
A Novel Intronic Splice—Site Mutation of the CYP11A1 Gene Linked to Adrenal Insufficiency with 46,XY Disorder of Sex Development

 , , and
, , and 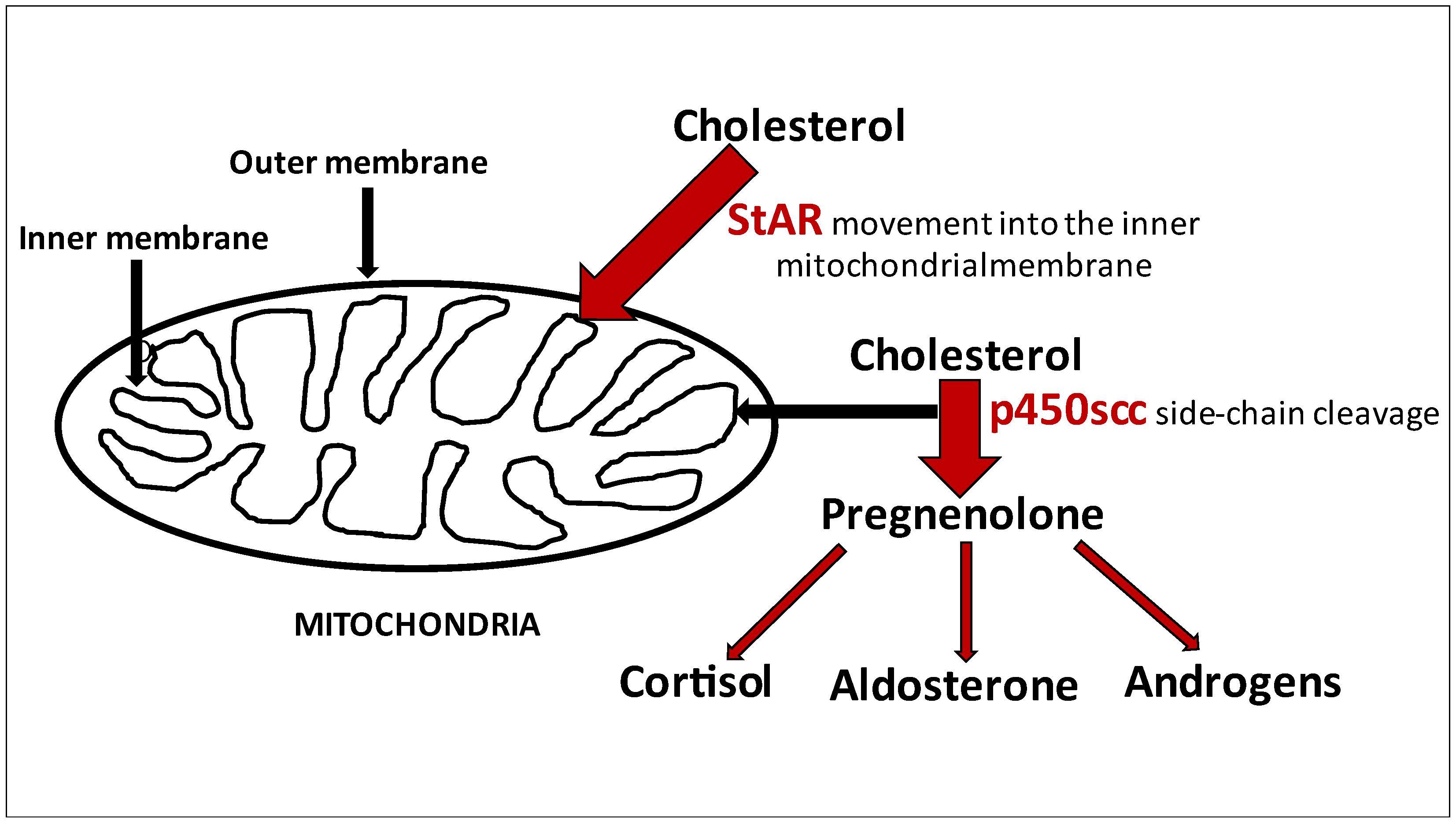{kind=link}
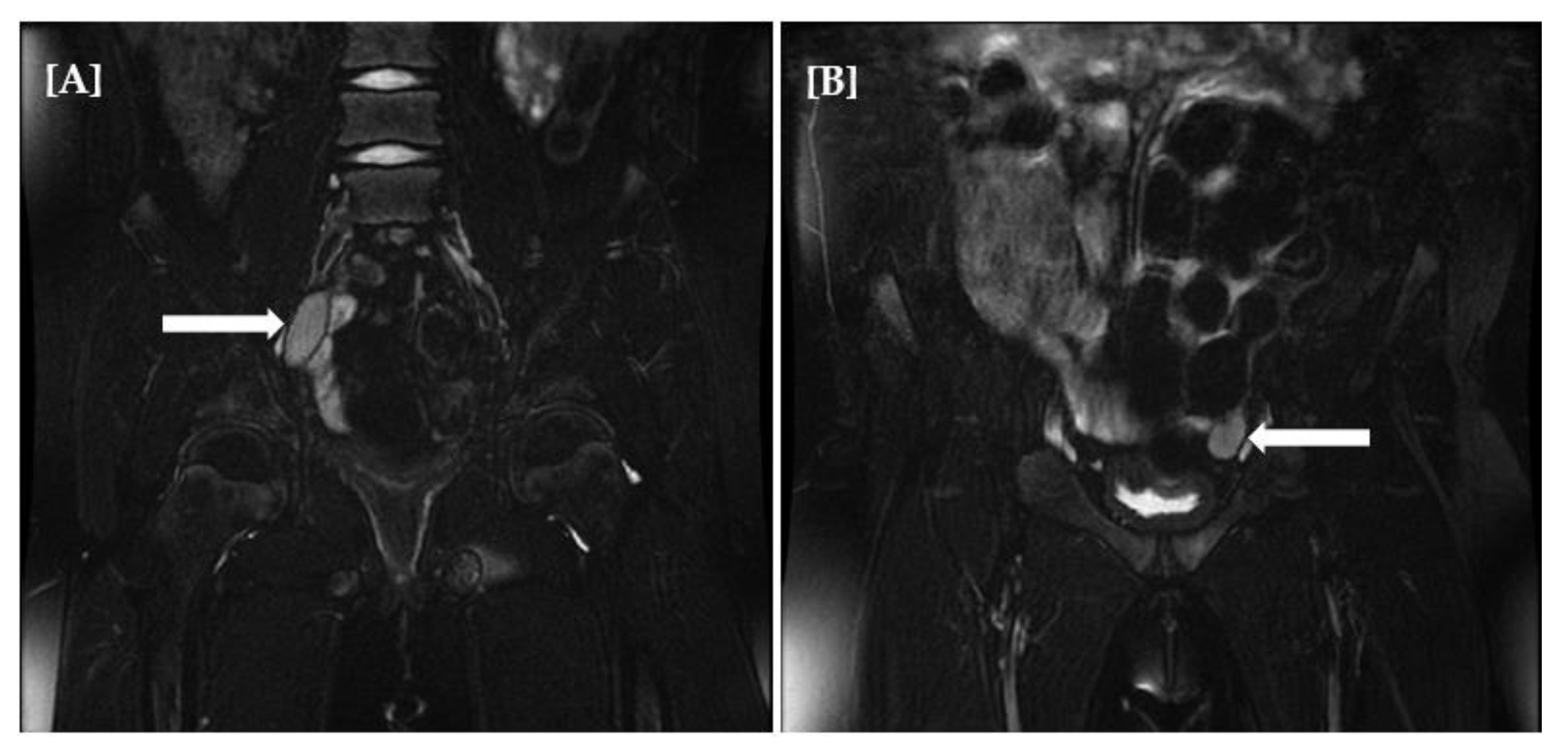{kind=link}
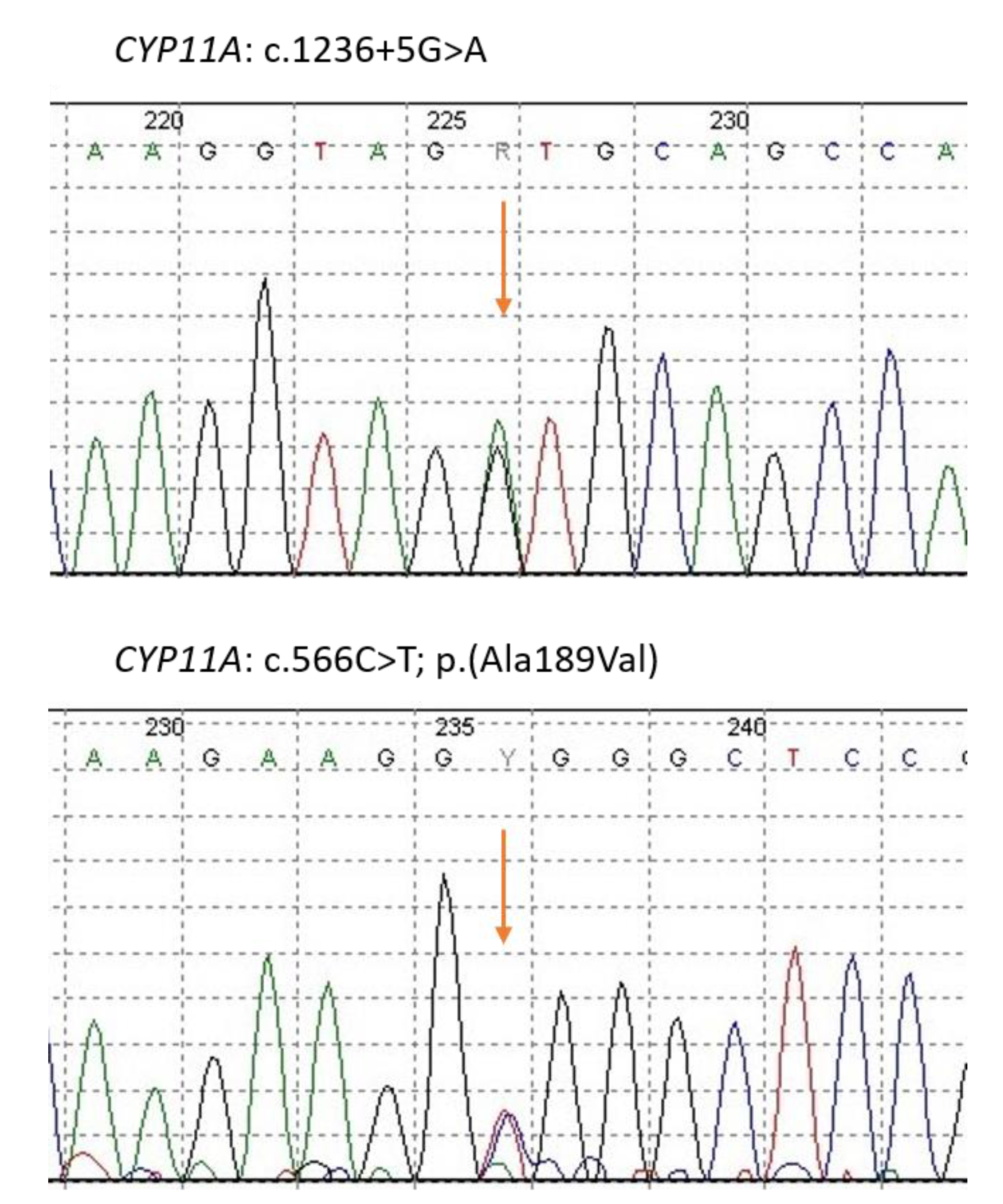{kind=link}
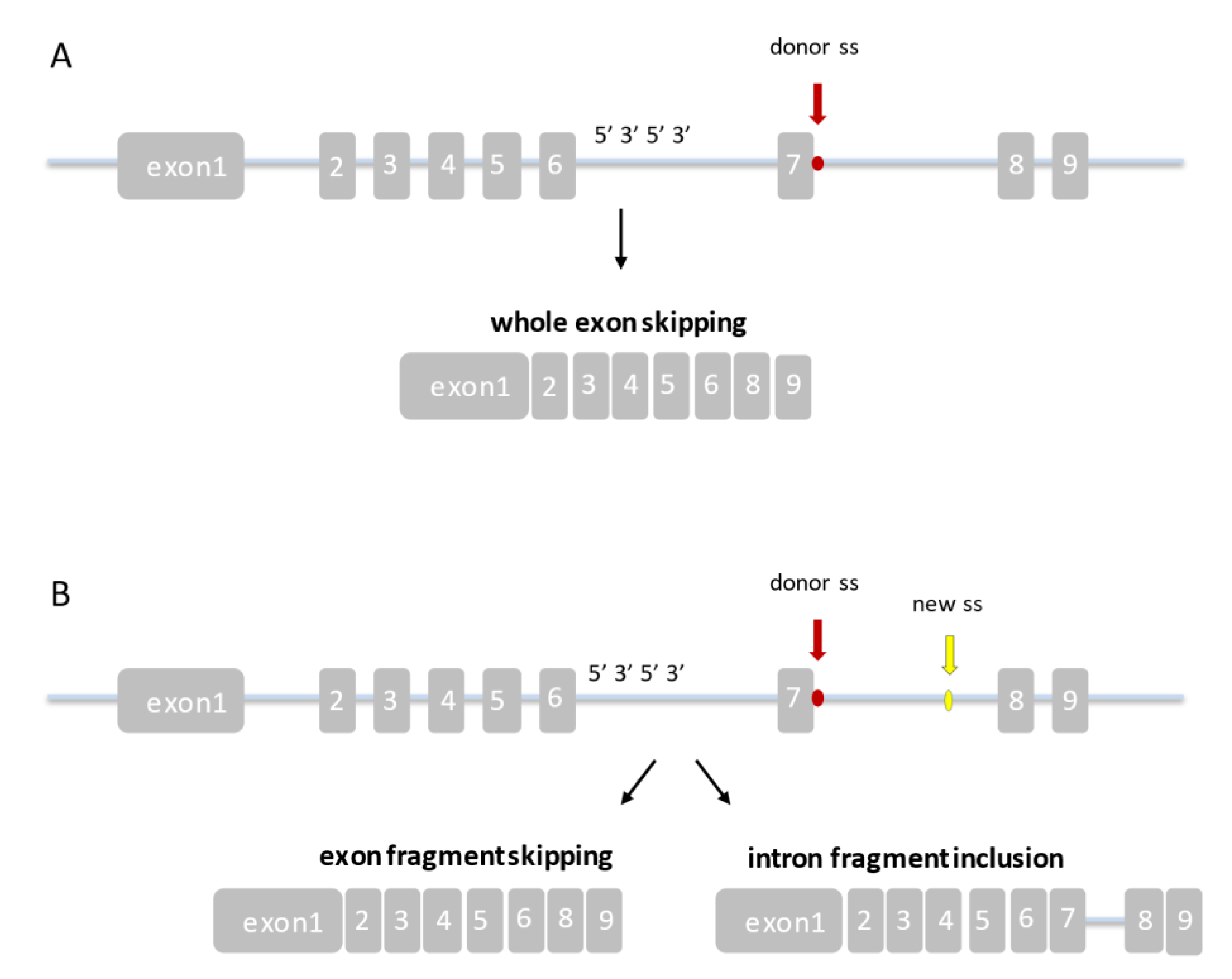{kind=link}
Abstract
:1. Introduction
2. Case Presentation
3. Methods
4. Results
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Miller, W.L.; Bose, H.S. Early steps in steroidogenesis: Intracellular cholesterol trafficking Thematic Review Series: Genetics of Human Lipid Diseases. J. Lipid Res. 2011, 52, 2111–2135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanukoglu, I. Steroidogenic enzymes: Structure, function, and role in regulation of steroid hormone biosynthesis. J. Steroid Biochem. Mol. Biol. 1992, 43, 779–804. [Google Scholar] [CrossRef] [Green Version]
- Katsumata, N.; Ohtake, M.; Hojo, T.; Ogawa, E.; Hara, T.; Sato, N.; Tanaka, T. Compound heterozygous mutations in the cholesterol side-chain cleavage enzyme gene (CYP11A) cause congenital adrenal insufficiency in humans. J. Clin. Endocrinol. Metab. 2002, 87, 3808–3813. [Google Scholar] [CrossRef]
