Role of NLRP3 Inflammasome Activation in Obesity-Mediated Metabolic Disorders



Abstract
1. Introduction
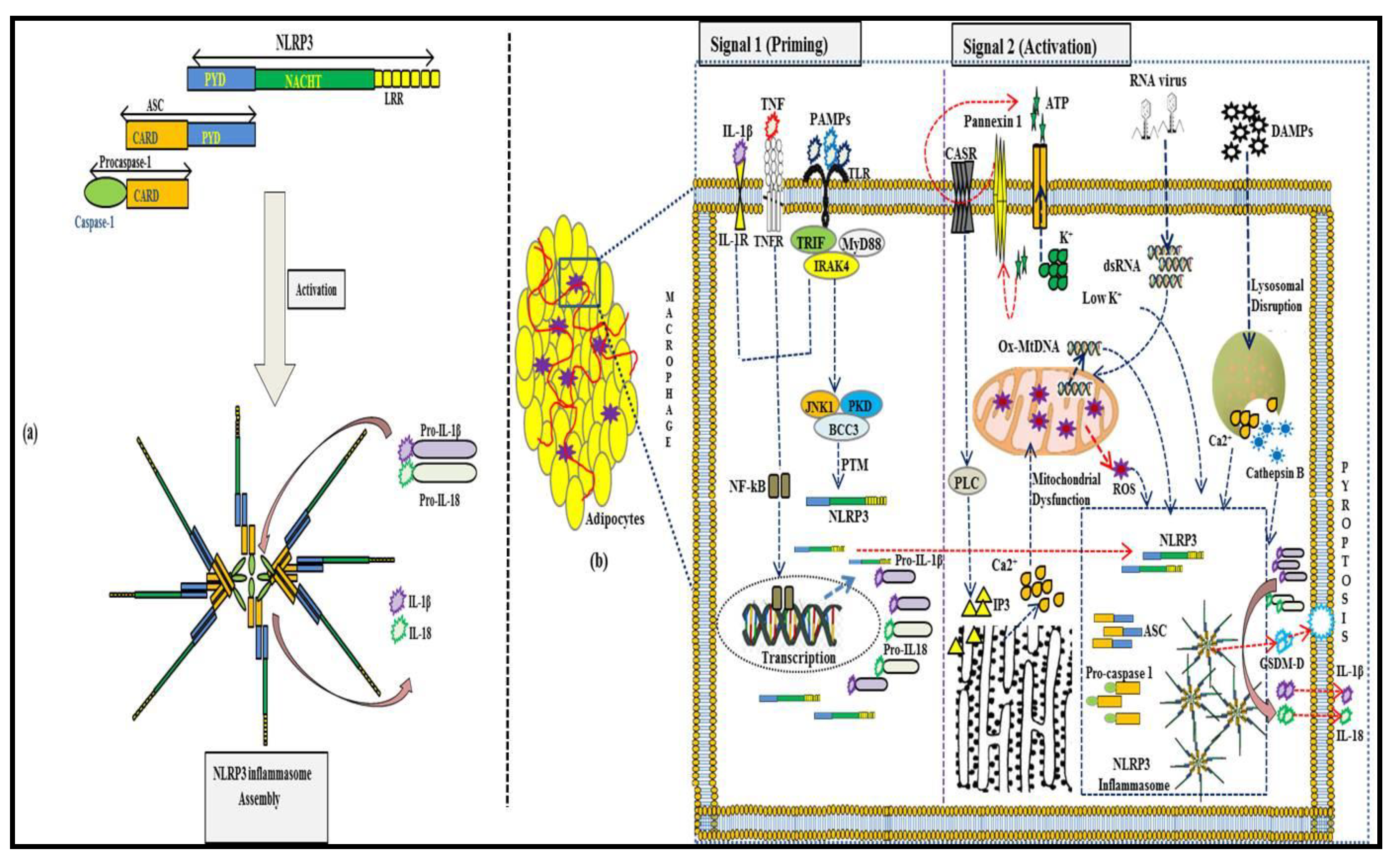
2. NLRP3 Inflammasome Activation in Adipose Tissue
2.1. Signal 1: Priming the NLRP3 Inflammasome
2.2. Signal 2: Activating the NLRP3 Inflammasome
2.2.1. Conditions Causing Permeability to Ions
K+ Efflux
Ca2+ Signaling
Na+ Influx and Cl– Efflux
2.2.2. Reactive Oxygen Species (ROS) and Mitochondrial Dysfunction
2.2.3. Lysosomal Damage
2.3. Post-Translational Modifications of NLRP3 and Associated Proteins as Regulators of NLRP3-Inflammasome Activation
2.4. NLRP3-Interacting Proteins and Their Effect on Inflammasome Activation
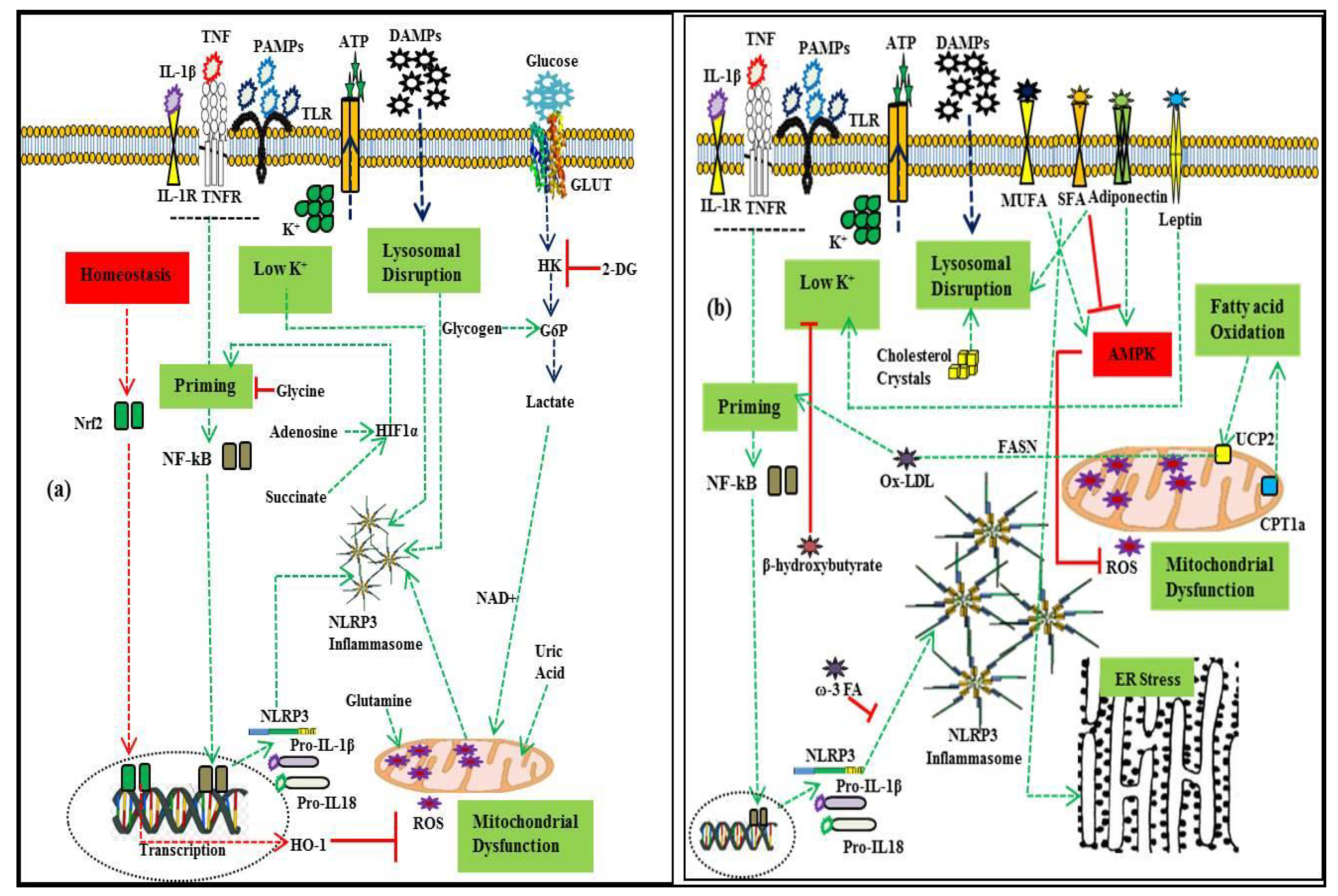
3. NLRP3 Inflammasome in Obesity-Associated Metabolic Syndrome
3.1. Metabolic Regulators of NLRP3 Inflammasome Activation
3.1.1. Lipids
3.1.2. Carbohydrates
3.1.3. Amino Acid and Nucleotide Metabolism
3.1.4. Lipopolysaccharides (LPS)
3.1.5. Adipokines
4. NLRP3 Inflammasome and Disorder in Metabolic Homeostasis
5. Inhibition of NLRP3 Inflammasome Activation
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Coyne, C.B.; Zeh, H.J.; Lotze, M.T. Pamp s and damp s: Signal 0s that spur autophagy and immunity. Immunol. Rev. 2012, 249, 158–175. [Google Scholar] [CrossRef] [PubMed]
- Vandanmagsar, B.; Youm, Y.-H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The nlrp3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proil-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Davis, B.K.; Wen, H.; Ting, J.P.-Y. The inflammasome nlrs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 2011, 29, 707–735. [Google Scholar] [CrossRef]
- Henao-Mejia, J.; Elinav, E.; Strowig, T.; Flavell, R.A. Inflammasomes: Far beyond inflammation. Nat. Immunol. 2012, 13, 321–324. [Google Scholar] [CrossRef]
- Amin, J.; Boche, D.; Rakic, S. What do we know about the inflammasome in humans? Brain Pathol. 2017, 27, 192–204. [Google Scholar] [CrossRef]
- Meunier, E.; Broz, P. Evolutionary convergence and divergence in nlr function and structure. Trends Immunol. 2017, 38, 744–757. [Google Scholar] [CrossRef]
- Hughes, M.M.; O’Neill, L.A. Metabolic regulation of nlrp 3. Immunol. Rev. 2018, 281, 88–98. [Google Scholar] [CrossRef]
- Nishimura, S.; Manabe, I.; Nagai, R. Adipose tissue inflammation in obesity and metabolic syndrome. Discov. Med. 2009, 8, 55–60. [Google Scholar]
- Trim, W.; Turner, J.E.; Thompson, D. Parallels in immunometabolic adipose tissue dysfunction with ageing and obesity. Front. Immunol. 2018, 9, 169. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Wollam, J.; Olefsky, J.M. An integrated view of immunometabolism. Cell 2018, 172, 22–40. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, S.E.; Gee, L.L.; Wachtel, M.S.; Frezza, E.E. Adipose tissue: The new endocrine organ? A review article. Dig. Dis. Sci. 2009, 54, 1847–1856. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar] [CrossRef]
- Castoldi, A.; Naffah de Souza, C.; Câmara, N.O.S.; Moraes-Vieira, P.M. The macrophage switch in obesity development. Front. Immunol. 2016, 6, 637. [Google Scholar] [CrossRef]
- Barra, N.G.; Henriksbo, B.D.; Anhê, F.F.; Schertzer, J.D. The nlrp3 inflammasome regulates adipose tissue metabolism. Biochem. J. 2020, 477, 1089–1107. [Google Scholar] [CrossRef]
- Choe, S.S.; Huh, J.Y.; Hwang, I.J.; Kim, J.I.; Kim, J.B. Adipose tissue remodeling: Its role in energy metabolism and metabolic disorders. Front. Endocrinol. 2016, 7, 30. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Saltiel, A.R. Inflammatory links between obesity and metabolic disease. J. Clin. Investig. 2011, 121, 2111–2117. [Google Scholar] [CrossRef]
- Lee, J. Adipose tissue macrophages in the development of obesity-induced inflammation, insulin resistance and type 2 diabetes. Arch. Pharmacal Res. 2013, 36, 208–222. [Google Scholar] [CrossRef]
- Johnson, A.R.; Justin Milner, J.; Makowski, L. The inflammation highway: Metabolism accelerates inflammatory traffic in obesity. Immunol. Rev. 2012, 249, 218–238. [Google Scholar] [CrossRef]
- Chimenti, M.; Triggianese, P.; Conigliaro, P.; Candi, E.; Melino, G.; Perricone, R. The interplay between inflammation and metabolism in rheumatoid arthritis. Cell Death Dis. 2015, 6, e1887. [Google Scholar] [CrossRef] [PubMed]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of nafld and obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Ballak, D.B.; Stienstra, R.; Tack, C.J.; Dinarello, C.A.; van Diepen, J.A. Il-1 family members in the pathogenesis and treatment of metabolic disease: Focus on adipose tissue inflammation and insulin resistance. Cytokine 2015, 75, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: Nf-kb activating pattern recognition and cytokine receptors license nlrp3 inflammasome activation by regulating nlrp3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The nlrp3 inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Ball, D.P.; Taabazuing, C.Y.; Griswold, A.R.; Orth, E.L.; Rao, S.D.; Kotliar, I.B.; Vostal, L.E.; Johnson, D.C.; Bachovchin, D.A. Caspase-1 interdomain linker cleavage is required for pyroptosis. Life Sci. Alliance 2020, 3. [Google Scholar] [CrossRef]
- Gurung, P.; Anand, P.K.; Malireddi, R.S.; Walle, L.V.; van Opdenbosch, N.; Dillon, C.P.; Weinlich, R.; Green, D.R.; Lamkanfi, M.; Kanneganti, T.-D.; et al. Fadd and caspase-8 mediate priming and activation of the canonical and noncanonical nlrp3 inflammasomes. J. Immunol. 2014, 192, 1835–1846. [Google Scholar] [CrossRef]
- Juliana, C.; Fernandes-Alnemri, T.; Kang, S.; Farias, A.; Qin, F.; Alnemri, E.S. Non-transcriptional priming and deubiquitination regulate nlrp3 inflammasome activation. J. Biol. Chem. 2012, 287, 36617–36622. [Google Scholar] [CrossRef]
- Lin, K.-M.; Hu, W.; Troutman, T.D.; Jennings, M.; Brewer, T.; Li, X.; Nanda, S.; Cohen, P.; Thomas, J.A.; Pasare, C.; et al. Irak-1 bypasses priming and directly links tlrs to rapid nlrp3 inflammasome activation. Proc. Natl. Acad. Sci. USA 2014, 111, 775–780. [Google Scholar] [CrossRef]
