Early Life Exposure to Aflatoxin B1 in Rats: Alterations in Lipids, Hormones, and DNA Methylation among the Offspring


 and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Chemicals
2.3. Experimental Design
2.4. Hormone and Lipid Analysis
2.5. DNA Methylation Analysis
2.6. Statistical Analysis
3. Results
3.1. Weight Gain/Weight Status of the Rodent Population
3.2. Lipids
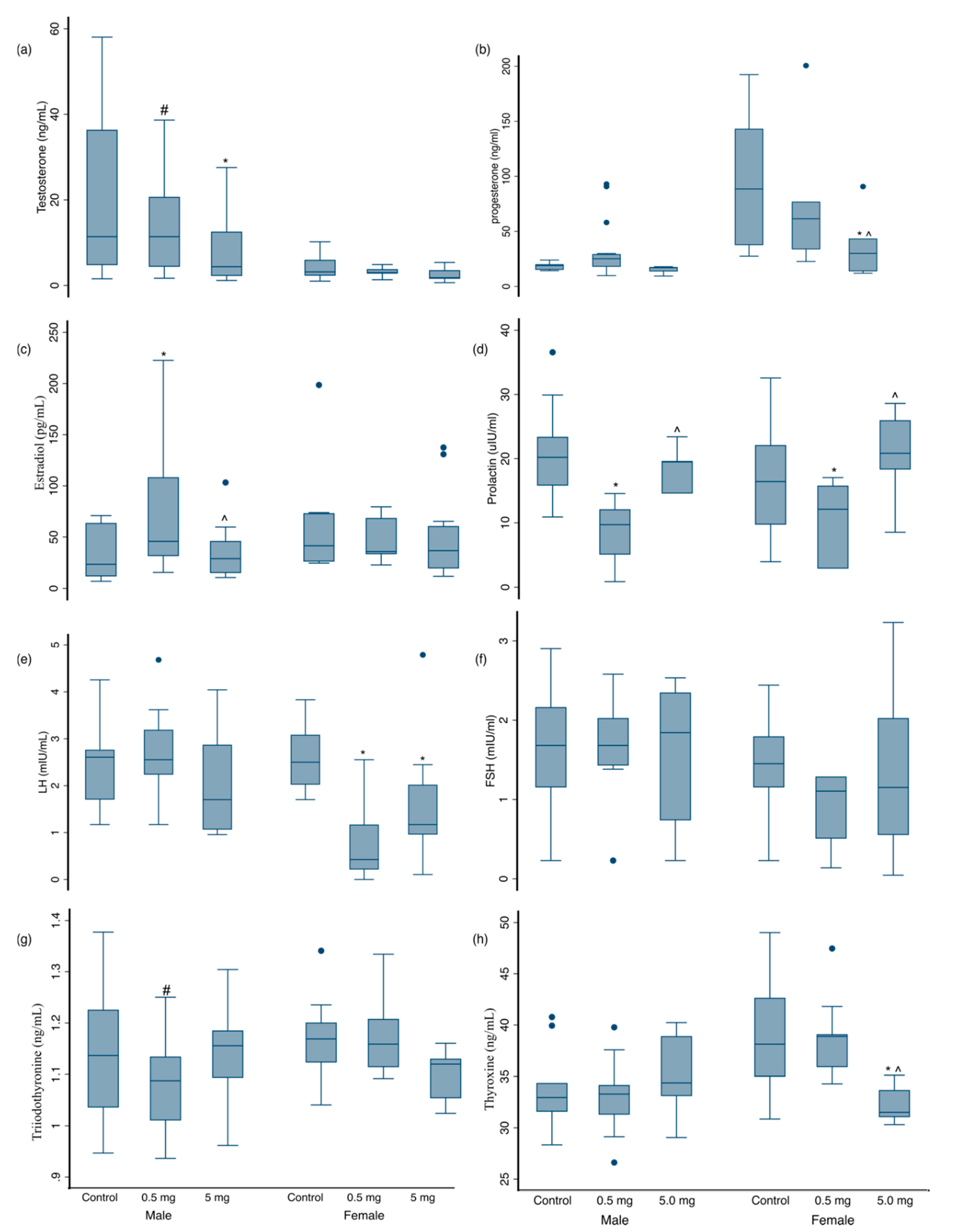
3.3. Hormonal Changes at 3 Months of Age
3.4. DNA Methylation in Liver and Blood
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gluckman, P.D.; Hanson, M.A.; Cooper, C.; Thornburg, K.L. Effect of in utero and early-life conditions on adult health and disease. N. Engl. J. Med. 2008, 359, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J.; Osmond, C.; Forsen, T.J.; Kajantie, E.; Eriksson, J.G. Trajectories of growth among children who have coronary events as adults. N. Engl. J. Med. 2005, 353, 1802–1809. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.L.; Ho, S.-M. Developmental reprogramming of cancer susceptibility. Nat. Rev. Cancer 2012, 12, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kohli, C.; Johnson, L.; Bennet, L.; Brusselaers, N.; Nilsson, P.M. Birth size and cancer prognosis: A systematic review and meta-analysis. J. Dev. Orig. Health Dis. 2020, 11, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Spector, L.G.; Puumala, S.E.; Carozza, S.E.; Chow, E.J.; Fox, E.E.; Horel, S.; Johnson, K.J.; McLaughlin, C.C.; Reynolds, P.; Von Behren, J. Cancer risk among children with very low birth weights. Pediatrics 2009, 124, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, H.; Matsuyama, S.; Tanimura, M. Association between hepatoblastoma and very low birth weight: A trend or a chance? J. Pediatr. 1997, 130, 557–560. [Google Scholar] [CrossRef]
- Zimmermann, E.; Berentzen, T.L.; Gamborg, M.; Sørensen, T.I.; Baker, J.L. Sex-specific associations between birth weight and adult primary liver cancer in a large cohort of D anish children. Int. J. Cancer 2016, 138, 1410–1415. [Google Scholar] [CrossRef]
- Sandboge, S.; Perälä, M.-M.; Salonen, M.K.; Blomstedt, P.A.; Osmond, C.; Kajantie, E.; Barker, D.J.; Eriksson, J.G. Early growth and non-alcoholic fatty liver disease in adulthood—the NAFLD liver fat score and equation applied on the Helsinki Birth Cohort Study. Ann. Med. 2013, 45, 430–437. [Google Scholar] [CrossRef]
- Li, E. Chromatin modification and epigenetic reprogramming in mammalian development. Nat. Rev. Genet. 2002, 3, 662–673. [Google Scholar] [CrossRef]
- Reik, W.; Dean, W.; Walter, J. Epigenetic reprogramming in mammalian development. Science 2001, 293, 1089–1093. [Google Scholar] [CrossRef]
- Silva Lima, B.; Van der Laan, J.W. Mechanisms of Nongenotoxic Carcinogenesis and Assessment of the Human Hazard. Regul. Toxicol. Pharm. 2000, 32, 135–143. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Hivert, M.F.; Perron, P.; Poirier, P.; Guay, S.P.; Brisson, D.; Bouchard, L. IGF2 DNA methylation is a modulator of newborn’s fetal growth and development. Epigenet. Off. J. DNA Methylation Soc. 2012, 7, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Amarger, V.; Giudicelli, F.; Pagniez, A.; Parnet, P. Perinatal high methyl donor alters gene expression in IGF system in male offspring without altering DNA methylation. Future Sci. OA 2017, 3, Fso164. [Google Scholar] [CrossRef] [PubMed]
