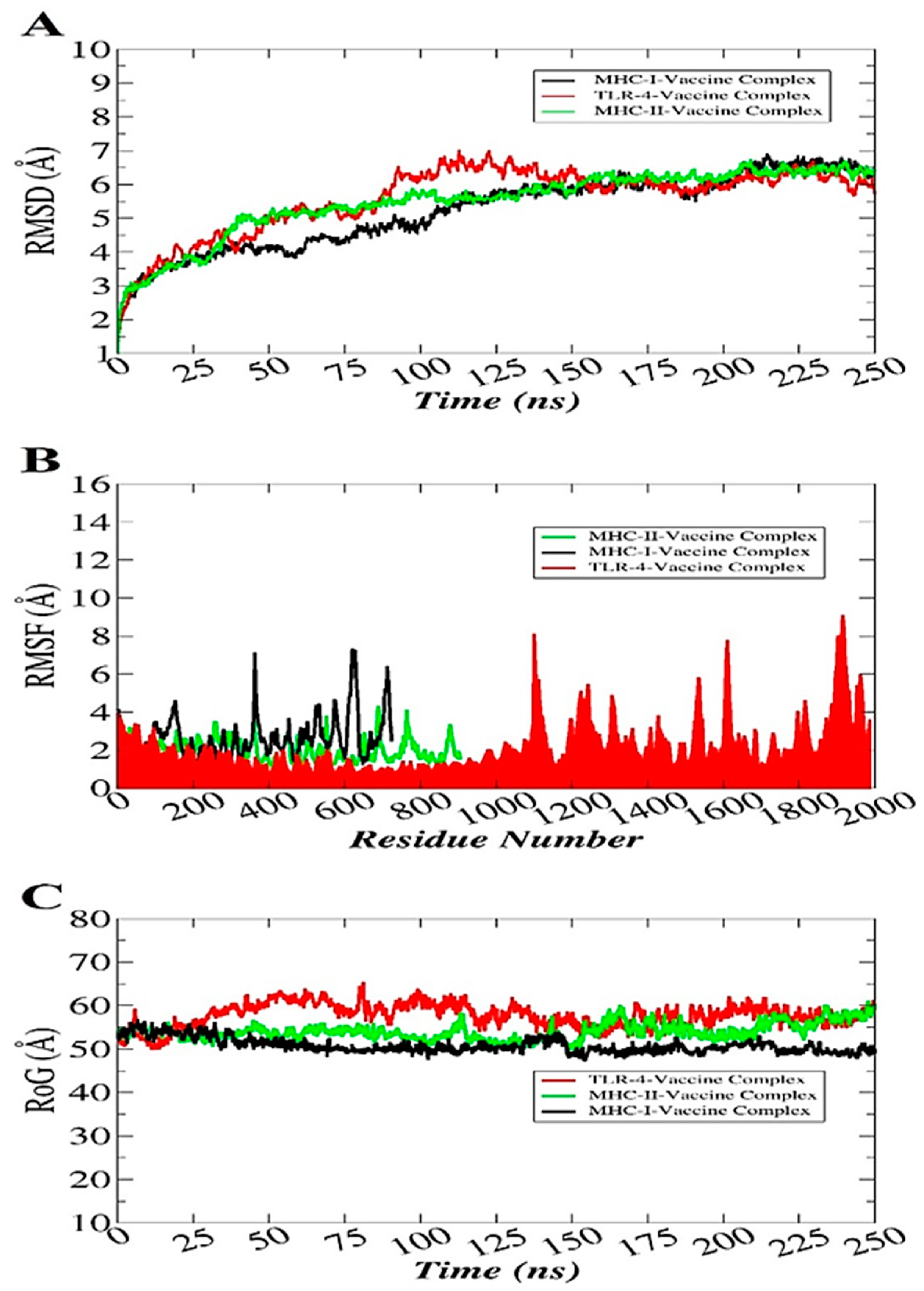
3.12. Molecular Dynamics Simulation
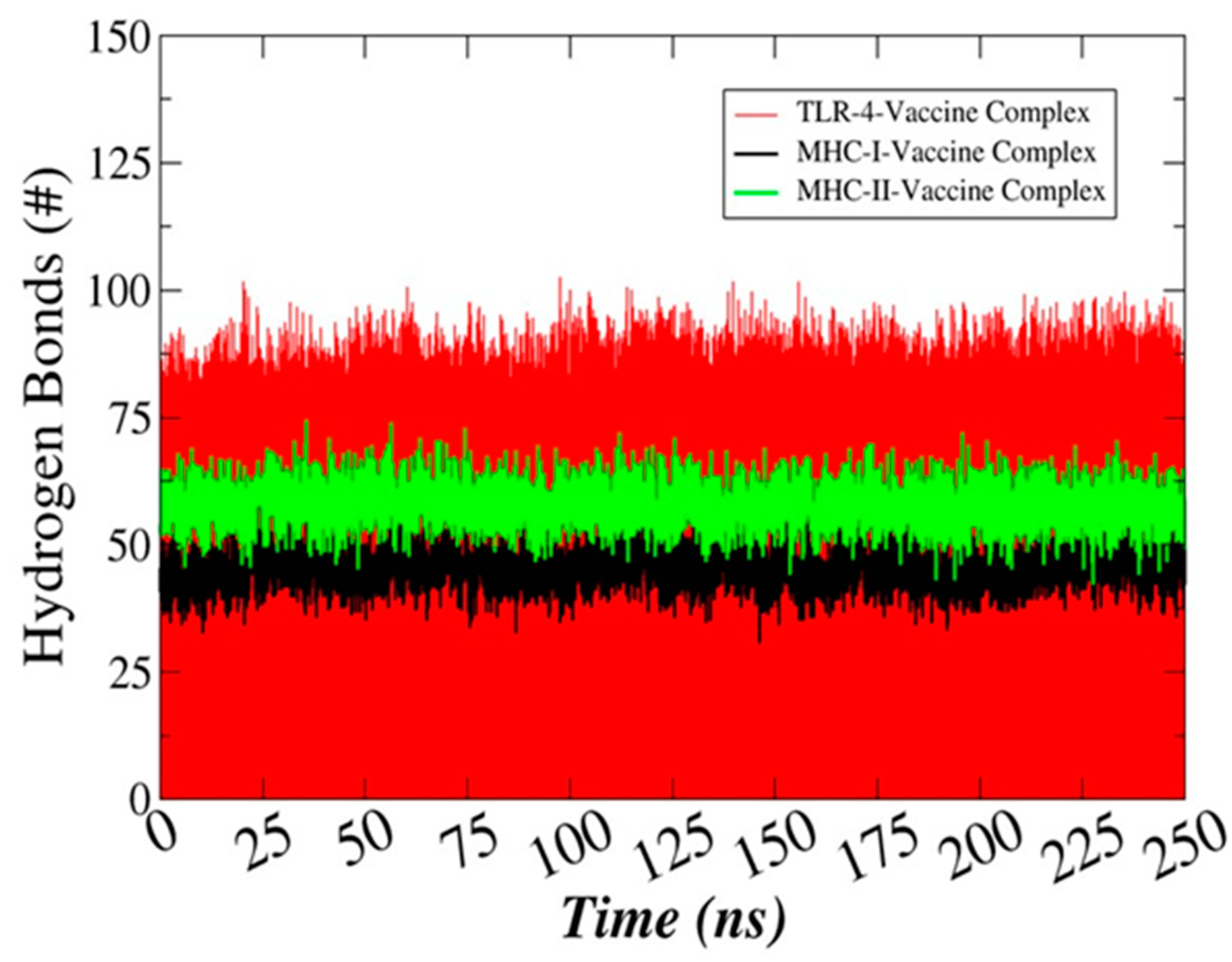
3.13. Hydrogen Bonds Analysis
3.14. Determination of the Binding Free Energies
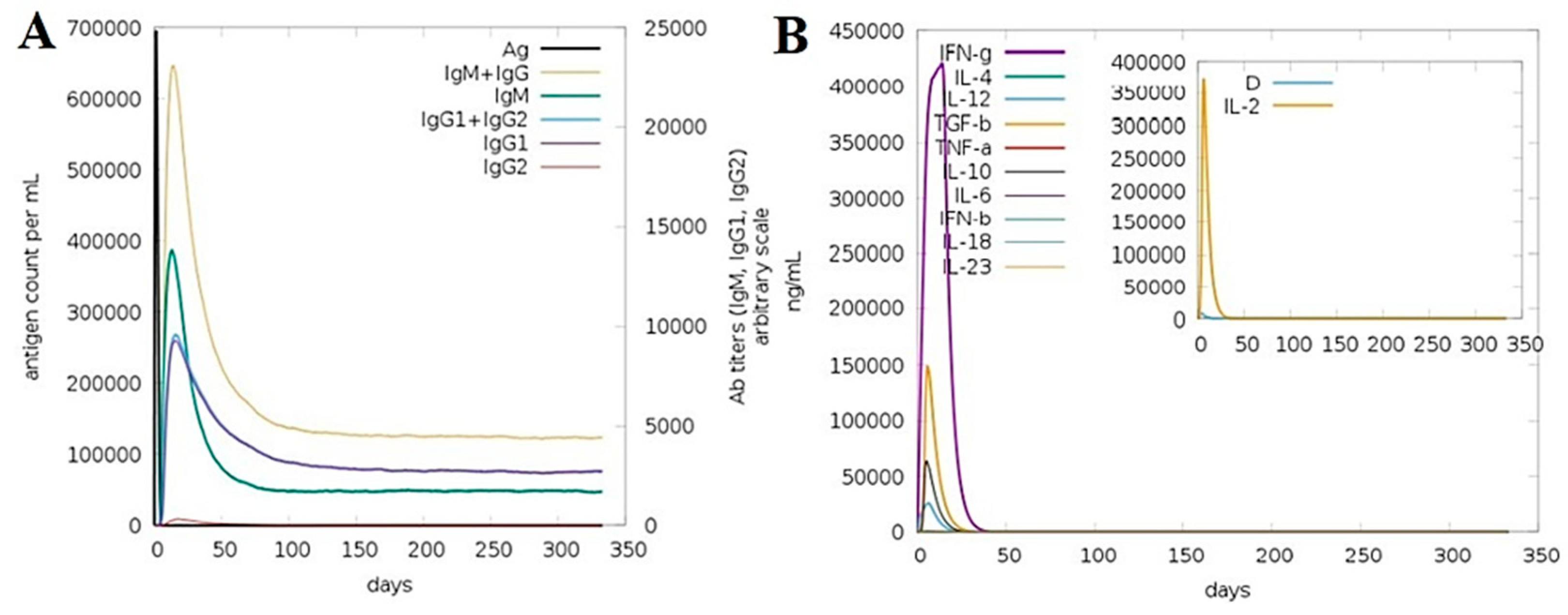
3.15. Computationally Immune Simulations
4. Discussion
5. Conclusions and Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Edwards, M.; Hamilton, R.; Oliver, N.; Fitzgibbon, S.; Samarasekera, R. Antibiotic Resistance: Modelling the Impact on Mortality and Morbidity. Institute and Faculty of Actuaries: London, UK, 2019. [Google Scholar]
- Sifri, Z.; Chokshi, A.; Cennimo, D.; Horng, H. Global contributors to antibiotic resistance. J. Glob. Infect. Dis. 2019, 11, 36–42. [Google Scholar] [CrossRef]
- Ventola, C.L. The antibiotic resistance crisis: Part 2: Management strategies and new agents. Pharm. Ther. 2015, 40, 344. [Google Scholar]
- Ventola, C.L. The antibiotic resistance crisis: Part 1: Causes and threats. Pharm. Ther. 2015, 40, 277. [Google Scholar]
- Andreano, E.; D’Oro, U.; Rappuoli, R.; Finco, O. Vaccine Evolution and Its Application to Fight Modern Threats. Front. Immunol. 2019, 10, 1722. [Google Scholar] [CrossRef] [PubMed]
- Moriel, D.G.; Scarselli, M.; Serino, L.; Mora, M.; Rappuoli, R.; Masignani, V. Genome-based vaccine development: A short cut for the future. Hum. Vaccines 2008, 4, 184–188. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Baseer, S.; Ahmad, S.; Ranaghan, K.E.; Azam, S.S. Towards a peptide-based vaccine against Shigella sonnei: A subtractive reverse vaccinology based approach. Biology 2017, 50, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Mora, M.; Veggi, D.; Santini, L.; Pizza, M.; Rappuoli, R. Reverse vaccinology. Drug Discov. Today 2003, 8, 459–464. [Google Scholar] [CrossRef]
- Qamar, M.T.U.; Ismail, S.; Ahmad, S.; Mirza, M.U.; Abbasi, S.W.; Ashfaq, U.A.; Chen, L.-L. Development of a Novel Multi-Epitope Vaccine Against Crimean-Congo Hemorrhagic Fever Virus: An Integrated Reverse Vaccinology, Vaccine Informatics and Biophysics Approach. Front. Immunol. 2021, 12, 12. [Google Scholar] [CrossRef]
- Ali, A.; Naz, A.; Soares, S.C.; Bakhtiar, M.; Tiwari, S.; Hassan, S.S.; Hanan, F.; Ramos, R.; Pereira, U.; Barh, D.; et al. Pan-Genome Analysis of Human Gastric Pathogen H. pylori: Comparative Genomics and Pathogenomics Approaches to Identify Regions Associated with Pathogenicity and Prediction of Potential Core Therapeutic Targets. BioMed Res. Int. 2015, 2015, 1–17. [Google Scholar] [CrossRef]
- Adu-Bobie, J.; Capecchi, B.; Serruto, D.; Rappuoli, R.; Pizza, M. Two years into reverse vaccinology. Vaccine 2003, 21, 605–610. [Google Scholar] [CrossRef]
- Fernandes, L.G.V.; Teixeira, A.F.; Filho, A.F.; Souza, G.O.; Vasconcellos, S.A.; Heinemann, M.B.; Romero, E.C.; Nascimento, A.L.T.O. Immune response and protective profile elicited by a multi-epitope chimeric protein derived from Leptospira interrogans. Int. J. Infect. Dis. 2017, 57, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Naz, K.; Naz, A.; Ashraf, S.T.; Rizwan, M.; Ahmad, J.; Baumbach, J.; Ali, A. PanRV: Pangenome-reverse vaccinology approach for identifications of potential vaccine candidates in microbial pangenome. BMC Bioinform. 2019, 20, 123. [Google Scholar] [CrossRef] [PubMed]