- Triantafilou, K.; Hughes, T.R.; Triantafilou, M.; Morgan, B.P. The complement membrane attack complex triggers intracellular ca2+ fluxes leading to nlrp3 inflammasome activation. J. Cell Sci. 2013, 126, 2903–2913. [Google Scholar] [CrossRef] [PubMed]
- Perregaux, D.; Gabel, C.A. Interleukin-1 beta maturation and release in response to atp and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J. Biol. Chem. 1994, 269, 15195–15203. [Google Scholar] [PubMed]
- Petrilli, V.; Papin, S.; Dostert, C.; Mayor, A.; Martinon, F.; Tschopp, J. Activation of the nalp3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007, 14, 1583–1589. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Planillo, R.; Kuffa, P.; Martínez-Colón, G.; Smith, B.L.; Rajendiran, T.M.; Núñez, G. K+ efflux is the common trigger of nlrp3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef] [PubMed]
- Compan, V.; Baroja-Mazo, A.; López-Castejón, G.; Gomez, A.I.; Martínez, C.M.; Angosto, D.; Montero, M.T.; Herranz, A.S.; Bazán, E.; Reimers, D.; et al. Cell volume regulation modulates nlrp3 inflammasome activation. Immunity 2012, 37, 487–500. [Google Scholar] [CrossRef]
- Lee, G.-S.; Subramanian, N.; Kim, A.I.; Aksentijevich, I.; Goldbach-Mansky, R.; Sacks, D.B.; Germain, R.N.; Kastner, D.L.; Chae, J.J. The calcium-sensing receptor regulates the nlrp3 inflammasome through ca 2+ and camp. Nature 2012, 492, 123–127. [Google Scholar] [CrossRef]
- Vandecaetsbeek, I.; Vangheluwe, P.; Raeymaekers, L.; Wuytack, F.; Vanoevelen, J. The ca2+ pumps of the endoplasmic reticulum and golgi apparatus. Cold Spring Harb. Perspect. Biol. 2011, 3, a004184. [Google Scholar] [CrossRef]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the nlrp3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef]
- Camello-Almaraz, C.; Gomez-Pinilla, P.J.; Pozo, M.J.; Camello, P.J. Mitochondrial reactive oxygen species and ca2+ signaling. Am. J. Physiol. Cell Physiol. 2006, 291, C1082–C1088. [Google Scholar] [CrossRef]
- Katsnelson, M.; Dubyak, G. Cytosolic k+ and extracellular na+ as regulators of nlrp3 inflammasome activation and the il-1β secretion response of macrophages to crystalline stimuli. J. Immunol. 2013, 188. [Google Scholar] [CrossRef]
- Verhoef, P.A.; Kertesy, S.B.; Lundberg, K.; Kahlenberg, J.M.; Dubyak, G.R. Inhibitory effects of chloride on the activation of caspase-1, il-1β secretion, and cytolysis by the p2x7 receptor. J. Immunol. 2005, 175, 7623–7634. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Lang, X.; Xu, C.; Wang, X.; Gong, T.; Yang, Y.; Cui, J.; Bai, L.; Wang, J.; Jiang, W.; et al. Clics-dependent chloride efflux is an essential and proximal upstream event for nlrp3 inflammasome activation. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in nlrp3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Sorbara, M.T.; Girardin, S.E. Mitochondrial ros fuel the inflammasome. Cell Res. 2011, 21, 558–560. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the nlrp3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.-H.; Brickey, W.J.; Ting, J.P. Fatty acid–induced nlrp3-asc inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef]
- He, Y.; Zeng, M.Y.; Yang, D.; Motro, B.; Núñez, G. Nek7 is an essential mediator of nlrp3 activation downstream of potassium efflux. Nature 2016, 530, 354–357. [Google Scholar] [CrossRef]
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial cardiolipin is required for nlrp3 inflammasome activation. Immunity 2013, 39, 311–323. [Google Scholar] [CrossRef]
- Subramanian, N.; Natarajan, K.; Clatworthy, M.R.; Wang, Z.; Germain, R.N. The adaptor mavs promotes nlrp3 mitochondrial localization and inflammasome activation. Cell 2013, 153, 348–361. [Google Scholar] [CrossRef]
- Park, S.; Juliana, C.; Hong, S.; Datta, P.; Hwang, I.; Fernandes-Alnemri, T.; Yu, J.-W.; Alnemri, E.S. The mitochondrial antiviral protein mavs associates with nlrp3 and regulates its inflammasome activity. J. Immunol. 2013, 191, 4358–4366. [Google Scholar] [CrossRef]
- Orlowski, G.M.; Colbert, J.D.; Sharma, S.; Bogyo, M.; Robertson, S.A.; Rock, K.L. Multiple cathepsins promote pro–il-1β synthesis and nlrp3-mediated il-1β activation. J. Immunol. 2015, 195, 1685–1697. [Google Scholar] [CrossRef] [PubMed]
- Spalinger, M.R.; Kasper, S.; Gottier, C.; Lang, S.; Atrott, K.; Vavricka, S.R.; Scharl, S.; Gutte, P.M.; Grütter, M.G.; Beer, H.-D.; et al. Nlrp3 tyrosine phosphorylation is controlled by protein tyrosine phosphatase ptpn22. J. Clin. Investig. 2016, 126, 1783–1800. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Xie, S.; Chi, Z.; Zhang, J.; Liu, Y.; Zhang, L.; Zheng, M.; Zhang, X.; Xia, D.; Ke, Y.; et al. Bile acids control inflammation and metabolic disorder through inhibition of nlrp3 inflammasome. Immunity 2016, 45, 802–816. [Google Scholar] [CrossRef] [PubMed]
- Mortimer, L.; Moreau, F.; MacDonald, J.A.; Chadee, K. Nlrp3 inflammasome inhibition is disrupted in a group of auto-inflammatory disease caps mutations. Nat. Immunol. 2016, 17, 1176–1186. [Google Scholar] [CrossRef]
- Stutz, A.; Kolbe, C.-C.; Stahl, R.; Horvath, G.L.; Franklin, B.S.; van Ray, O.; Brinkschulte, R.; Geyer, M.; Meissner, F.; Latz, E.; et al. Nlrp3 inflammasome assembly is regulated by phosphorylation of the pyrin domain. J. Exp. Med. 2017, 214, 1725–1736. [Google Scholar] [CrossRef]
- Song, N.; Liu, Z.-S.; Xue, W.; Bai, Z.-F.; Wang, Q.-Y.; Dai, J.; Liu, X.; Huang, Y.-J.; Cai, H.; Zhan, X.-Y.; et al. Nlrp3 phosphorylation is an essential priming event for inflammasome activation. Mol. Cell 2017, 68, 185–197. [Google Scholar] [CrossRef]
- Zhang, Z.; Meszaros, G.; He, W.-T.; Xu, Y.; de Fatima Magliarelli, H.; Mailly, L.; Mihlan, M.; Liu, Y.; Puig Gámez, M.; Goginashvili, A.; et al. Protein kinase d at the golgi controls nlrp3 inflammasome activation. J. Exp. Med. 2017, 214, 2671–2693. [Google Scholar] [CrossRef]
- Py, B.F.; Kim, M.-S.; Vakifahmetoglu-Norberg, H.; Yuan, J. Deubiquitination of nlrp3 by brcc3 critically regulates inflammasome activity. Mol. Cell 2013, 49, 331–338. [Google Scholar] [CrossRef]
- Han, S.; Lear, T.B.; Jerome, J.A.; Rajbhandari, S.; Snavely, C.A.; Gulick, D.L.; Gibson, K.F.; Zou, C.; Chen, B.B.; Mallampalli, R.K.; et al. Lipopolysaccharide primes the nalp3 inflammasome by inhibiting its ubiquitination and degradation mediated by the scffbxl2 e3 ligase. J. Biol. Chem. 2015, 290, 18124–18133. [Google Scholar] [CrossRef]