- Agrogiannis, G.D.; Sifakis, S.; Patsouris, E.S.; Konstantinidou, A.E. Insulin-like growth factors in embryonic and fetal growth and skeletal development (Review). Mol. Med. Rep. 2014, 10, 579–584. [Google Scholar] [CrossRef]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Dolinoy, D.C.; Huang, D. Jirtle, R.L. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc. Natl. Acad. Sci. USA 2007, 104, 13056–13061. [Google Scholar] [CrossRef]
- Montrose, L.; Padmanabhan, V.; Goodrich, J.M.; Domino, S.E.; Treadwell, M.C.; Meeker, J.D.; Watkins, D.J.; Dolinoy, D.C. Maternal levels of endocrine disrupting chemicals in the first trimester of pregnancy are associated with infant cord blood DNA methylation. Epigenetics 2018, 13, 301–309. [Google Scholar] [CrossRef]
- Neier, K.; Cheatham, D.; Bedrosian, L.D.; Dolinoy, D.C. Perinatal exposures to phthalates and phthalate mixtures result in sex-specific effects on body weight, organ weights and intracisternal A-particle (IAP) DNA methylation in weanling mice. J. Dev. Orig. Health Dis. 2019, 10, 176–187. [Google Scholar] [CrossRef]
- Hernandez-Vargas, H.; Castelino, J.; Silver, M.J.; Dominguez-Salas, P.; Cros, M.P.; Durand, G.; Le Calvez-Kelm, F.; Prentice, A.M.; Wild, C.P.; Moore, S.E.; et al. Exposure to aflatoxin B1 in utero is associated with DNA methylation in white blood cells of infants in The Gambia. Int. J. Epidemiol. 2015, 44, 1238–1248. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Rossner, P., Jr.; Chen, Y.; Agrawal, M.; Wang, Q.; Wang, L.; Ahsan, H.; Yu, M.W.; Lee, P.H.; Santella, R.M. Aflatoxin B1 and polycyclic aromatic hydrocarbon adducts, p53 mutations and p16 methylation in liver tissue and plasma of hepatocellular carcinoma patients. Int. J. Cancer 2006, 119, 985–991. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Wu, H.C.; Yazici, H.; Yu, M.W.; Lee, P.H.; Santella, R.M. Global hypomethylation in hepatocellular carcinoma and its relationship to aflatoxin B(1) exposure. World J. Hepatol. 2012, 4, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Livingstone, M.C.; Johnson, N.M.; Roebuck, B.D.; Kensler, T.W.; Groopman, J.D. Profound changes in miRNA expression during cancer initiation by aflatoxin B(1) and their abrogation by the chemopreventive triterpenoid CDDO-Im. Mol. Carcinog. 2017, 56, 2382–2390. [Google Scholar] [CrossRef]
- Wu, H.C.; Wang, Q.; Yang, H.I.; Tsai, W.Y.; Chen, C.J.; Santella, R.M. Global DNA methylation in a population with aflatoxin B1 exposure. Epigenetics 2013, 8, 962–969. [Google Scholar] [CrossRef] [PubMed]
- Kumi, J.; Mitchell, N.; Asare, G.; Dotse, E.; Kwaa, F.; Phillips, T.; Ankrah, N. Aflatoxins and fumonisins contamination of home-made food (weanimix) from cereal-legume blends for children. Ghana Med. J. 2014, 48, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Benkerroum, N. Aflatoxins: Producing-Molds, Structure, Health Issues and Incidence in Southeast Asian and Sub-Saharan African Countries. Int. J. Environ. Res. Public Health. 2020, 17, 1215. [Google Scholar] [CrossRef]