- Nuccitelli, A.; Rinaudo, C.D.; Maione, D. Group B Streptococcus vaccine: State of the art. Ther. Adv. Vaccines 2015, 3, 76–90. [Google Scholar] [CrossRef]
- Mbelle, N.; Sekyere, J.O.; Feldman, C.; Maningi, N.; Modipane, L.; Essack, S. Genomic analysis of two drug-resistant clinical Morganella morganii strains isolated from UTI patients in Pretoria, South Africa. Lett. Appl. Microbiol. 2020, 70, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Coordinators, N.R. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2017, 45, D12–D17. [Google Scholar] [CrossRef]
- Chaudhari, N.M.; Gupta, V.; Dutta, C. BPGA- an ultra-fast pan-genome analysis pipeline. Sci. Rep. 2016, 6, 24373. [Google Scholar] [CrossRef]
- Qamar, M.T.U.; Zhu, X.; Khan, M.S.; Xing, F.; Chen, L. Pan-genome: A promising resource for noncoding RNA discovery in plants. Plant. Genome 2020, 13, e20046. [Google Scholar] [CrossRef]
- Ismail, S.; Shahid, F.; Khan, A.; Bhatti, S.; Ahmad, S.; Naz, A.; Almatroudi, A.; Qamar, M.T.U. Pan-vaccinomics approach towards a universal vaccine candidate against WHO priority pathogens to address growing global antibiotic resistance. Comput. Biol. Med. 2021, 136, 104705. [Google Scholar] [CrossRef]
- Ahmad, S.; Raza, S.; Uddin, R.; Azam, S.S. Comparative subtractive proteomics based ranking for antibiotic targets against the dirtiest superbug: Acinetobacter baumannii. J. Mol. Graph. Model. 2018, 82, 74–92. [Google Scholar] [CrossRef]
- Qamar, M.T.U.; Ahmad, S.; Fatima, I.; Ahmad, F.; Shahid, F.; Naz, A.; Abbasi, S.W.; Khan, A.; Mirza, M.U.; Ashfaq, U.A.; et al. Designing multi-epitope vaccine against Staphylococcus aureus by employing subtractive proteomics, reverse vaccinology and immuno-informatics approaches. Comput. Biol. Med. 2021, 132, 104389. [Google Scholar] [CrossRef]
- Yu, N.; Wagner, J.R.; Laird, M.; Melli, G.; Rey, S.; Lo, R.; Dao, P.; Sahinalp, S.C.; Ester, M.; Foster, L.J.; et al. PSORTb 3.0: Improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 2010, 26, 1608–1615. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v. 2—A server for in silico prediction of allergens. J. Mol. Model. 2014, 20, 2278. [Google Scholar] [CrossRef]
- Blast, N. Basic local alignment search tool. Natl. Libr. Med. Natl. Cent. Biotechnol. Inf. 2015, 43(D1), D6–D17. [Google Scholar]
- Chen, Y.; Yu, P.; Luo, J.; Jiang, Y. Secreted protein prediction system combining CJ-SPHMM, TMHMM, and PSORT. Mamm. Genome 2003, 14, 859–865. [Google Scholar] [CrossRef]
- Ahmad, S.; Ranaghan, K.E.; Azam, S.S. Combating tigecycline resistant Acinetobacter baumannii: A leap forward towards multi-epitope based vaccine discovery. Eur. J. Pharm. Sci. 2019, 132, 1–17. [Google Scholar] [CrossRef]
- ProtParam, E. ExPASy-ProtParam Tool 2017.
- Nezafat, N.; Karimi, Z.; Eslami, M.; Mohkam, M.; Zandian, S.; Ghasemi, Y. Designing an efficient multi-epitope peptide vaccine against Vibrio cholerae via combined immunoinformatics and protein interaction based approaches. Comput. Biol. Chem. 2016, 62, 82–95. [Google Scholar] [CrossRef]