- Song, H.; Liu, B.; Huai, W.; Yu, Z.; Wang, W.; Zhao, J.; Han, L.; Jiang, G.; Zhang, L.; Gao, C.; et al. The e3 ubiquitin ligase trim31 attenuates nlrp3 inflammasome activation by promoting proteasomal degradation of nlrp3. Nat. Commun. 2016, 7, 1–11. [Google Scholar] [CrossRef]
- Yan, Y.; Jiang, W.; Liu, L.; Wang, X.; Ding, C.; Tian, Z.; Zhou, R. Dopamine controls systemic inflammation through inhibition of nlrp3 inflammasome. Cell 2015, 160, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, A.; Karasawa, T.; Tago, K.; Kimura, H.; Kamata, R.; Usui-Kawanishi, F.; Watanabe, S.; Ohta, S.; Funakoshi-Tago, M.; Yanagisawa, K.; et al. Arih2 ubiquitinates nlrp3 and negatively regulates nlrp3 inflammasome activation in macrophages. J. Immunol. 2017, 199, 3614–3622. [Google Scholar] [CrossRef] [PubMed]
- Humphries, F.; Bergin, R.; Jackson, R.; Delagic, N.; Wang, B.; Yang, S.; Dubois, A.V.; Ingram, R.J.; Moynagh, P.N. The e3 ubiquitin ligase pellino2 mediates priming of the nlrp3 inflammasome. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Guan, K.; Wei, C.; Zheng, Z.; Song, T.; Wu, F.; Zhang, Y.; Cao, Y.; Ma, S.; Chen, W.; Xu, Q.; et al. Mavs promotes inflammasome activation by targeting asc for k63-linked ubiquitination via the e3 ligase traf3. J. Immunol. 2015, 194, 4880–4890. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Seo, D.; You, J.; Chung, S.; Park, J.S.; Lee, J.H.; Jung, S.M.; Lee, Y.S.; Park, S.H. The deubiquitinating enzyme, ubiquitin-specific peptidase 50, regulates inflammasome activation by targeting the asc adaptor protein. FEBS Lett. 2017, 591, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Boisson, B.; Casanova, J.-L. Lubac: A New Function in Immunity; The Rockefeller University Press: New York, NY, USA, 2014. [Google Scholar]
- Labbé, K.; McIntire, C.R.; Doiron, K.; Leblanc, P.M.; Saleh, M. Cellular inhibitors of apoptosis proteins ciap1 and ciap2 are required for efficient caspase-1 activation by the inflammasome. Immunity 2011, 35, 897–907. [Google Scholar] [CrossRef]
- Duong, B.H.; Onizawa, M.; Oses-Prieto, J.A.; Advincula, R.; Burlingame, A.; Malynn, B.A.; Ma, A. A20 restricts ubiquitination of pro-interleukin-1β protein complexes and suppresses nlrp3 inflammasome activity. Immunity 2015, 42, 55–67. [Google Scholar] [CrossRef]
- He, Y.; Varadarajan, S.; Muñoz-Planillo, R.; Burberry, A.; Nakamura, Y.; Núñez, G. 3, 4-methylenedioxy-β-nitrostyrene inhibits nlrp3 inflammasome activation by blocking assembly of the inflammasome. J. Biol. Chem. 2014, 289, 1142–1150. [Google Scholar] [CrossRef]
- Hernandez-Cuellar, E.; Tsuchiya, K.; Hara, H.; Fang, R.; Sakai, S.; Kawamura, I.; Akira, S.; Mitsuyama, M. Cutting edge: Nitric oxide inhibits the nlrp3 inflammasome. J. Immunol. 2012, 189, 5113–5117. [Google Scholar] [CrossRef]
- Schmid-Burgk, J.L.; Chauhan, D.; Schmidt, T.; Ebert, T.S.; Reinhardt, J.; Endl, E.; Hornung, V. A genome-wide crispr (clustered regularly interspaced short palindromic repeats) screen identifies nek7 as an essential component of nlrp3 inflammasome activation. J. Biol. Chem. 2016, 291, 103–109. [Google Scholar] [CrossRef]
- Shi, H.; Wang, Y.; Li, X.; Zhan, X.; Tang, M.; Fina, M.; Su, L.; Pratt, D.; Bu, C.H.; Hildebrand, S.; et al. Nlrp3 activation and mitosis are mutually exclusive events coordinated by nek7, a new inflammasome component. Nat. Immunol. 2016, 17, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Cullati, S.N.; Kabeche, L.; Kettenbach, A.N.; Gerber, S.A. A bifurcated signaling cascade of nima-related kinases controls distinct kinesins in anaphase. J. Cell Biol. 2017, 216, 2339–2354. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.; Nakajima, S.; Wang, Q.; Sun, H.; Xue, J.; Wu, J.; Hellwig, S.; Zeng, X.; Yates, N.A.; Smithgall, T.E.; et al. Nek7 protects telomeres from oxidative DNA damage by phosphorylation and stabilization of trf1. Mol. Cell 2017, 65, 818–831. [Google Scholar] [CrossRef] [PubMed]
- Kooi, I.E.; Mol, B.M.; Massink, M.P.; de Jong, M.C.; de Graaf, P.; van der Valk, P.; Meijers-Heijboer, H.; Kaspers, G.J.; Moll, A.C.; te Riele, H.; et al. A meta-analysis of retinoblastoma copy numbers refines the list of possible driver genes involved in tumor progression. PLoS ONE 2016, 11, e0153323. [Google Scholar] [CrossRef]
- Li, X.; Thome, S.; Ma, X.; Amrute-Nayak, M.; Finigan, A.; Kitt, L.; Masters, L.; James, J.R.; Shi, Y.; Meng, G.; et al. Mark4 regulates nlrp3 positioning and inflammasome activation through a microtubule-dependent mechanism. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef]
- Wang, J.; Shen, X.; Liu, J.; Chen, W.; Wu, F.; Wu, W.; Meng, Z.; Zhu, M.; Miao, C. High glucose mediates nlrp3 inflammasome activation via upregulation of elf3 expression. Cell Death Dis. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Mayor, A.; Martinon, F.; De Smedt, T.; Pétrilli, V.; Tschopp, J. A crucial function of sgt1 and hsp90 in inflammasome activity links mammalian and plant innate immune responses. Nat. Immunol. 2007, 8, 497–503. [Google Scholar] [CrossRef]
- Martine, P.; Rébé, C. Heat shock proteins and inflammasomes. Int. J. Mol. Sci. 2019, 20, 4508. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef]
- Shenoy, A.R.; Wellington, D.A.; Kumar, P.; Kassa, H.; Booth, C.J.; Cresswell, P.; MacMicking, J.D. Gbp5 promotes nlrp3 inflammasome assembly and immunity in mammals. Science 2012, 336, 481–485. [Google Scholar] [CrossRef]
- Lu, B.; Nakamura, T.; Inouye, K.; Li, J.; Tang, Y.; Lundbäck, P.; Valdes-Ferrer, S.I.; Olofsson, P.S.; Kalb, T.; Roth, J.; et al. Novel role of pkr in inflammasome activation and hmgb1 release. Nature 2012, 488, 670–674. [Google Scholar] [CrossRef] [PubMed]