- Abramowicz, A.; Gos, M. Splicing mutations in human genetic disorders: Examples, detection, and confirmation. J. Appl. Genet. 2018, 59, 231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiort, O.; Holterhus, P.M.; Werner, R.; Marschke, C.; Hoppe, U.; Partsch, C.J.; Riepe, F.G.; Achermann, J.C.; Struve, D. Homozygous Disruption of P450 Side-Chain Cleavage (CYP11A1) Is Associated with Prematurity, Complete 46,XY Sex Reversal, and Severe Adrenal Failure. J. Clin. Endocrinol. Metab. 2005, 90, 538–541. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.J.; Lin, L.; Huang, N.; Quigley, C.A.; AvRuskin, T.W.; Achermann, J.C.; Miller, W.L. Severe Combined Adrenal and Gonadal Deficiency Caused by Novel Mutations in the Cholesterol Side Chain Cleavage Enzyme, P450scc. J. Clin. Endocrinol. Metab. 2008, 93, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Al Kandari, H.; Katsumata, N.; Alexander, S.; Rasoul, M.A. Homozygous Mutation of P450 Side-Chain Cleavage Enzyme Gene (CYP11A1) in 46, XY Patient with Adrenal Insufficiency, Complete Sex Reversal, and Agenesis of Corpus Callosum. J. Clin. Endocrinol. Metab. 2006, 91, 2821–2826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubtsov, P.; Karmanov, M.; Sverdlova, P.; Spirin, P.; Tiulpakov, A. A Novel Homozygous Mutation in CYP11A1 Gene Is Associated with Late-Onset Adrenal Insufficiency and Hypospadias in a 46,XY Patient. J. Clin. Endocrinol. Metab. 