- Mahapatra, S.R.; Sahoo, S.; Dehury, B.; Raina, V.; Patro, S.; Misra, N.; Suar, M. Designing an efficient multi-epitope vaccine displaying interactions with diverse HLA molecules for an efficient humoral and cellular immune response to prevent COVID-19 infection. Expert Rev. Vaccines 2020, 19, 871–885. [Google Scholar] [CrossRef] [PubMed]
- Sajjad, R.; Ahmad, S.; Azam, S.S. In silico screening of antigenic B-cell derived T-cell epitopes and designing of a multi-epitope peptide vaccine for Acinetobacter nosocomialis. J. Mol. Graph. Model. 2020, 94, 107477. [Google Scholar] [CrossRef]
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: Improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017, 45, W24–W29. [Google Scholar] [CrossRef] [PubMed]
- Vashi, Y.; Jagrit, V.; Kumar, S. Understanding the B and T cell epitopes of spike protein of severe acute respiratory syndrome coronavirus-2: A computational way to predict the immunogens. Infect. Genet. Evol. 2020, 84, 104382. [Google Scholar] [CrossRef]
- Vita, R.; Overton, J.A.; Greenbaum, J.A.; Ponomarenko, J.; Clark, J.D.; Cantrell, J.R.; Wheeler, D.K.; Gabbard, J.L.; Hix, D.; Sette, A.; et al. The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 2015, 43, D405–D412. [Google Scholar] [CrossRef] [PubMed]
- Ismail, S.; Ahmad, S.; Azam, S.S. Immunoinformatics characterization of SARS-CoV-2 spike glycoprotein for prioritization of epitope based multivalent peptide vaccine. J. Mol. Liq. 2020, 314, 113612. [Google Scholar] [CrossRef]
- Naz, A.; Awan, F.M.; Obaid, A.; Muhammad, S.A.; Paracha, R.Z.; Ahmad, J.; Ali, A. Identification of putative vaccine candidates against Helicobacter pylori exploiting exoproteome and secretome: A reverse vaccinology based approach. Infect. Genet. Evol. 2015, 32, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Raghava, G.P.S.; Consortium, O.S.D.D. Others in silico approach for predicting toxicity of peptides and proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef]
- Hon, J.; Marusiak, M.; Martinek, T.; Kunka, A.; Zendulka, J.; Bednar, D.; Damborsky, J. SoluProt: Prediction of soluble protein expression in Escherichia coli. Bioinformatics 2021, 37, 23–28. [Google Scholar] [CrossRef]
- Li, W.; Joshi, M.D.; Singhania, S.; Ramsey, K.H.; Murthy, A.K. Peptide Vaccine: Progress and Challenges. Vaccines 2014, 2, 515–536. [Google Scholar] [CrossRef]
- Zhang, L. Multi-epitope vaccines: A promising strategy against tumors and viral infections. Cell. Mol. Immunol. 2018, 15, 182–184. [Google Scholar] [CrossRef]
- Jafari, E.; Mahmoodi, S. Design, expression, and purification of a multi-epitope vaccine against Helicobacter pylori based on Melittin as an adjuvant. Microb. Pathog. 2021, 157, 104970. [Google Scholar] [CrossRef]
- Stratmann, T. Cholera Toxin Subunit B as Adjuvant—An Accelerator in Protective Immunity and a Break in Autoimmunity. Vaccines 2015, 3, 579–596. [Google Scholar] [CrossRef]
- Cheng, J.; Randall, A.Z.; Sweredoski, M.J.; Baldi, P. SCRATCH: A protein structure and structural feature prediction server. Nucleic Acids Res. 2005, 33, W72–W76. [Google Scholar] [CrossRef]