- Yim, H.C.; Wang, D.; Yu, L.; White, C.L.; Faber, P.W.; Williams, B.R.; Sadler, A.J. The kinase activity of pkr represses inflammasome activity. Cell Res. 2016, 26, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Rheinheimer, J.; de Souza, B.M.; Cardoso, N.S.; Bauer, A.C.; Crispim, D. Current role of the nlrp3 inflammasome on obesity and insulin resistance: A systematic review. Metabolism 2017, 74, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Esser, N.; L’homme, L.; De Roover, A.; Kohnen, L.; Scheen, A.J.; Moutschen, M.; Piette, J.; Legrand-Poels, S.; Paquot, N. Obesity phenotype is related to nlrp3 inflammasome activity and immunological profile of visceral adipose tissue. Diabetologia 2013, 56, 2487–2497. [Google Scholar] [CrossRef] [PubMed]
- Moschen, A.R.; Molnar, C.; Enrich, B.; Geiger, S.; Ebenbichler, C.F.; Tilg, H. Adipose and liver expression of interleukin (il)-1 family members in morbid obesity and effects of weight loss. Mol. Med. 2011, 17, 840–845. [Google Scholar] [CrossRef] [PubMed]
- Mocanu, A.O.; Mulya, A.; Huang, H.; Dan, O.; Shimizu, H.; Batayyah, E.; Brethauer, S.A.; Dinischiotu, A.; Kirwan, J.P. Effect of roux-en-y gastric bypass on the nlrp3 inflammasome in adipose tissue from obese rats. PLoS ONE 2015, 10, e0139764. [Google Scholar] [CrossRef]
- Ringling, R.E.; Gastecki, M.L.; Woodford, M.L.; Lum-Naihe, K.J.; Grant, R.W.; Pulakat, L.; Vieira-Potter, V.J.; Padilla, J. Loss of nlrp3 does not protect mice from western diet-induced adipose tissue inflammation and glucose intolerance. PLoS ONE 2016, 11, e0161939. [Google Scholar] [CrossRef]
- Finucane, O.M.; Lyons, C.L.; Murphy, A.M.; Reynolds, C.M.; Klinger, R.; Healy, N.P.; Cooke, A.A.; Coll, R.C.; McAllan, L.; Nilaweera, K.N.; et al. Monounsaturated fatty acid–enriched high-fat diets impede adipose nlrp3 inflammasome–mediated il-1β secretion and insulin resistance despite obesity. Diabetes 2015, 64, 2116–2128. [Google Scholar] [CrossRef]
- Karasawa, T.; Kawashima, A.; Usui-Kawanishi, F.; Watanabe, S.; Kimura, H.; Kamata, R.; Shirasuna, K.; Koyama, Y.; Sato-Tomita, A.; Matsuzaka, T.; et al. Saturated fatty acids undergo intracellular crystallization and activate the nlrp3 inflammasome in macrophages. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 744–756. [Google Scholar] [CrossRef]
- Zeng, X.; Zhu, M.; Liu, X.; Chen, X.; Yuan, Y.; Li, L.; Liu, J.; Lu, Y.; Cheng, J.; Chen, Y.; et al. Oleic acid ameliorates palmitic acid induced hepatocellular lipotoxicity by inhibition of er stress and pyroptosis. Nutr. Metab. 2020, 17, 1–14. [Google Scholar]
- Yan, Y.; Jiang, W.; Spinetti, T.; Tardivel, A.; Castillo, R.; Bourquin, C.; Guarda, G.; Tian, Z.; Tschopp, J.; Zhou, R.; et al. Omega-3 fatty acids prevent inflammation and metabolic disorder through inhibition of nlrp3 inflammasome activation. Immunity 2013, 38, 1154–1163. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-Y.; Dong, Y.-Q.; Wang, P.; Zhang, X.; Yan, Y.; Sun, L.; Liu, B.; Zhang, D.; Zhang, H.; Liu, H.; et al. Adipocyte-derived lysophosphatidylcholine activates adipocyte and adipose tissue macrophage nod-like receptor protein 3 inflammasomes mediating homocysteine-induced insulin resistance. EBioMedicine 2018, 31, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. Nlrp3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [PubMed]
- Sheedy, F.J.; Grebe, A.; Rayner, K.J.; Kalantari, P.; Ramkhelawon, B.; Carpenter, S.B.; Becker, C.E.; Ediriweera, H.N.; Mullick, A.E.; Golenbock, D.T.; et al. Cd36 coordinates nlrp3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 2013, 14, 812–820. [Google Scholar] [CrossRef] [PubMed]
- Zang, Y.-H.; Chen, D.; Zhou, B.; Chen, A.-D.; Wang, J.-J.; Gao, X.-Y.; Chen, Q.; Li, Y.-H.; Kang, Y.-M.; Zhu, G.-Q.; et al. Fndc5 inhibits foam cell formation and monocyte adhesion in vascular smooth muscle cells via suppressing nfκb-mediated nlrp3 upregulation. Vasc. Pharmacol. 2019, 121. [Google Scholar] [CrossRef]
- Koenen, T.B.; Stienstra, R.; Van Tits, L.J.; De Graaf, J.; Stalenhoef, A.F.; Joosten, L.A.; Tack, C.J.; Netea, M.G. Hyperglycemia activates caspase-1 and txnip-mediated il-1β transcription in human adipose tissue. Diabetes 2011, 60, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Zhou, W.; Zhang, Y.; Li, J.; Zhao, Y.; Pan, L.; Shen, Z.; Chen, W.; Hui, J. Aminooxyacetic acid attenuates post-infarct cardiac dysfunction by balancing macrophage polarization through modulating macrophage metabolism in mice. J. Cell. Mol. Med. 2020, 24, 2593–2609. [Google Scholar] [CrossRef]
- Moon, J.-S.; Hisata, S.; Park, M.-A.; DeNicola, G.M.; Ryter, S.W.; Nakahira, K.; Choi, A.M. Mtorc1-induced hk1-dependent glycolysis regulates nlrp3 inflammasome activation. Cell Rep. 2015, 12, 102–115. [Google Scholar] [CrossRef]
- Xie, M.; Yu, Y.; Kang, R.; Zhu, S.; Yang, L.; Zeng, L.; Sun, X.; Yang, M.; Billiar, T.R.; Wang, H.; et al. Pkm2-dependent glycolysis promotes nlrp3 and aim2 inflammasome activation. Nat. Commun. 2016, 7, 1–13. [Google Scholar] [CrossRef]
- Thwe, P.M.; Fritz, D.I.; Snyder, J.P.; Smith, P.R.; Curtis, K.D.; O’Donnell, A.; Galasso, N.A.; Sepaniac, L.A.; Adamik, B.J.; Hoyt, L.R.; et al. Syk-dependent glycolytic reprogramming in dendritic cells regulates il-1β production to β-glucan ligands in a TLR-independent manner. J. Leukoc. Biol. 2019, 106, 1325–1335. [Google Scholar] [CrossRef]
- Yang, S.-J.; Han, A.R.; Kim, E.-A.; Yang, J.W.; Ahn, J.-Y.; Na, J.-M.; Cho, S.-W. Khg21834 attenuates glutamate-induced mitochondrial damage, apoptosis, and nlrp3 inflammasome activation in sh-sy5y human neuroblastoma cells. Eur. J. Pharmacol. 2019, 856. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ma, X.; Jiang, D.; Chen, J.; Jia, H.; Wu, Z.; Kim, I.H.; Yang, Y. Glycine attenuates lipopolysaccharide-induced acute lung injury by regulating nlrp3 inflammasome and nrf2 signaling. Nutrients 2020, 12, 611. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; Ghani, A.; Malik, A.; Wilder, T.; Colegio, O.R.; Flavell, R.A.; Cronstein, B.N.; Mehal, W.Z. Adenosine is required for sustained inflammasome activation via the a 2a receptor and the hif-1α pathway. Nat. Commun. 2013, 4, 1–9. [Google Scholar] [CrossRef]