2009, 94, 936–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahakitrungruang, T.; Tee, M.K.; Blackett, P.R.; Miller, W.L. Partial Defect in the Cholesterol Side-Chain Cleavage Enzyme P450scc (CYP11A1) Resembling Nonclassic Congenital Lipoid Adrenal Hyperplasia. J. Clin. Endocrinol. Metab. 2011, 96, 792–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tajima, T.; Fujieda, K.; Kouda, N.; Nakae, J.; Miller, W.L. Heterozygous Mutation in the Cholesterol Side Chain Cleavage Enzyme (P450scc) Gene in a Patient with 46,XY Sex Reversal and Adrenal Insufficiency. J. Clin. Endocrinol. Metab. 2001, 86, 3820–3825. [Google Scholar] [CrossRef] [PubMed]
- Bose, H.S.; Sugawara, T.; Strauss, J.F.; Miller, W.L. The Pathophysiology and Genetics of Congenital Lipoid Adrenal Hyperplasia. N. Engl. J. Med. 1996, 335, 1870–1879. [Google Scholar] [CrossRef] [PubMed]
- Nakae, J.; Tajima, T.; Sugawara, T.; Arakane, F.; Hanaki, K.; Hotsubo, T.; Igarashi, N.; Igarashi, Y.; Ishii, T.; Koda, N.; et al. Analysis of the Steroidogenic Acute Regulatory Protein (StAR) Gene in Japanese Patients with Congenital Lipoid Adrenal Hyperplasia. Hum. Mol. Genet. 