- Giardine, B.; Riemer, C.; Hardison, R.; Burhans, R.; Elnitski, L.; Shah, P.; Zhang, Y.; Blankenberg, D.; Albert, I.; Taylor, J.; et al. Galaxy: A platform for interactive large-scale genome analysis. Genome Res. 2005, 15, 1451–1455. [Google Scholar] [CrossRef]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013, 41, W384–W388. [Google Scholar] [CrossRef]
- Craig, D.B.; Dombkowski, A.A. Disulfide by Design 2.0: A web-based tool for disulfide engineering in proteins. BMC Bioinform. 2013, 14, 346. [Google Scholar] [CrossRef]
- Angov, E. Codon usage: Nature’s roadmap to expression and folding of proteins. Biotechnol. J. 2011, 6, 650–659. [Google Scholar] [CrossRef]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, B.; Hempel, D.C.; Jahn, D. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005, 33, W526–W531. [Google Scholar] [CrossRef] [PubMed]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [PubMed]
- Mashiach, E.; Schneidman-Duhovny, D.; Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: A web server for fast interaction refinement in molecular docking. Nucleic Acids Res. 2008, 36, W229–W232. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Belfon, K.; Ben-Shalom, I.; Brozell, S.R.; Cerutti, D.; Cheatham, T.; Cruzeiro, V.W.D.; Darden, T.; Duke, R.E.; Giambasu, G.; et al. Amber; University of California: San Francisco, CA, USA, 2020; Volume 2020. [Google Scholar]
- Ismail, S.; Ahmad, S.; Azam, S.S. Vaccinomics to design a novel single chimeric subunit vaccine for broad-spectrum immunological applications targeting nosocomial Enterobacteriaceae pathogens. Eur. J. Pharm. Sci. 2020, 146, 105258. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Antechamber: An accessory software package for molecular mechanical calculations. J. Am. Chem. Soc. 2001, 222, U403. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Izaguirre, J.A.; Catarello, D.P.; Wozniak, J.M.; Skeel, R.D. Langevin stabilization of molecular dynamics. J. Chem. Phys. 2001, 114, 2090–2098. [Google Scholar] [CrossRef]
- van Gunsteren, W.F.; Kräutler, V.; Hünenberger, P.H. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 2001, 22, 501–508. [Google Scholar] [CrossRef]
- Aslyamov, T.; Akhatov, I. Zeros of partition functions in the N P T ensemble. Phys. Rev. E 2019, 100, 52118. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the Performance of the MM_PBSA and MM_GBSA Methods. 1. The Accuracy.pdf. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Miller, I.B.R.; McGee, J.T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational Immunology Meets Bioinformatics: The Use of Prediction Tools for Molecular Binding in the Simulation of the Immune System. PLoS ONE 2010, 5, e9862. [Google Scholar] [CrossRef]
- Sanober, G.; Ahmad, S.; Azam, S.S. Identification of plausible drug targets by investigating the druggable genome of MDR Staphylococcus epidermidis. Gene Rep. 2017, 7, 147–153. [Google Scholar] [CrossRef]