- Nomura, J.; Kobayashi, T.; So, A.; Busso, N. Febuxostat, a xanthine oxidoreductase inhibitor, decreases nlrp3-dependent inflammation in macrophages by activating the purine salvage pathway and restoring cellular bioenergetics. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Eleftheriadis, T.; Pissas, G.; Antoniadi, G.; Makri, P.; Liakopoulos, V.; Stefanidis, I. Urate crystals induce nlrp3 inflammasome-dependent il-1β secretion and proliferation in isolated primary human t-cells. Hippokratia 2015, 19, 41. [Google Scholar]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef]
- Wang, F.; Wang, L.; Liu, Y.; Yuan, J.; Mo, Z. Adiponectin attenuates nlrp3 inflammasome by modulating ampk-ros pathway. Am Diabetes Assoc. 2018, 67. [Google Scholar] [CrossRef]
- Lagathu, C.; Yvan-Charvet, L.; Bastard, J.-P.; Maachi, M.; Quignard-Boulange, A.; Capeau, J.; Caron, M. Long-term treatment with interleukin-1β induces insulin resistance in murine and human adipocytes. Diabetologia 2006, 49, 2162–2173. [Google Scholar] [CrossRef]
- Fu, S.; Liu, L.; Han, L.; Yu, Y. Leptin promotes il-18 secretion by activating the nlrp3 inflammasome in raw 264.7 cells. Mol. Med. Rep. 2017, 16, 9770–9776. [Google Scholar] [CrossRef][Green Version]
- Bauernfeind, F.; Niepmann, S.; Knolle, P.A.; Hornung, V. Aging-associated tnf production primes inflammasome activation and nlrp3-related metabolic disturbances. J. Immunol. 2016, 197, 2900–2908. [Google Scholar] [CrossRef]
- Steen, K.A.; Xu, H.; Bernlohr, D.A. Fabp4/ap2 regulates macrophage redox signaling and inflammasome activation via control of ucp2. Mol. Cell. Biol. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Vlaicu, S.I.; Tatomir, A.; Boodhoo, D.; Vesa, S.; Mircea, P.A.; Rus, H. The role of complement system in adipose tissue-related inflammation. Immunol. Res. 2016, 64, 653–664. [Google Scholar] [CrossRef]
- Wang, J.; Wen, Y.; Lv, L.-L.; Liu, H.; Tang, R.-N.; Ma, K.-L.; Liu, B.-C. Involvement of endoplasmic reticulum stress in angiotensin ii-induced nlrp3 inflammasome activation in human renal proximal tubular cells in vitro. Acta Pharmacol. Sin. 2015, 36, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Chi, Y.; Chai, J.; Xu, C.; Luo, H.; Zhang, Q. Apelin inhibits the activation of the nucleotide-binding domain and the leucine-rich, repeat-containing family, pyrin-containing 3 (nlrp3) inflammasome and ameliorates insulin resistance in severely burned rats. Surgery 2015, 157, 1142–1152. [Google Scholar] [CrossRef] [PubMed]
- Bian, F.; Yang, X.-Y.; Xu, G.; Zheng, T.; Jin, S. Crp-induced nlrp3 inflammasome activation increases ldl transcytosis across endothelial cells. Front. Pharmacol. 2019, 10, 40. [Google Scholar] [CrossRef] [PubMed]
- Ahechu, P.; Zozaya, G.; Martí, P.; Hernández-Lizoáin, J.L.; Baixauli, J.; Unamuno, X.; Frühbeck, G.; Catalán, V. Nlrp3 inflammasome: A possible link between obesity-associated low-grade chronic inflammation and colorectal cancer development. Front. Immunol. 2018, 9, 2918. [Google Scholar] [CrossRef] [PubMed]
- Kusunoki, H.; Taniyama, Y.; Otsu, R.; Rakugi, H.; Morishita, R. Anti-inflammatory effects of hepatocyte growth factor on the vicious cycle of macrophages and adipocytes. Hypertens. Res. 2014, 37, 500–506. [Google Scholar] [CrossRef]
- Frank, P.G.; Lisanti, M.P. Icam-1: Role in inflammation and in the regulation of vascular permeability. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H926–H927. [Google Scholar] [CrossRef]
- Hasham, S.N.; Pillarisetti, S. Vascular lipases, inflammation and atherosclerosis. Clin. Chim. Acta 2006, 372, 179–183. [Google Scholar] [CrossRef]
- McAllister, M.; Chemaly, M.; Eakin, A.J.; Gibson, D.S.; McGilligan, V.E. Nlrp3 as a potentially novel biomarker for the management of osteoarthritis. Osteoarthr. Cartil. 2018, 26, 612–619. [Google Scholar] [CrossRef]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte chemoattractant protein-1 (mcp-1): An overview. J. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhang, Z.; Qian, G.; Zhou, J. Omentin-1 attenuates adipose tissue inflammation via restoration of txnip/nlrp3 signaling in high-fat diet-induced obese mice. Fundam. Clin. Pharmacol. 2020, 34. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, G.; Xu, C.; Liu, L.; Zhang, Q.; Xu, Q.; Jia, H.; Li, X.; Li, X. Perilipin 1 mediates lipid metabolism homeostasis and inhibits inflammatory cytokine synthesis in bovine adipocytes. Front. Immunol. 2018, 9, 467. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.-M. Oxidative stress, plasminogen activator inhibitor 1, and lung fibrosis. Antioxid. Redox Signal. 2008, 10, 303–320. [Google Scholar] [CrossRef]
- Niemi, K.; Teirilä, L.; Lappalainen, J.; Rajamäki, K.; Baumann, M.H.; Öörni, K.; Wolff, H.; Kovanen, P.T.; Matikainen, S.; Eklund, K.K.; et al. Serum amyloid a activates the nlrp3 inflammasome via p2×7 receptor and a cathepsin b-sensitive pathway. J. Immunol. 2011, 186, 6119–6128. [Google Scholar] [CrossRef]
- Xu, L.; Wang, Z.; Deng, L. Effect of vaspin on myocardial ischemia-reperfusion injury rats and expression of nlr family pyrin domain containing 3 (nlrp3). J. Biomater. Tissue Eng. 2020, 10, 895–900. [Google Scholar] [CrossRef]
- Romacho, T.; Valencia, I.; Ramos-González, M.; Vallejo, S.; López-Esteban, M.; Lorenzo, O.; Cannata, P.; Romero, A.; San Hipólito-Luengo, A.; Gómez-Cerezo, J.F.; et al. Visfatin/enampt induces endothelial dysfunction in vivo: A role for toll-like receptor 4 and nlrp3 inflammasome. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Kursawe, R.; Dixit, V.D.; Scherer, P.E.; Santoro, N.; Narayan, D.; Gordillo, R.; Giannini, C.; Lopez, X.; Pierpont, B.; Nouws, J.; et al. A role of the inflammasome in the low storage capacity of the abdominal subcutaneous adipose tissue in obese adolescents. Diabetes 2016, 65, 610–618. [Google Scholar] [CrossRef]
- Sun, K.; Scherer, P.E. Adipose Tissue Dysfunction: A Multistep Process. In Novel Insights into Adipose Cell Functions; Springer: Berlin/Heidelberg, Germany, 2010; pp. 67–75. [Google Scholar]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef]