- Xue, K.S.; Tang, L.; Shen, C.L.; Pollock, B.H.; Guerra, F.; Phillips, T.D.; Wang, J.-S. Increase in aflatoxin exposure in two populations residing in East and West Texas, United States. Int. J. Hyg. Environ. Health 2020, 231, 113662. [Google Scholar] [CrossRef]
- Nazhand, A.; Durazzo, A.; Lucarini, M.; Souto, E.B.; Santini, A. Characteristics, Occurrence, Detection and Detoxification of Aflatoxins in Foods and Feeds. Foods 2020, 9, 644. [Google Scholar] [CrossRef]
- Gruber-Dorninger, C.; Jenkins, T.; Schatzmayr, G. Global mycotoxin occurrence in feed: A ten-year survey. Toxins 2019, 11, 375. [Google Scholar] [CrossRef]
- Zhang, N.-Y.; Qi, M.; Gao, X.; Zhao, L.; Liu, J.; Gu, C.-Q.; Song, W.-J.; Krumm, C.S.; Sun, L.-H.; Qi, D.-S. Response of the hepatic transcriptome to aflatoxin B 1 in ducklings. Toxicon 2016, 111, 69–76. [Google Scholar] [CrossRef]
- Deng, J.; Zhao, L.; Zhang, N.-Y.; Karrow, N.A.; Krumm, C.S.; Qi, D.-S.; Sun, L.-H. Aflatoxin B1 metabolism: Regulation by phase I and II metabolizing enzymes and chemoprotective agents. Mutat. Res. Rev. Mutat. Res. 2018, 778, 79–89. [Google Scholar] [CrossRef]
- Benkerroum, N. Chronic and acute toxicities of aflatoxins: Mechanisms of action. Int. J. Environ. Res. Public Health 2020, 17, 423. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Rieswijk, L.; Claessen, S.M.; Bekers, O.; van Herwijnen, M.; Theunissen, D.H.; Jennen, D.G.; de Kok, T.M.; Kleinjans, J.C.; van Breda, S.G. Aflatoxin B1 induces persistent epigenomic effects in primary human hepatocytes associated with hepatocellular carcinoma. Toxicology 2016, 350, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Khlangwiset, P.; Shephard, G.S.; Wu, F. Aflatoxins and growth impairment: A review. Crit. Rev. Toxicol. 2011, 41, 740–755. [Google Scholar] [CrossRef] [PubMed]
- Fujitake, S.; Hibi, K.; Okochi, O.; Kodera, Y.; Ito, K.; Akiyama, S.; Nakao, A. Aberrant methylation of SOCS-1 was observed in younger colorectal cancer patients. J. Gastroenterol. 2004, 39, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Steele, R.; Lane, D. P53 in cancer: A paradigm for modern management of cancer. Surgeon 2005, 3, 197–205. [Google Scholar] [CrossRef]
- Rotimi, O.A.; Rotimi, S.O.; Oluwafemi, F.; Ademuyiwa, O.; Balogun, E.A. Coexistence of aflatoxicosis with protein malnutrition worsens hepatic oxidative damage in rats. J. Biochem. Mol. Toxicol. 2016, 30, 269–276. [Google Scholar] [CrossRef]
- Charni-Natan, M.; Aloni-Grinstein, R.; Osher, E.; Rotter, V. Liver and Steroid Hormones—Can a Touch of p53 Make a Difference? Front. Endocrinol. 2019, 10, 374. [Google Scholar] [CrossRef]
- Goldstein, I.; Ezra, O.; Rivlin, N.; Molchadsky, A.; Madar, S.; Goldfinger, N.; Rotter, V. p53, a novel regulator of lipid metabolism pathways. J. Hepatol. 2012, 56, 656–662. [Google Scholar] [CrossRef]
- Lacroix, M.; Riscal, R.; Arena, G.; Linares, L.K.; Le Cam, L. Metabolic functions of the tumor suppressor p53: Implications in normal physiology, metabolic disorders, and cancer. Mol. Metab. 2020, 33, 2–22. [Google Scholar] [CrossRef]