- Barh, D.; Barve, N.; Gupta, K.; Chandra, S.; Jain, N.; Tiwari, S.; Leon-Sicairos, N.; Canizalez-Roman, A.; Santos, A.; Hassan, S.S.; et al. Exoproteome and Secretome Derived Broad Spectrum Novel Drug and Vaccine Candidates in Vibrio cholerae Targeted by Piper betel Derived Compounds. PLoS ONE 2013, 8, e52773. [Google Scholar] [CrossRef]
- Wadood, A.; Jamal, A.; Riaz, M.; Khan, A.; Uddin, R.; Jelani, M.; Azam, S.S. Subtractive genome analysis for in silico identification and characterization of novel drug targets in Streptococcus pneumonia strain JJA. Microb. Pathog. 2018, 115, 194–198. [Google Scholar] [CrossRef]
- Ebihara, T. Dichotomous Regulation of Acquired Immunity by Innate Lymphoid Cells. Cells 2020, 9, 1193. [Google Scholar] [CrossRef]
- Bonilla, F.A.; Oettgen, H.C. Adaptive immunity. J. Allergy Clin. Immunol. 2010, 125, S33–S40. [Google Scholar] [CrossRef]
- Lee, S.; Choi, Y.-K.; Goo, Y.-K. Humoral and cellular immune response to Plasmodium vivax VIR recombinant and synthetic antigens in individuals naturally exposed to P. vivax in the Republic of Korea. Malar. J. 2021, 20, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Trincado, J.L.; Gomez-Perosanz, M.; Reche, P.A. Fundamentals and Methods for T- and B-Cell Epitope Prediction. J. Immunol. Res. 2017, 2017, 2680160. [Google Scholar] [CrossRef] [PubMed]
- Dombkowski, A.A.; Sultana, K.Z.; Craig, D.B. Protein disulfide engineering. FEBS Lett. 2014, 588, 206–212. [Google Scholar] [CrossRef]
- Nieuwkoop, T.; Claassens, N.J.; Van Der Oost, J. Improved protein production and codon optimization analyses in Escherichia coli by bicistronic design. Microb. Biotechnol. 2018, 12, 173–179. [Google Scholar] [CrossRef]
- Maiorov, V.N.; Crippen, G.M. Significance of Root-Mean-Square Deviation in Comparing Three-dimensional Structures of Globular Proteins. J. Mol. Biol. 1994, 235, 625–634. [Google Scholar] [CrossRef]
- Ahmad, S.; Raza, S.; Uddin, R.; Azam, S.S. Binding mode analysis, dynamic simulation and binding free energy calculations of the MurF ligase from Acinetobacter baumannii. J. Mol. Graph. Model. 2017, 77, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Lobanov, M.Y.; Bogatyreva, N.S.; Galzitskaya, O. V Radius of gyration as an indicator of protein structure compactness. Mol. Biol. 2008, 42, 623–628. [Google Scholar] [CrossRef]
- Chen, L.; Yuan, J.; Xu, Y.; Zhang, F.; Chen, Z. Comparison of clinical manifestations and antibiotic resistances among three genospecies of the Acinetobacter calcoaceticus-Acinetobacter baumannii complex. PLoS ONE 2018, 13, e0191748. [Google Scholar] [CrossRef]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Micoli, F.; Bagnoli, F.; Rappuoli, R.; Serruto, D. The role of vaccines in combatting antimicrobial resistance. Nat. Rev. Microbiol. Genet. 2021, 19, 287–302. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Khan, A.; Rehman, A.U.; Ahmad, I.; Ullah, S.; Khan, A.A.; Ali, S.S.; Afridi, S.G.; Wei, D.-Q. Immunoinformatics and structural vaccinology driven prediction of multi-epitope vaccine against Mayaro virus and validation through in-silico expression. Infect. Genet. Evol. 2019, 73, 390–400. [Google Scholar] [CrossRef]
