
An Assessment on Ethanol-Blended Gasoline/Diesel Fuels on Cancer Risk and Mortality


Abstract
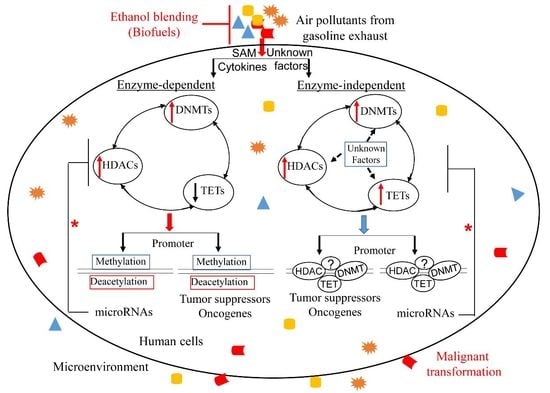
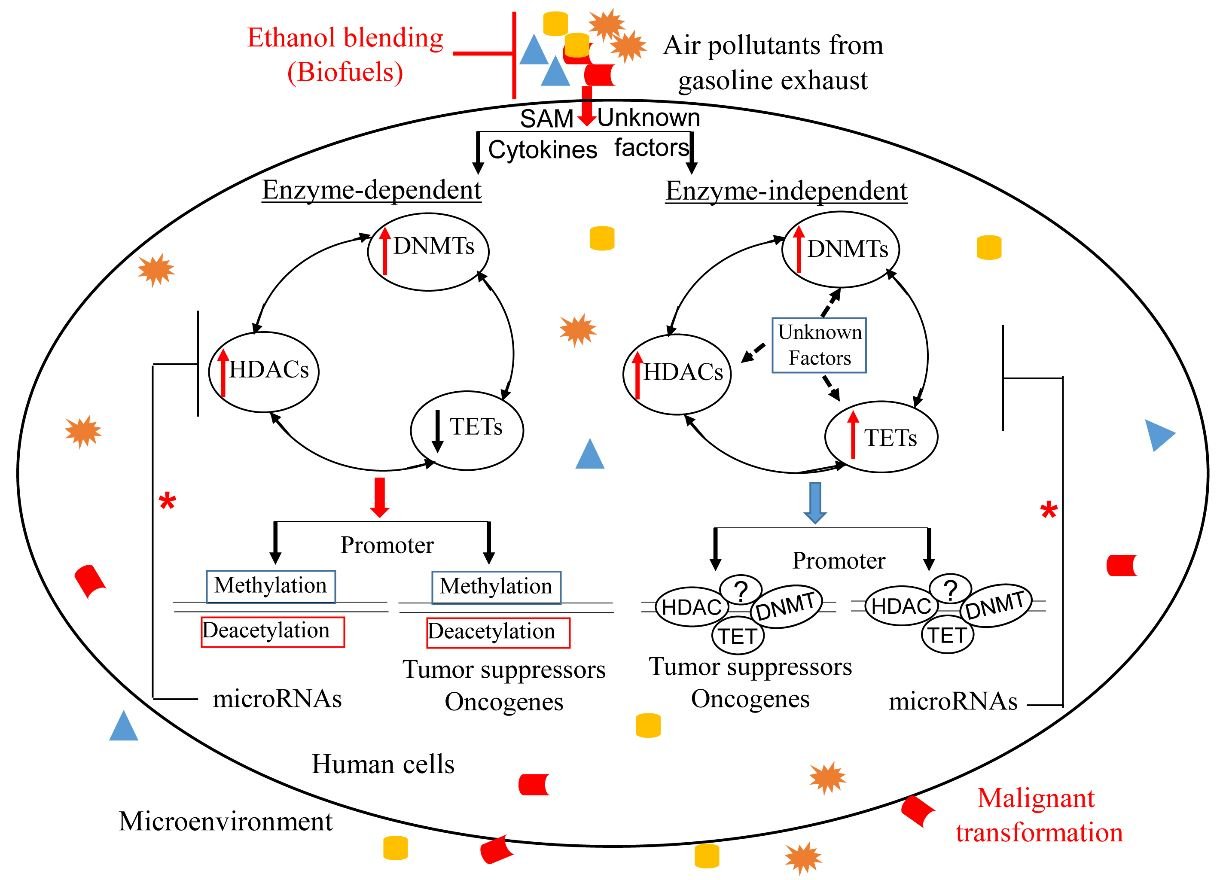