- Koumenis, C.; Naczki, C.; Koritzinsky, M.; Rastani, S.; Diehl, A.; Sonenberg, N.; Koromilas, A.; Wouters, B.G. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase perk and phosphorylation of the translation initiation factor eif2α. Mol. Cell. Biol. 2002, 22, 7405–7416. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ebenezer, P.J.; Dasuri, K.; Fernandez-Kim, S.O.; Francis, J.; Mariappan, N.; Gao, Z.; Ye, J.; Bruce-Keller, A.J.; Keller, J.N.; et al. Aging is associated with hypoxia and oxidative stress in adipose tissue: Implications for adipose function. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E599–E607. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Lam, K.S.; Wang, Y.; Wu, D.; Lam, M.C.; Shen, J.; Wong, L.; Hoo, R.L.; Zhang, J.; Xu, A.; et al. Hypoxia dysregulates the production of adiponectin and plasminogen activator inhibitor-1 independent of reactive oxygen species in adipocytes. Biochem. Biophys. Res. Commun. 2006, 341, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Ambrosini, G.; Nath, A.K.; Sierra-Honigmann, M.R.; Flores-Riveros, J. Transcriptional activation of the human leptin gene in response to hypoxia involvement of hypoxia-inducible factor 1. J. Biol. Chem. 2002, 277, 34601–34609. [Google Scholar] [CrossRef]
- Trayhurn, P. Hypoxia and adipose tissue function and dysfunction in obesity. Physiol. Rev. 2013, 93, 1–21. [Google Scholar] [CrossRef]
- Abranches, M.V.; de Oliveira, F.C.E.; da Conceição, L.L.; Peluzio, M.D.C.G. Obesity and diabetes: The link between adipose tissue dysfunction and glucose homeostasis. Nutr. Res. Rev. 2015, 28, 121–132. [Google Scholar] [CrossRef]
- Schinner, S.; Scherbaum, W.; Bornstein, S.; Barthel, A. Molecular mechanisms of insulin resistance. Diabet. Med. 2005, 22, 674–682. [Google Scholar] [CrossRef]
- Stienstra, R.; Joosten, L.A.; Koenen, T.; van Tits, B.; van Diepen, J.A.; van Den Berg, S.A.; Rensen, P.C.; Voshol, P.J.; Fantuzzi, G.; Hijmans, A.; et al. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab. 2010, 12, 593–605. [Google Scholar] [CrossRef]
- Gao, D.; Madi, M.; Ding, C.; Fok, M.; Steele, T.; Ford, C.; Hunter, L.; Bing, C. Interleukin-1β mediates macrophage-induced impairment of insulin signaling in human primary adipocytes. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E289–E304. [Google Scholar] [CrossRef]
- Newsholme, P.; Cruzat, V.F.; Keane, K.N.; Carlessi, R.; de Bittencourt, P.I.H., Jr. Molecular mechanisms of ros production and oxidative stress in diabetes. Biochem. J. 2016, 473, 4527–4550. [Google Scholar] [CrossRef]
- Ackermann, J.A.; Hofheinz, K.; Zaiss, M.M.; Krönke, G. The double-edged role of 12/15-lipoxygenase during inflammation and immunity. Biochim. Biophys. Acta 2017, 1862, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Nunes, S.; Rolo, A.P.; Palmeira, C.M.; Reis, F. Diabetic cardiomyopathy: Focus on oxidative stress, mitochondrial dysfunction and inflammation. In Cardiomyopathies-Types and Treatments; Intech: London, UK, 2017; pp. 235–257. [Google Scholar]
- Rani, V.; Deep, G.; Singh, R.K.; Palle, K.; Yadav, U.C. Oxidative stress and metabolic disorders: Pathogenesis and therapeutic strategies. Life Sci. 2016, 148, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Manna, P.; Jain, S.K. Obesity, oxidative stress, adipose tissue dysfunction, and the associated health risks: Causes and therapeutic strategies. Metab. Syndr. Relat. Disord. 2015, 13, 423–444. [Google Scholar] [CrossRef] [PubMed]
- Camell, C.D.; Sander, J.; Spadaro, O.; Lee, A.; Nguyen, K.Y.; Wing, A.; Goldberg, E.L.; Youm, Y.-H.; Brown, C.W.; Elsworth, J.; et al. Inflammasome-driven catecholamine catabolism in macrophages blunts lipolysis during ageing. Nature 2017, 550, 119–123. [Google Scholar] [CrossRef]
- Jiang, H.; He, H.; Chen, Y.; Huang, W.; Cheng, J.; Ye, J.; Wang, A.; Tao, J.; Wang, C.; Liu, Q.; et al. Identification of a selective and direct nlrp3 inhibitor to treat inflammatory disorders. J. Exp. Med. 2017, 214, 3219–3238. [Google Scholar] [CrossRef]
- Huang, Y.; Jiang, H.; Chen, Y.; Wang, X.; Yang, Y.; Tao, J.; Deng, X.; Liang, G.; Zhang, H.; Jiang, W.; et al. Tranilast directly targets nlrp 3 to treat inflammasome-driven diseases. EMBO Mol. Med. 2018, 10, e8689. [Google Scholar] [CrossRef]
- Marchetti, C.; Swartzwelter, B.; Gamboni, F.; Neff, C.P.; Richter, K.; Azam, T.; Carta, S.; Tengesdal, I.; Nemkov, T.; D’Alessandro, A.; et al. Olt1177, a β-sulfonyl nitrile compound, safe in humans, inhibits the nlrp3 inflammasome and reverses the metabolic cost of inflammation. Proc. Natl. Acad. Sci. USA 2018, 115, E1530–E1539. [Google Scholar] [CrossRef]
- He, H.; Jiang, H.; Chen, Y.; Ye, J.; Wang, A.; Wang, C.; Liu, Q.; Liang, G.; Deng, X.; Jiang, W. Oridonin is a covalent nlrp3 inhibitor with strong anti-inflammasome activity. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Shim, D.-W.; Shin, W.-Y.; Yu, S.-H.; Kim, B.-H.; Ye, S.-K.; Koppula, S.; Won, H.-S.; Kang, T.-B.; Lee, K.-H. Bot-4-one attenuates nlrp3 inflammasome activation: Nlrp3 alkylation leading to the regulation of its atpase activity and ubiquitination. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Cocco, M.; Pellegrini, C.; Martínez-Banaclocha, H.; Giorgis, M.; Marini, E.; Costale, A.; Miglio, G.; Fornai, M.; Antonioli, L.; Lopez-Castejon, G.; et al. Development of an acrylate derivative targeting the nlrp3 inflammasome for the treatment of inflammatory bowel disease. J. Med. Chem. 2017, 60, 3656–3671. [Google Scholar] [CrossRef]
- Juliana, C.; Fernandes-Alnemri, T.; Wu, J.; Datta, P.; Solorzano, L.; Yu, J.-W.; Meng, R.; Quong, A.A.; Latz, E.; Scott, C.P.; et al. Anti-inflammatory compounds parthenolide and bay 11–7082 are direct inhibitors of the inflammasome. J. Biol. Chem. 2010, 285, 9792–9802. [Google Scholar] [CrossRef] [PubMed]
- Rudolphi, K.; Gerwin, N.; Verzijl, N.V.D.; van der Kraan, P.V.D.; van den Berg, W. Pralnacasan, an inhibitor of interleukin-1β converting enzyme, reduces joint damage in two murine models of osteoarthritis. Osteoarthr. Cartil. 2003, 11, 738–746. [Google Scholar] [CrossRef]