- Berger, C.; Qian, Y.; Chen, X. The p53-estrogen receptor loop in cancer. Curr. Mol. Med. 2013, 13, 1229–1240. [Google Scholar] [CrossRef] [PubMed]
- Dinda, S.; Sanchez, A.; Moudgil, V. Estrogen-like effects of thyroid hormone on the regulation of tumor suppressor proteins, p53 and retinoblastoma, in breast cancer cells. Oncogene 2002, 21, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Rotimi, O.A.; Rotimi, S.O.; Duru, C.U.; Ebebeinwe, O.J.; Abiodun, A.O.; Oyeniyi, B.O.; Faduyile, F.A. Acute aflatoxin B1–Induced hepatotoxicity alters gene expression and disrupts lipid and lipoprotein metabolism in rats. Toxicol. Rep. 2017, 4, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Rotimi, O.A.; Rotimi, S.O.; Goodrich, J.M.; Adelani, I.B.; Agbonihale, E.; Talabi, G. Time-course effects of acute aflatoxin B1 exposure on hepatic mitochondrial lipids and oxidative stress in rats. Front. Pharmacol. 2019, 10, 467. [Google Scholar] [CrossRef]
- Ugbaja, R.N.; Okedairo, O.M.; Oloyede, A.R.; Ugwor, E.I.; Akinloye, D.I.; Ojo, O.P.; Ademuyiwa, O. Probiotics consortium synergistically ameliorates aflatoxin B1-induced disruptions in lipid metabolism of female albino rats. Toxicon 2020, 186, 109–119. [Google Scholar] [CrossRef]
- Supriya, C.; Girish, B.; Reddy, P.S. Aflatoxin B1-induced reproductive toxicity in male rats: Possible mechanism of action. Int. J. Toxicol. 2014, 33, 155–161. [Google Scholar] [CrossRef]
- Adedara, I.A.; Nanjappa, M.K.; Farombi, E.O.; Akingbemi, B.T. Aflatoxin B1 disrupts the androgen biosynthetic pathway in rat Leydig cells. Food Chem. Toxicol. 2014, 65, 252–259. [Google Scholar] [CrossRef]
- Chen, X.; Li, C.; Chen, Y.; Ni, C.; Chen, X.; Zhang, L.; Xu, X.; Chen, M.; Ma, X.; Zhan, H. Aflatoxin B1 impairs leydig cells through inhibiting AMPK/mTOR-mediated autophagy flux pathway. Chemosphere 2019, 233, 261–272. [Google Scholar] [CrossRef]
- Cui, H.; Cruz-Correa, M.; Giardiello, F.M.; Hutcheon, D.F.; Kafonek, D.R.; Brandenburg, S.; Wu, Y.; He, X.; Powe, N.R.; Feinberg, A.P. Loss of IGF2 imprinting: A potential marker of colorectal cancer risk. Science 2003, 299, 1753–1755. [Google Scholar] [CrossRef]
- Yoshimura, H.; Matsuda, Y.; Yamamoto, M.; Kamiya, S.; Ishiwata, T. Expression and role of long non-coding RNA H19 in carcinogenesis. Front. Biosci. 2018, 23, 614–625. [Google Scholar] [CrossRef]
- Raveh, E.; Matouk, I.J.; Gilon, M.; Hochberg, A. The H19 Long non-coding RNA in cancer initiation, progression and metastasis—A proposed unifying theory. Mol. Cancer 2015, 14, 184. [Google Scholar] [CrossRef] [PubMed]
- Adetunji, M.; Atanda, O.; Ezekiel, C.N.; Sulyok, M.; Warth, B.; Beltrán, E.; Krska, R.; Obadina, O.; Bakare, A.; Chilaka, C.A. Fungal and bacterial metabolites of stored maize (Zea mays, L.) from five agro-ecological zones of Nigeria. Mycotoxin Res. 2014, 30, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Grunau, C.; Clark, S.J.; Rosenthal, A. Bisulfite genomic sequencing: Systematic investigation of critical experimental parameters. Nucleic Acids Res. 2001, 29, E65. [Google Scholar] [CrossRef] [PubMed]