- Zeb, A.; Ali, S.S.; Azad, A.K.; Safdar, M.; Anwar, Z.; Suleman, M.; Nizam-Uddin, N.; Khan, A.; Wei, D.-Q. Genome-wide screening of vaccine targets prioritization and reverse vaccinology aided design of peptides vaccine to enforce humoral immune response against Campylobacter jejuni. Comput. Biol. Med. 2021, 133, 104412. [Google Scholar] [CrossRef]
- Rappuoli, R. Reverse vaccinology, a genome-based approach to vaccine development. Vaccine 2001, 19, 2688–2691. [Google Scholar] [CrossRef]
- Dhar, R.; Slusky, J.S. Outer membrane protein evolution. Curr. Opin. Struct. Biol. 2021, 68, 122–128. [Google Scholar] [CrossRef]
- Nottelet, P.; Bataille, L.; Gourgues, G.; Anger, R.; Lartigue, C.; Sirand-Pugnet, P.; Marza, E.; Fronzes, R.; Arfi, Y. The mycoplasma surface proteins MIB and MIP promote the dissociation of the antibody-antigen interaction. Sci. Adv. 2021, 7, eabf2403. [Google Scholar] [CrossRef]
- Torisu, T.; Shikama, S.; Nakamura, K.; Enomoto, K.; Maruno, T.; Mori, A.; Uchiyama, S.; Satou, T. Physicochemical Characterization of Sabin Inactivated Poliovirus Vaccine for Process Development. J. Pharm. Sci. 2021, 110, 2121–2129. [Google Scholar] [CrossRef]
- Silva, F.M.; Barbosa, M.D.S.; Tiwari, S.; Seyffert, N.; Azevedo, V.A.D.C.; Nascimento, R.J.M.; Castro, T.L.D.P.; Marchioro, S.B. Immunoinformatic approach for the evaluation of sortase C and E proteins as vaccine targets against Caseous lymphadenitis. Inform. Med. Unlocked 2021, 26, 100718. [Google Scholar] [CrossRef]
- Morris, G.M.; Lim-Wilby, M. Molecular docking. In Molecular Modeling of Proteins; Springer: Heidelberg/Berlin, Germany, 2008; pp. 365–382. [Google Scholar]
- Hansson, T.; Oostenbrink, C.; van Gunsteren, W. Molecular dynamics simulations. Curr. Opin. Struct. Biol. 2002, 12, 190–196. [Google Scholar] [CrossRef]
- Hubbard, R.E.; Haider, M.K. Hydrogen Bonds in Proteins: Role and Strength. In eLS; Wiley: Hoboken, NJ, USA, 2010. [Google Scholar]
- Ehsan, N.; Ahmad, S.; Azam, S.S.; Rungrotmongkol, T.; Uddin, R. Proteome-wide identification of epitope-based vaccine candidates against multi-drug resistant Proteus mirabilis. Biology 2018, 55, 27–37. [Google Scholar] [CrossRef]
- Elhag, M.; Alaagib, R.M.; Ahmed, N.M.; Abubaker, M.; Haroun, E.M.; Albagi, S.O.A.; Hassan, M.A. Design of Epitope-Based Peptide Vaccine against Pseudomonas aeruginosa Fructose Bisphosphate Aldolase Protein Using Immunoinformatics. J. Immunol. Res. 2020, 2020, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Asad, Y.; Ahmad, S.; Rungrotmongkol, T.; Ranaghan, K.E.; Azam, S.S. Immuno-informatics driven proteome-wide investigation revealed novel peptide-based vaccine targets against emerging multiple drug resistant Providencia stuartii. J. Mol. Graph. Model. 2018, 80, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Dar, H.A.; Zaheer, T.; Shehroz, M.; Ullah, N.; Naz, K.; Muhammad, S.A.; Zhang, T.; Ali, A. Immunoinformatics-Aided Design and Evaluation of a Potential Multi-Epitope Vaccine against Klebsiella Pneumoniae. Vaccines 2019, 7, 88. [Google Scholar] [CrossRef] [PubMed]