- Altenhöfer, S.; Kleikers, P.W.; Radermacher, K.A.; Scheurer, P.; Hermans, J.R.; Schiffers, P.; Ho, H.; Wingler, K.; Schmidt, H.H. The nox toolbox: Validating the role of nadph oxidases in physiology and disease. Cell. Mol. Life Sci. 2012, 69, 2327–2343. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.-S.; Nakahira, K.; Chung, K.-P.; DeNicola, G.M.; Koo, M.J.; Pabón, M.A.; Rooney, K.T.; Yoon, J.-H.; Ryter, S.W.; Stout-Delgado, H.; et al. Nox4-dependent fatty acid oxidation promotes nlrp3 inflammasome activation in macrophages. Nat. Med. 2016, 22, 1002–1012. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yao, X.; Zhu, Y.; Zhang, H.; Wang, H.; Ma, Q.; Yan, F.; Yang, Y.; Zhang, J.; Shi, H.; et al. The caspase inhibitor z-vad-fmk alleviates endotoxic shock via inducing macrophages necroptosis and promoting mdscs-mediated inhibition of macrophages activation. Front. Immunol. 2019, 10, 1824. [Google Scholar] [CrossRef] [PubMed]
- Rathkey, J.K.; Zhao, J.; Liu, Z.; Chen, Y.; Yang, J.; Kondolf, H.C.; Benson, B.L.; Chirieleison, S.M.; Huang, A.Y.; Dubyak, G.R.; et al. Chemical disruption of the pyroptotic pore-forming protein gasdermin d inhibits inflammatory cell death and sepsis. Sci. Immunol. 2018, 3. [Google Scholar] [CrossRef]
- Ashcroft, F.M. Atp-sensitive potassium channelopathies: Focus on insulin secretion. J. Clin. Investig. 2005, 115, 2047–2058. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, C.; Chojnacki, J.; Toldo, S.; Mezzaroma, E.; Tranchida, N.; Rose, S.W.; Federici, M.; van Tassell, B.W.; Zhang, S.; Abbate, A.; et al. A novel pharmacologic inhibitor of the nlrp3 inflammasome limits myocardial injury following ischemia-reperfusion in the mouse. J. Cardiovasc. Pharmacol. 2014, 63, 316. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Sl# | PTM | Protein | Modification Site | Regulatory Enzyme | Role in Regulation of NLRP3 Inflammasome | Ref. |
|---|---|---|---|---|---|---|
| 1 | Phosphorylation | NLRP3 | Y861 (LRR domain) | Protein tyrosine phosphatase non-receptor 22 (PTPN22) | Dephosphorylation at Typ861 residue leads to efficient NLRP3 inflammasome activation and Il-1β secretion. ● | [52] |
| 2 | Ser291 or Ser295 (NACHT domain) | Protein Kinase A (PKA) | Cholesterol catabolism leads to an increase in intracellular cAMP levels, activating PKA, which phosphorylates NACHT domain at Ser291 or Ser295 site, leading to NLRP3 degradation and inhibiting inflammasome activation. ● | [53,54] | ||
| 3 | Ser5 (PYD domain) | Protein Phosphatase 2A (PP2A) | Dephosphorylation of Ser5 site at the PYD domain of NLRP3 protein by PP2A promotes NLRP3-ASC interaction, which is required for inflammasome assembly. ● | [55] | ||
| 4 | Ser194 (NACHT domain) | Jun N-Terminal Kinase (JNK) | TLR ligands phosphorylate the Ser194 residue in the NACHT domain of NLRP3 protein in a JNK1-dependent manner to facilitate inflammasome assembly. ● | [56] | ||
| 5 | Ser295 (NACHT domain) | Protein Kinase D (PKD) | Activated PKD phosphorylates the Ser293 residue in the NACHT domain, promoting its release from mitochondria-associated membranes (MAMs) to the cytoplasm, thereby facilitating inflammasome assembly and maturation. ● | [57] | ||
| 6 | Ubiquitination | NLRP3 | LRR domain at an unknown site | BRCA1/BRCA2-containing complex 3 (BRCC3) | LPS stimulation induces NLRP3 deubiquitination at an unknown site of the LRR domain in a BRCC3-mediated manner to promote activation. ● | [58] |
| 7 | Lys689 (LRR domain) | F-Box L2 (FBXL2) | FBXL2, a ubiquitin E3 ligase, interacts with NLRP3 protein at lysine689 residue of the LRR domain, leading to its degradation. ● | [59] | ||
| 8 | PYD domain at an unknown site | Tripartite motif-containing protein31 (TRIM31) | TRIM31, a ubiquitin E3 ligase, promotes its K48-linked ubiquitination at the PYD domain leading to proteosomal degradation, and is a part of the feedback suppressor of the inflammasome. ● | [60] | ||
| 9 | NACHT and LRR domains at unknown sites | Membrane-associated RING-CH-type finger protein 7 (MARCH 7) | MARCH 7, another ubiquitin E3 ligase, promotes ubiquitination and degradation of NLRP3 at both NACHT and LRR domains in response to stimulation of dopamine D1 receptor (DRD1), leading to NLRP3 inflammasome inhibition. ● | [61] | ||
| 10 | NACHT domain at an unknown site | Ariadne Homolog 2 (ARIH2) | ARIH2, another ubiquitin E3 ligase, induces K48 ubiquitination at the NACHT domain of NLRP3 and acts as an endogenous negative regulator of NLRP3 inflammasome activation. ● | [62] | ||
| 11 | Unknown domain | Pellino2 (PEL2) | Pellino2, an E3 ubiquitin ligase, facilitates the activation of NLRP3-inflammasome by promoting the ubiquitination of NLRP3 at an unknown domain during the priming stage. ● | [63] | ||
| 12 | ASC | CARD domain | Tumor necrosis factor receptor-associated factor 3 (TRAF3) | Ubiquitination of CARD domain of ASC at K174 residue with K63 chains stabilizes it and promotes NLRP3-inflammasome assembly. ● | [64] | |
| 13 | Unknown domain | Ubiquitin Specific Peptidase 50 (USP50) | USP50 deubiquitinates Lys-63 at an unknown domain of ASC and promotes speck formation and oligomerization, helping in NLRP3-inflammasome activation. ● | [65] | ||
| 14 | Unknown domain | Linear Ubiquitin Assembly Complex (LUBAC) | LUBAC has been implicated as a key driver of the nuclear translocation of NF-kB and hence plays an important role in NLRP3-inflammasome activation. ● | [66] | ||
| 15 | Caspase-1 | CARD domain | Cellular inhibitor of Apoptosis proteins 2 (clAP-2) | clAP-2 mediates the polyubiquitination of the CARD domain of caspase-1 leading to its activation. ● | [67] | |
| 16 | Pro-IL-1β | Unknown domain | A20 | A20, a ubiquitin modifying enzyme, is an NFkB inhibitor that reduces pro-IL-1β K63 ubiquitination and maturation and hence inhibits NLRP3-inflammasome activation. ● | [68] | |
| 17 | Alkylation | Unknown domain | 3,4-methylenedioxy-β-nitrostyrene (MNS) | NLRP3-alkylating agents like MNS reduce the ATP binding affinity of NLRP3, which is required for NLRP3-ASC association and hence negatively regulates NLRP3 inflammasome activation. ● | [69] | |
| 18 | S-Nitrosylation | LRR domain | Inducible nitric oxide synthase (iNOS) | Expression of iNOS by prolonged exposure to LPS leads to the production of NO. This leads to the S-nitrosylation of the LRR region of NLRP3, preventing its oligomerization. ● | [70] |
| Sl# | Adipokine | Regulation of Inflammasome Activation in Metabolic Disorder | Ref. |
|---|---|---|---|