- Tost, J.; Gut, I.G. Analysis of gene-specific DNA methylation patterns by pyrosequencing technology. Methods Mol. Biol. 2007, 373, 89–102. [Google Scholar] [CrossRef]
- He, X.; Xie, Z.; Dong, Q.; Li, J.; Li, W.; Chen, P. Effect of Folic Acid Supplementation on Renal Phenotype and Epigenotype in Early Weanling Intrauterine Growth Retarded Rats. Kidney Blood Press Res. 2015, 40, 395–402. [Google Scholar] [CrossRef]
- Goodrich, J.M.; Sanchez, B.N.; Dolinoy, D.C.; Zhang, Z.; Hernandez-Avila, M.; Hu, H.; Peterson, K.E.; Tellez-Rojo, M.M. Quality Control and Statistical Modeling for Environmental Epigenetics: A Study on in Utero Lead Exposure and DNA Methylation at Birth. Epigenetics 2015, 10, 19–30. [Google Scholar] [CrossRef]
- Kihara, T.; Matsuo, T.; Sakamoto, M.; Yasuda, Y.; Yamamoto, Y.; Tanimura, T. Effects of prenatal aflatoxin B1 exposure on behaviors of rat offspring. Toxicol. Sci. 2000, 53, 392–399. [Google Scholar] [CrossRef]
- Supriya, C.; Akhila, B.; Pratap Reddy, K.; Girish, B.; Sreenivasula Reddy, P. Effects of maternal exposure to aflatoxin B1 during pregnancy on fertility output of dams and developmental, behavioral and reproductive consequences in female offspring using a rat model. Toxicol. Mech. Methods 2016, 26, 202–210. [Google Scholar] [CrossRef]
- Supriya, C.; Reddy, P.S. Prenatal exposure to aflatoxin B1: Developmental, behavioral, and reproductive alterations in male rats. Sci. Nat. 2015, 102, 26. [Google Scholar] [CrossRef]
- Turner, P.C.; Collinson, A.C.; Cheung, Y.B.; Gong, Y.; Hall, A.J.; Prentice, A.M.; Wild, C.P. Aflatoxin exposure in utero causes growth faltering in Gambian infants. Int. J. Epidemiol. 2007, 36, 1119–1125. [Google Scholar] [CrossRef]
- Lee, B.J.; Kim, J.Y. Identification of metabolic syndrome using phenotypes consisting of triglyceride levels with anthropometric indices in Korean adults. BMC Endocr. Disord. 2020, 20, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, C.; Sparks, D.L. Hepatic lipase, high density lipoproteins, and hypertriglyceridemia. Am. J. Pathol. 2011, 178, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zeng, F.F.; Liu, Z.M.; Zhang, C.X.; Ling, W.H.; Chen, Y.M. Effects of blood triglycerides on cardiovascular and all-cause mortality: A systematic review and meta-analysis of 61 prospective studies. Lipids Health Dis. 2013, 12, 159. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahhab, M.A.; El-Nekeety, A.A.; Hathout, A.S.; Salman, A.S.; Abdel-Aziem, S.H.; Sabry, B.A.; Hassan, N.S.; Abdel-Aziz, M.S.; Aly, S.E.; Jaswir, I. Bioactive compounds from Aspergillus niger extract enhance the antioxidant activity and prevent the genotoxicity in aflatoxin B1-treated rats. Toxicon 2020, 181, 57–68. [Google Scholar] [CrossRef]
- El-Nekeety, A.A.; Abdel-Azeim, S.H.; Hassan, A.M.; Hassan, N.S.; Aly, S.E.; Abdel-Wahhab, M.A. Quercetin inhibits the cytotoxicity and oxidative stress in liver of rats fed aflatoxin-contaminated diet. Toxicol. Rep. 2014, 1, 319–329. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, Z.; Shen, W.-J.; Azhar, S. Cellular cholesterol delivery, intracellular processing and utilization for biosynthesis of steroid hormones. Nutr. Metab. 2010, 7, 47. [Google Scholar] [CrossRef]