- Enayatkhani, M.; Hasaniazad, M.; Faezi, S.; Gouklani, H.; Davoodian, P.; Ahmadi, N.; Einakian, M.A.; Karmostaji, A.; Ahmadi, K. Reverse vaccinology approach to design a novel multi-epitope vaccine candidate against COVID-19: An in silico study. J. Biomol. Struct. Dyn. 2021, 39, 2857–2872. [Google Scholar] [CrossRef] [PubMed]
- Nain, Z.; Abdullah, F.; Rahman, M.M.; Karim, M.M.; Khan, S.A.; Bin Sayed, S.; Mahmud, S.; Rahman, S.M.R.; Sheam, M.; Haque, Z.; et al. Proteome-wide screening for designing a multi-epitope vaccine against emerging pathogen Elizabethkingia anophelis using immunoinformatic approaches. J. Biomol. Struct. Dyn. 2020, 38, 4850–4867. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Selected Epitopes | DRB*0101 Binding Affinity, (IC50 Score < 100 nM) | Antigenicity (Threshold > 0.5) |
|---|---|---|
| RSDSRQLDS | 5.09 | 1.8181 |
| EGKATSADA | 6.24 | 2.0036 |
| GGETDYNNL | 98.4 | 1.2907 |
| TLRLQSQQS | 12.3 | 1.1516 |
| RPSYKDADY | 70.47 | 0.7769 |
| RTDDTVYGF | 21.98 | 0.6496 |
| SEKITGADH | 63.24 | 1.0815 |
| KQDQKISLR | 5.5 | 1.8436 |
| GLDSIDLQG | 38.19 | 1.1785 |
| TDVSVPQLR | 67.61 | 1.1671 |
| PLPPADIDL | 18.16 | 1.5027 |
| DARASAKGE | 30.13 | 2.119 |
| AGQDAAGNV | 35.48 | 1.587 |
| DGNGKLLQM | 15.81 | 1.5201 |
| KVTESSVSK | 42.46 | 1.3916 |
| PVSEKTSLK | 18.2 | 0.7 |
| MHC-I | ||||||
|---|---|---|---|---|---|---|
| Rank | Solution Number | Global Energy | Attractive van der Waals (VdW) | Repulsive van der Waals (VdW) | Atomic Contact Energy (ACE) | Hydrogen bond (HB) Energy |
| ↓ | ||||||
| 1 | 9 | −8.87 | −20.43 | 8.30 | −3.02 | −4.00 |
| 2 | 10 | −4.10 | −6.98 | 4.98 | 1.53 | −2.42 |
| 3 | 8 | 6.48 | −24.98 | 30.22 | 8.47 | −3.40 |
| 4 | 2 | 163.07 | −48.68 | 240.39 | 18.67 | −5.80 |
| 5 | 6 | 224.43 | −17.03 | 277.99 | 10.17 | −1.41 |
| 6 | 1 | 364.31 | −11.47 | 411.75 | 9.48 | −3.45 |
| 7 | 3 | 894.31 | −37.86 | 1119.67 | 8.40 | −8.31 |
| 8 | 5 | 1529.17 | −66.06 | 2019.83 | 15.80 | −14.94 |
| 9 | 4 | 2952.67 | −39.19 | 3697.93 | 14.61 | −6.84 |
| 10 | 7 | 8856.55 | −105.59 | 11,240.42 | 15.43 | −13.44 |
| MHC-II | ||||||
| ↓ | ||||||
| 1 | 9 | 6.51 | −4.12 | 1.47 | 0.59 | 0.00 |
| 2 | 8 | 811.10 | −46.86 | 1073.56 | 13.36 | −7.62 |
| 3 | 3 | 1016.15 | −32.82 | 1318.20 | 9.36 | −1.85 |
| 4 | 2 | 1726.20 | −27.51 | 2140.30 | 14.33 | −3.32 |
| 5 | 4 | 2034.57 | −29.52 | 2587.07 | 7.44 | −6.16 |
| 6 | 6 | 4647.55 | −89.28 | 5973.20 | −2.24 | −6.55 |
| 7 | 10 | 5123.90 | −70.81 | 6542.45 | 0.54 | −16.75 |
| 8 | 5 | 6044.06 | −68.06 | 7616.67 | 19.36 | −8.52 |
| 9 | 7 | 7053.34 | −88.86 | 8952.50 | 7.28 | −12.32 |
| 10 | 1 | 10,090.45 | −65.97 | 12,720.01 | 1.81 | −11.01 |
| TLR−4 | ||||||
| ↓ | ||||||
| 1 | 9 | 7.32 | −0.88 | 0.00 | 0.62 | 0.00 |
| 2 | 3 | 11.99 | −2.25 | 0.74 | 1.10 | −0.47 |
| 3 | 5 | 18.18 | −3.61 | 0.00 | 5.64 | −0.57 |
| 4 | 7 | 93.89 | −37.58 | 152.19 | 10.19 | −5.76 |
| 5 | 8 | 219.99 | −50.52 | 358.55 | 19.21 | −13.52 |
| 6 | 4 | 487.01 | −48.02 | 604.75 | 17.53 | −8.03 |
| 7 | 1 | 831.97 | −31.40 | 1082.97 | 2.03 | −5.53 |
| 8 | 2 | 849.36 | −39.75 | 1052.49 | 25.45 | −8.24 |
| 9 | 6 | 1628.72 | −64.12 | 2074.26 | 21.83 | −9.82 |
| 10 | 10 | 1635.90 | −32.51 | 2069.25 | 17.61 | −7.09 |