| 1. | Adipocyte fatty acid-binding protein 4 (FABP-4) | Control NLRP3 inflammasome activation through downregulating mitochondrial uncoupling protein-2 (UCP2) expression | [112] |
| 2. | Adipsin | Critical as a complement system component in vascular complications of metabolic disorder | [113] |
| 3. | Angiotensinogen and Angiotensin II | Linked to ER stress-induced NLRP3 inflammasome activation | [114] |
| 4. | Apelin | Inhibits NF-kB pathway and inflammasome activation helping in vasodilation | [115] |
| 5. | C-reactive protein (CRP) | Up-regulates NF-κB activity, thereby promoting IL-1β mediated atherosclerosis | [116] |
| 6. | Fibroblast growth factor 2 (FGF-2) | Enhances endothelial adhesion molecule (EAM) expression involved in NLRP3-mediated endothelial dysfunction | [117] |
| 7. | Hepatocyte growth factor (HGF) | Promotes inhibition through up-regulation of adiponectin in adipocytes | [118] |
| 8. | Intercellular adhesion molecule 1 (ICAM-1) | Involved in NF-κB mediated TNF-α signaling pathway and endothelial inflammation | [119] |
| 9. | Lipoprotein lipase (LPL) | Plays a central role in triglyceride and phospholipid hydrolysis, the products of which could elicit pro- or anti-inflammatory responses in endothelial cells | [120] |
| 10. | Matrix metalloproteinases (MMPs) | Involved in cartilage degeneration and NLRP3-mediated synovial inflammation in osteoarthritis | [121] |
| 11. | Monocyte chemoattractant protein 1 (MCP-1) | Key chemokine that regulates the migration and infiltration of adipose tissue via by monocyte/macrophages | [122] |
| 12. | Omentin 1 | Involved in inhibition of the TXNIP/NLRP3 signaling pathways in adipose tissue | [123] |
| 13. | Perilipin 1 | Involved in lipid metabolism homeostasis and inhibits the NF-κB inflammatory pathway | [124] |
| 14. | Plasminogen activator inhibitor 1 (PAI-1) | Plays an important role in regulating ROS-mediated fibrinolysis | [125] |
| 15. | Serum amyloid A | Promotes NLRP3 inflammasome activation via the cathepsin-sensitive pathway | [126] |
| 16. | Vaspin | A visceral adipose tissue-derived serpin that can regulate the PI3K/AKT signaling pathway and improve myocardial function by inhibiting NLRP3 expression | [127] |
| 17. | Visfatin | Visfatin, a pre-B-cell colony-enhancing factor, is involved in TL4-mediated endothelial dysfunction and vascular inflammation | [128] |
| Sl# | Compound | NLRP3 Inflammasome Inhibition | Inflammasome Target | Target Disease | Ref. |
|---|---|---|---|---|---|
| 1. | MCC950 | Sulfonylurea compound that block nigericin-induced NLRP3 inflammasome activation by inhibiting chloride efflux | NACHT domain of NLRP3 | Atherosclerosis, myocardial infarction, colitis, and skin and airway inflammation | [148] |
| 2. | Tranilast | Tryptophan derivative that inhibits NLRP3-NLRP3 interaction and subsequent ASC oligomerization | Gouty arthritis and cryopyrin-associated periodic syndrome (CAPS) | [149] | |
| 3. | OLT1177 | β-sulfonyl nitrile compound that inhibits NLRP3-NLRP3 interaction | Gouty arthritis and cryopyrin-associated periodic syndrome (CAPS) | [150] | |
| 4. | Oridonin | Prevents NLRP3-NEK7 interaction by binding to Cys 279 residues at the NACHT domain | Alzheimer’s disease and cancer | [151] | |
| 5. | CY-09 | Analog of a cystic fibrosis transmembrane conductance regulator (CFTR) channel inhibitor that impairs NLRP3 ATPase. | Gout, atherosclerosis, and neurodegenerative diseases | [148] | |
| 6. | MNS | Impairs ATPase activity of NLRP3 by covalently modifying Cys residues at the NACHT domain | Gout, atherosclerosis, and neurodegenerative diseases | [69] | |
| 7. | BOT-4-one | NLRP3 alkylation by BOT-4-one leading to impaired ATPase activity in NACHT domain | Inflammatory skin diseases | [152] | |
| 8. | IFN39 | Impairs ATPase activity of NLRP3 by binding at the NACHT domain | Inflammatory bowel disease | [153] | |
| 9. | Bay 11-7082 and Parthenolide | Inhibit ATPase activity of NLRP3, which is required for activation of Caspase-1 | Caspase-1 | Systemic lupus erythematosus | [154] |
| 10. | Pralnacasan and Belnacasan | Selectively inhibit Caspase-1 protease activity | Rheumatoid arthritis | [155] | |
| 11. | GKT137831 and VAS-2870 | NOX4-mediated inhibition of caspase-1 activation | Systemic sclerosis and pulmonary fibrosis | [156] | |
| 12. | Etomoxir | CPT1A-mediated inhibition of caspase-1 activation | Congestive heart failure and psoriasis | [157] | |
| 13. | Z-VAD-FMK | Binds to the catalytic site of caspase proteases and inhibits their activity | Granulosa cell apoptosis | [158] | |
| 14. | Necrosulfonamide | Alkylating compound binds to Gasdermin D, thereby preventing pyroptotic pore formation and cell lysis | Gasderimin D | Hemorrhagic necrosis | [159] |
| 15. | Glyburide | Sulfonylurea-containing compound inhibits ATP-sensitive potassium channels in pancreatic β-cells | NLRP3 (indirectly) | Type 2 diabetes | [160] |
| 16. | 16673-34-0 | Sulfonyl compound in glyburide synthesis pathway that inhibits inflammasome activity in the heart | Heart diseases | [161] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wani, K.; AlHarthi, H.; Alghamdi, A.; Sabico, S.; Al-Daghri, N.M. Role of NLRP3 Inflammasome Activation in Obesity-Mediated Metabolic Disorders. Int. J. Environ. Res. Public Health 2021, 18, 511. https://doi.org/10.3390/ijerph18020511
Wani K, AlHarthi H, Alghamdi A, Sabico S, Al-Daghri NM. Role of NLRP3 Inflammasome Activation in Obesity-Mediated Metabolic Disorders. International Journal of Environmental Research and Public Health. 2021; 18(2):511. https://doi.org/10.3390/ijerph18020511
Chicago/Turabian StyleWani, Kaiser, Hind AlHarthi, Amani Alghamdi, Shaun Sabico, and Nasser M. Al-Daghri. 2021. "Role of NLRP3 Inflammasome Activation in Obesity-Mediated Metabolic Disorders" International Journal of Environmental Research and Public Health 18, no. 2: 511. https://doi.org/10.3390/ijerph18020511
APA StyleWani, K., AlHarthi, H., Alghamdi, A., Sabico, S., & Al-Daghri, N. M. (2021). Role of NLRP3 Inflammasome Activation in Obesity-Mediated Metabolic Disorders. International Journal of Environmental Research and Public Health, 18(2), 511. https://doi.org/10.3390/ijerph18020511