- Giatti, S.; Diviccaro, S.; Serafini, M.M.; Caruso, D.; Garcia-Segura, L.M.; Viviani, B.; Melcangi, R.C. Sex differences in steroid levels and steroidogenesis in the nervous system: Physiopathological role. Front. Neuroendocrinol. 2020, 56, 100804. [Google Scholar] [CrossRef]
- Trejter, M.; Hochol, A.; Tyczewska, M.; Ziolkowska, A.; Jopek, K.; Szyszka, M.; Malendowicz, L.K.; Rucinski, M. Sex-related gene expression profiles in the adrenal cortex in the mature rat: Microarray analysis with emphasis on genes involved in steroidogenesis. Int. J. Mol. Med. 2015, 35, 702–714. [Google Scholar] [CrossRef]
- Hasanzadeh, S.; Hosseini, E.; Rezazadeh, L. Effects of Aflatoxin B1 on Profiles of Gonadotropic (FSH and LH), Steroid (Testosterone and 17β-Estradiol) and Prolactin Hormones in Adult Male Rat. Iran. J. Vet. Res. 2011, 12, 332–336. [Google Scholar]
- Abu El-Saad, A.S.; Mahmoud, H.M. Phytic acid exposure alters aflatoxinB1-induced reproductive and oxidative toxicity in albino rats (Rattus norvegicus). Evid. Based Complement. Altern. Med. 2009, 6, 107398. [Google Scholar] [CrossRef]
- Amin, Y.; Mohamed, R.; Zakaria, A.; Wehrend, A.; Hussein, H.A. Effects of aflatoxins on some reproductive hormones and composition of buffalo’s milk. Comp. Clin. Pathol. 2019, 28, 1191–1196. [Google Scholar] [CrossRef]
- Onuzulu, C.D.; Rotimi, O.A.; Rotimi, S.O. Epigenetic modifications associated with in utero exposure to endocrine disrupting chemicals BPA, DDT and Pb. Rev. Environ. Health 2019, 34, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357, eaal2380. [Google Scholar] [CrossRef] [PubMed]
- Darwiche, N. Epigenetic mechanisms and the hallmarks of cancer: An intimate affair. Am. J. Cancer Res. 2020, 10, 1954–1978. [Google Scholar]
- Wang, S.; He, Z.; Li, D.; Zhang, B.; Li, M.; Li, W.; Zhu, W.; Xing, X.; Zeng, X.; Wang, Q.; et al. Aberrant methylation of RUNX3 is present in Aflatoxin B(1)-induced transformation of the L02R cell line. Toxicology 2017, 385, 1–9. [Google Scholar] [CrossRef]
- Marrone, A.K.; Tryndyak, V.; Beland, F.A.; Pogribny, I.P. MicroRNA Responses to the Genotoxic Carcinogens Aflatoxin B1 and Benzo[a]pyrene in Human HepaRG Cells. Toxicol. Sci. 2016, 149, 496–502. [Google Scholar] [CrossRef]
- Tryndyak, V.; Kindrat, I.; Dreval, K.; Churchwell, M.I.; Beland, F.A.; Pogribny, I.P. Effect of aflatoxin B(1), benzo[a]pyrene, and methapyrilene on transcriptomic and epigenetic alterations in human liver HepaRG cells. Food Chem. Toxicol. 2018, 121, 214–223. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Chen, Y.; Ahsan, H.; Lunn, R.M.; Chen, S.Y.; Lee, P.H.; Chen, C.J.; Santella, R.M. Silencing of glutathione S-transferase P1 by promoter hypermethylation and its relationship to environmental chemical carcinogens in hepatocellular carcinoma. Cancer Lett. 2005, 221, 135–143. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Ahsan, H.; Chen, Y.; Lunn, R.M.; Wang, L.Y.; Chen, S.Y.; Lee, P.H.; Chen, C.J.; Santella, R.M. High frequency of promoter hypermethylation of RASSF1A and p16 and its relationship to aflatoxin B1-DNA adduct levels in human hepatocellular carcinoma. Mol. Carcinog. 2002, 35, 85–92. [Google Scholar] [CrossRef]