1997, 6, 571–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bose, H.S.; Sato, S.; Aisenberg, J.; Shalev, S.A.; Matsuo, N.; Miller, W.L. Mutations in the Steroidogenic Acute Regulatory Protein (StAR) in Six Patients with Congenital Lipoid Adrenal Hyperplasia. J. Clin. Endocrinol. Metab. 2000, 85, 3636–3639. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.T.; Li, W.; Kim, T.K.; Semak, I.; Wang, J.; Zjawiony, J.K.; Tuckey, R.C. Novel activities of CYP11A1 and their potential physiological significance. J. Steroid Biochem. Mol. Biol. 2015, 151, 25–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kara, O.; Gorukmez, O.; Ekici, A.; Celik, F. A Novel Homozygous Mutation in CYP11A1 Gene is Associated with Severe Adrenal Insufficiency in 46, XX Patient. Fetal Pediatr. Pathol. 2020, 31, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Splittstösser, V.; Schreiner, F.; Gohlke, B.; Welzel, M.; Holterhus, P.M.; Woelfle, J. A novel mutation of the StAR gene with congenital adrenal hyperplasia and its association with heterochromia iridis: A case report. BMC Endocr. Disord. 2019, 19, 116. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matusik, P.; Gach, A.; Zajdel-Cwynar, O.; Pinkier, I.; Kudela, G.; Gawlik, A. A Novel Intronic Splice—Site Mutation of the CYP11A1 Gene Linked to Adrenal Insufficiency with 46,XY Disorder of Sex Development. Int. J. Environ. Res. Public Health 2021, 18, 7186. https://doi.org/10.3390/ijerph18137186
Matusik P, Gach A, Zajdel-Cwynar O, Pinkier I, Kudela G, Gawlik A. A Novel Intronic Splice—Site Mutation of the CYP11A1 Gene Linked to Adrenal Insufficiency with 46,XY Disorder of Sex Development. International Journal of Environmental Research and Public Health. 2021; 18(13):7186. https://doi.org/10.3390/ijerph18137186
Chicago/Turabian StyleMatusik, Pawel, Agnieszka Gach, Olimpia Zajdel-Cwynar, Iwona Pinkier, Grzegorz Kudela, and Aneta Gawlik. 2021. "A Novel Intronic Splice—Site Mutation of the CYP11A1 Gene Linked to Adrenal Insufficiency with 46,XY Disorder of Sex Development" International Journal of Environmental Research and Public Health 18, no. 13: 7186. https://doi.org/10.3390/ijerph18137186
APA StyleMatusik, P., Gach, A., Zajdel-Cwynar, O., Pinkier, I., Kudela, G., & Gawlik, A. (2021). A Novel Intronic Splice—Site Mutation of the CYP11A1 Gene Linked to Adrenal Insufficiency with 46,XY Disorder of Sex Development. International Journal of Environmental Research and Public Health, 18(13), 7186. https://doi.org/10.3390/ijerph18137186