| Vaccine Complex | Interactive Residues |
|---|---|
| MHC-I | Arg81, Asn83, Leu87, Ser88, Gln89, Pro90, Lys91, Ile92, Lys94, Asp220, Gln255, Arg256 |
| MHC-II | Asp43, Val44, Glu47, Arg48, Glu52, Arg55, Thr95, Tyr101, Asn103, Thr104, Arg105, Glu106, Tyr111, Lys112, Lys120, Ala130, Tyr136, Lys138, Thr145, Asp147, Ile148, Pro150, Pro156, Phe158, Glu177, Gly179, Gly180, Lys183, Phe188, Ser199, Thr200, Val201, Arg220, Arg254, Thr18 |
| TLR4 | Asn26, Ser28, Val30, Glu31, Val32, Cys37, Asp38, Lys39, Asn49, Pro 50, Cys51, Glu53, Asn58, Pro68, Asn77, Tyr79, Val82, Asn83, Thy84, Met85, Asn86, Leu87, Lys89, Arg90, Lys128, Lys130, Glu136, Glu144, Cys148, Gln156, Trp232, Glu434, Arg456, Ala462, Phe463, Asp490, Ser491, Phe492, Thy493, Glu509, Ser512, Thr514, Asn575, Thr577, Gln578, Glu603, Glu605, Arg606 |
| Energy Parameter | TLR-4–Vaccine Complex | MHC-I–Vaccine Complex | MHC-II–Vaccine Complex |
|---|---|---|---|
| MM-GBSA | |||
| VDWAALS | −75.48 | −66.85 | −60.74 |
| EEL | −65.47 | −55.17 | −52.43 |
| EGB | 71.91 | 52.14 | 68.48 |
| ESURF | −9.36 | −11.56 | −10.32 |
| Delta G gas | −140.95 | −122.02 | −113.17 |
| Delta G solv | 62.55 | 40.58 | 58.16 |
| Delta Total | −78.4 | −81.44 | −55.01 |
| MM-PBSA | |||
| VDWAALS | −75.48 | −66.85 | −60.74 |
| EEL | −65.47 | −55.17 | −52.43 |
| EPB | 73.45 | 43.87 | 50.98 |
| ENPOLAR | −5.28 | −6.54 | −8.61 |
| Delta G gas | −140.95 | −122.02 | −113.17 |
| Delta G solv | 68.17 | 37.33 | 42.37 |
| Delta Total | −72.78 | −84.69 | −70.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ullah, A.; Ahmad, S.; Ismail, S.; Afsheen, Z.; Khurram, M.; Tahir ul Qamar, M.; AlSuhaymi, N.; Alsugoor, M.H.; Allemailem, K.S. Towards A Novel Multi-Epitopes Chimeric Vaccine for Simulating Strong Immune Responses and Protection against Morganella morganii. Int. J. Environ. Res. Public Health 2021, 18, 10961. https://doi.org/10.3390/ijerph182010961
Ullah A, Ahmad S, Ismail S, Afsheen Z, Khurram M, Tahir ul Qamar M, AlSuhaymi N, Alsugoor MH, Allemailem KS. Towards A Novel Multi-Epitopes Chimeric Vaccine for Simulating Strong Immune Responses and Protection against Morganella morganii. International Journal of Environmental Research and Public Health. 2021; 18(20):10961. https://doi.org/10.3390/ijerph182010961
Chicago/Turabian StyleUllah, Asad, Sajjad Ahmad, Saba Ismail, Zobia Afsheen, Muhammad Khurram, Muhammad Tahir ul Qamar, Naif AlSuhaymi, Mahdi H. Alsugoor, and Khaled S. Allemailem. 2021. "Towards A Novel Multi-Epitopes Chimeric Vaccine for Simulating Strong Immune Responses and Protection against Morganella morganii" International Journal of Environmental Research and Public Health 18, no. 20: 10961. https://doi.org/10.3390/ijerph182010961
APA StyleUllah, A., Ahmad, S., Ismail, S., Afsheen, Z., Khurram, M., Tahir ul Qamar, M., AlSuhaymi, N., Alsugoor, M. H., & Allemailem, K. S. (2021). Towards A Novel Multi-Epitopes Chimeric Vaccine for Simulating Strong Immune Responses and Protection against Morganella morganii. International Journal of Environmental Research and Public Health, 18(20), 10961. https://doi.org/10.3390/ijerph182010961