- Hussain, S.P.; Schwank, J.; Staib, F.; Wang, X.W.; Harris, C.C. TP53 mutations and hepatocellular carcinoma: Insights into the etiology and pathogenesis of liver cancer. Oncogene 2007, 26, 2166–2176. [Google Scholar] [CrossRef]
- Menendez, D.; Inga, A.; Resnick, M.A. The biological impact of the human master regulator p53 can be altered by mutations that change the spectrum and expression of its target genes. Mol. Cell. Biol. 2006, 26, 2297–2308. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hoyo, C.; Daltveit, A.K.; Iversen, E.; Benjamin-Neelon, S.E.; Fuemmeler, B.; Schildkraut, J.; Murtha, A.P.; Overcash, F.; Vidal, A.C.; Wang, F.; et al. Erythrocyte folate concentrations, CpG methylation at genomically imprinted domains, and birth weight in a multiethnic newborn cohort. Epigenetics 2014, 9, 1120–1130. [Google Scholar] [CrossRef] [PubMed]
- LaRocca, J.; Binder, A.M.; McElrath, T.F.; Michels, K.B. The impact of first trimester phthalate and phenol exposure on IGF2/H19 genomic imprinting and birth outcomes. Environ. Res. 2014, 133, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Barraza-Villarreal, A.; Biessy, C.; Duarte-Salles, T.; Sly, P.D.; Ramakrishnan, U.; Rivera, J.; Herceg, Z.; Romieu, I. Dietary supplementation with polyunsaturated fatty acid during pregnancy modulates DNA methylation at IGF2/H19 imprinted genes and growth of infants. Physiol. Genom. 2014, 46, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Soubry, A.; Murphy, S.; Huang, Z.; Murtha, A.; Schildkraut, J.; Jirtle, R.; Wang, F.; Kurtzberg, J.; Demark-Wahnefried, W.; Forman, M.; et al. The effects of depression and use of antidepressive medicines during pregnancy on the methylation status of the IGF2 imprinted control regions in the offspring. Clin. Epigenet. 2011, 3, 2. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.-L.; Huang, Z.; Wang, Q.; Chen, H.-H.; Ma, S.-N.; Wu, R.; Cai, W.-S. The association of polymorphisms in lncRNA-H19 with hepatocellular cancer risk and prognosis. Biosci. Rep. 2018, 38, BSR20171652. [Google Scholar] [CrossRef]


{kind=link}
| Exposure Group | Offspring | At Birth | 3 Weeks | 3 Months | |||
|---|---|---|---|---|---|---|---|
| Mean (SD) | N | Mean (SD) | N | Mean (SD) | N | ||
| Control | All | 6.3 (1.5) | 41 | 47.9 (5.1) | 19 | 191 (28.8) | 22 |
| Females | 6.2 (1.5) | 22 | 46.1 (3.6) | 11 | 167 (13.7) | 11 | |
| Males | 6.5 (1.6) | 19 | 50.0 (6.0) | 8 | 217 (12.7) | 11 | |
| 0.5 mg/kg | All | 6.3 (1.1) | 48 | 34.6 (6.7) ** | 19 | 155 (12.8) ** | 29 |
| Females | 6.4 (1.2) | 20 | 34.9 (7.4) * | 9 | 158 (11.9) | 11 | |
| Males | 6.3 (1.0) | 28 | 34.3 (6.5) ** | 10 | 153 (13.4) ** | 18 | |
| 5.0 mg/kg | All | 5.3 (0.7) ** ## | 36 | 32.7 (4.2) ** | 17 | 177 (33.6) # | 19 |
| Females | 5.4 (0.8) * # | 21 | 33.8 (4.5) ** | 10 | 153 (15.8) * | 11 | |
| Males | 5.2 (0.7) * ## | 15 | 31.1 (3.4) ** | 7 | 209 (23.2) ## | 8 |
| AFB1 Dose Group | Cholesterol (mg/dL) | Triglycerides (mg/dL) | ||
|---|---|---|---|---|
| 3 Weeks | 3 Months | 3 Weeks | 3 Months | |
| Control (no AFB1) | 299.16 ± 19.73 a | 184.60 ± 11.98 a# | 373.44 ± 36.72 a | 212.55 ± 12.39 a# |
| 0.5 mg/kg | 204.91 ± 17.45 b | 148.85 ± 9.83 b# | 425.93 ± 25.44 b | 200.75 ± 12.50 a# |
| 5.0 mg/kg | 190.61 ± 9.98 b | 150.22 ± 9.46 b# | 439.94 ± 23.52 b | 253.52 ± 13.85 a# |
| Tp53 | H19 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Exposure Group | Mean (SD) | N | ANOVA p-Value | Effect Estimate (SE) | p-Value | Mean (SD) | N | ANOVA p-Value | Effect Estimate (SE) | p-Value |
| Control | 5.99 (1.46) | 16 | 0.06 | ref | 49.56 (9.54) | 18 | 0.0041 | ref | ||
| Low—0.5 mg/kg | 4.82 (1.54) | 16 | −1.09 (0.34) | 0.002 | 55.33 (4.60) | 18 | 5.37 (2.56) | 0.04 | ||
| High—5.0 mg/kg | 5.12 (1.19) | 16 | −0.91 (0.34) | 0.01 | 56.80 (3.51) | 17 | 6.88 (2.64) | 0.0098 | ||
| Tp53 | H19 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Exposure Group | Mean (SD) | N | ANOVA p-Value | Effect Estimate (SE) | p-Value | Mean (SD) | N | ANOVA p-Value | Effect Estimate (SE) | p-Value |
| 3-weeks | ||||||||||
| Control | 3.10 (2.44) | 18 | 0.07 * | ref | 41.74 (2.68) | 18 | 0.08 | ref | ||
| Low—0.5 mg/kg | 4.55 (2.61) | 18 | 1.69 (0.73) | 0.02 | 40.79 (2.16) | 18 | −0.80 (1.04) | 0.44 | ||
| High—5.0 mg/kg | 6.15 (4.86) | 14 | 3.42 (0.78) | <0.0001 | 42.83 (2.88) | 16 | 1.07 (1.08) | 0.32 | ||
| 3-months | ||||||||||
| Control | 0.82 (1.20) | 20 | <0.001 | ref | 52.87 (16.08) | 17 | 0.038 | ref | ||
| Low—0.5 mg/kg | 2.48 (1.94) | 23 | 1.52 (0.38) | 0.0002 | 42.96 (6.40) | 23 | −10.7 (6.05) | 0.08 | ||
| High—5.0 mg/kg | 4.53 (2.24) | 13 | 3.11 (0.44) | <0.0001 | 41.18 (18.97) | 15 | −12.8 (6.59) | 0.05 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rotimi, O.A.; Onuzulu, C.D.; Dewald, A.L.; Ehlinger, J.; Adelani, I.B.; Olasehinde, O.E.; Rotimi, S.O.; Goodrich, J.M. Early Life Exposure to Aflatoxin B1 in Rats: Alterations in Lipids, Hormones, and DNA Methylation among the Offspring. Int. J. Environ. Res. Public Health 2021, 18, 589. https://doi.org/10.3390/ijerph18020589
Rotimi OA, Onuzulu CD, Dewald AL, Ehlinger J, Adelani IB, Olasehinde OE, Rotimi SO, Goodrich JM. Early Life Exposure to Aflatoxin B1 in Rats: Alterations in Lipids, Hormones, and DNA Methylation among the Offspring. International Journal of Environmental Research and Public Health. 2021; 18(2):589. https://doi.org/10.3390/ijerph18020589
Chicago/Turabian StyleRotimi, Oluwakemi A., Chinonye D. Onuzulu, Alisa L. Dewald, Jessa Ehlinger, Isaacson B. Adelani, Olutola E. Olasehinde, Solomon O. Rotimi, and Jaclyn M. Goodrich. 2021. "Early Life Exposure to Aflatoxin B1 in Rats: Alterations in Lipids, Hormones, and DNA Methylation among the Offspring" International Journal of Environmental Research and Public Health 18, no. 2: 589. https://doi.org/10.3390/ijerph18020589
APA StyleRotimi, O. A., Onuzulu, C. D., Dewald, A. L., Ehlinger, J., Adelani, I. B., Olasehinde, O. E., Rotimi, S. O., & Goodrich, J. M. (2021). Early Life Exposure to Aflatoxin B1 in Rats: Alterations in Lipids, Hormones, and DNA Methylation among the Offspring. International Journal of Environmental Research and Public Health, 18(2), 589. https://doi.org/10.3390/ijerph18020589