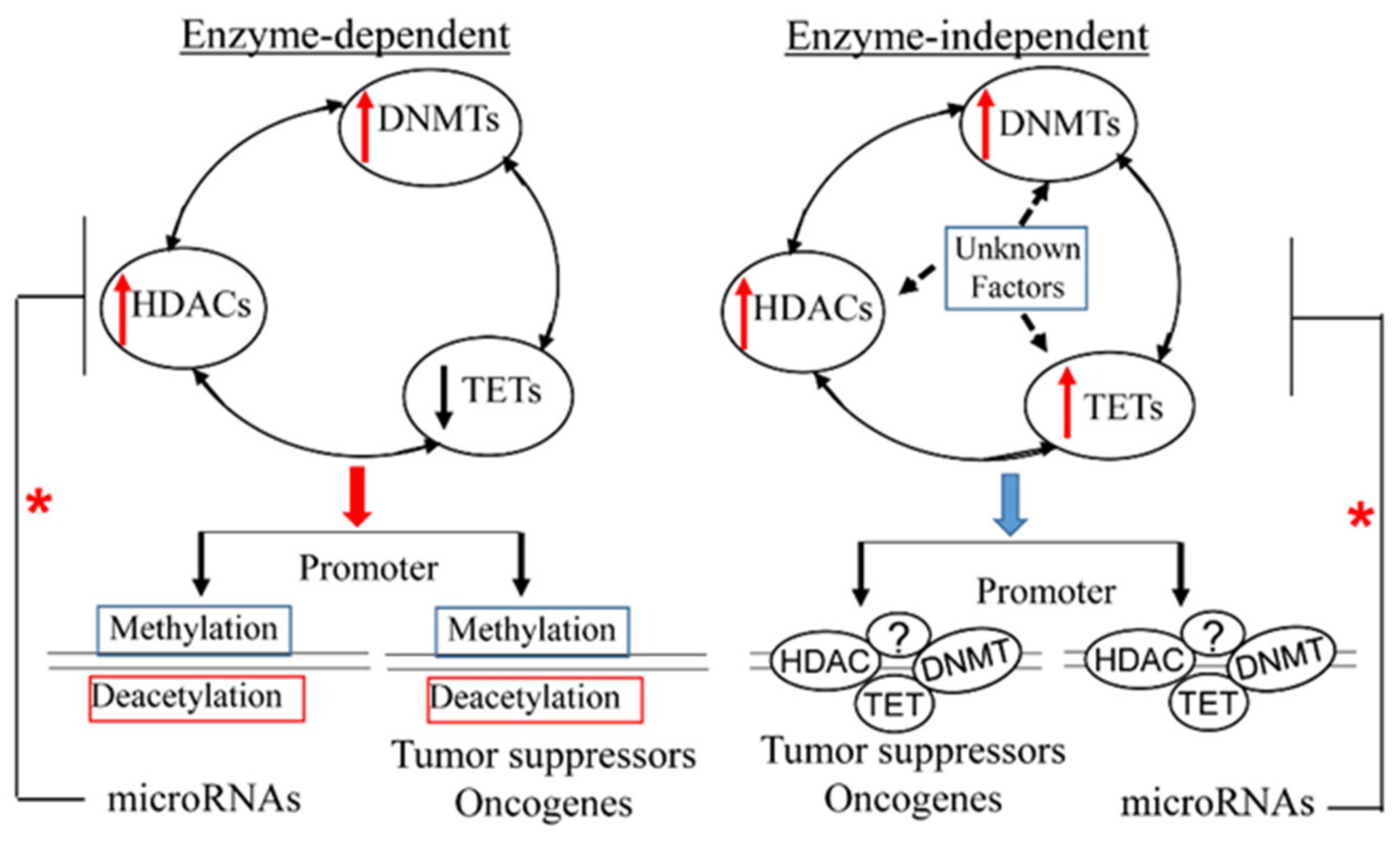
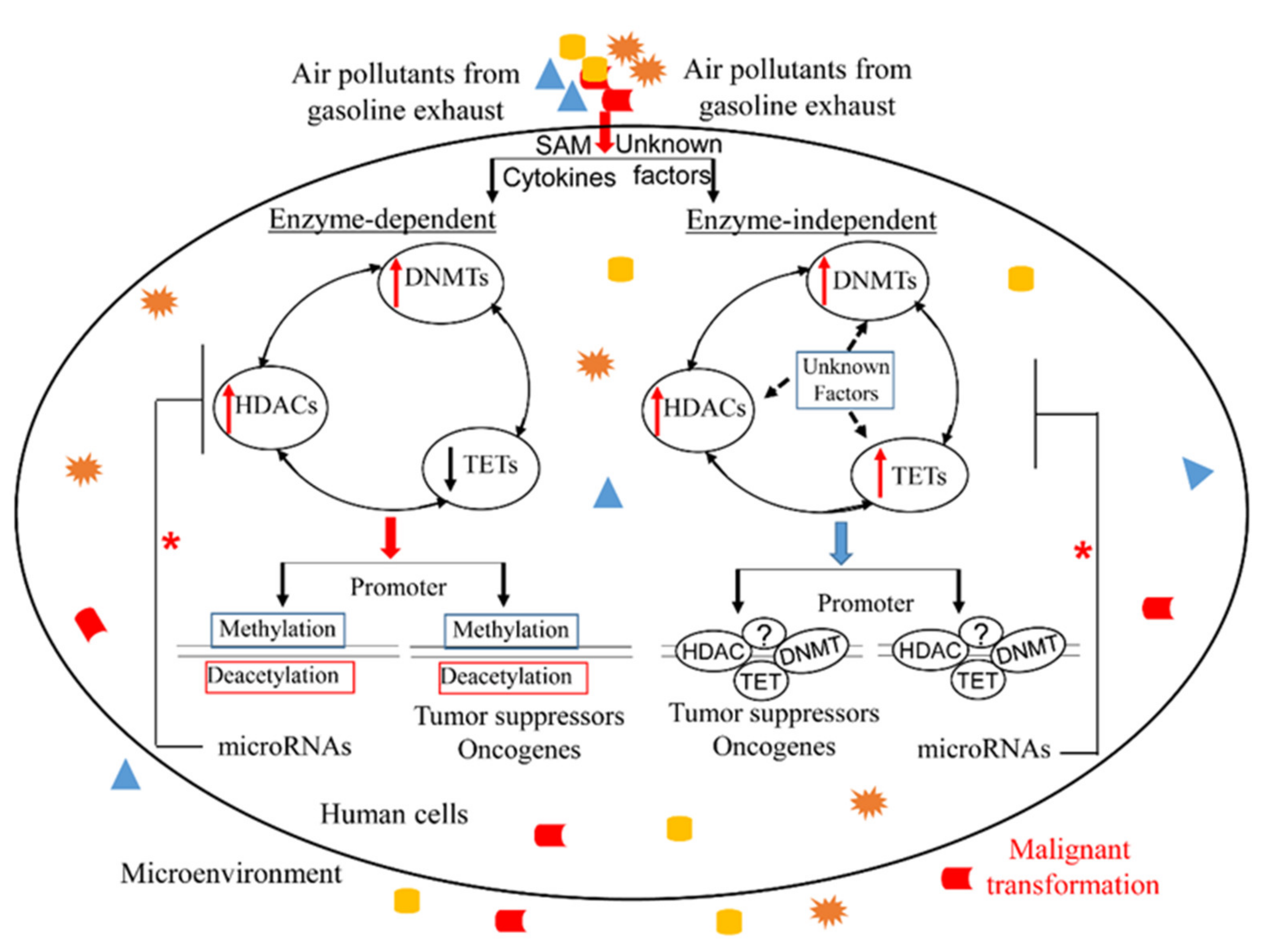
1. Introduction
2. Carcinogenic Potential of Chemicals Associated with Air Pollution from Gasoline
3. The Positive Effects on Human Health of Blending Ethanol into Gasoline
4. Overview of Epigenetic Mechanisms in Cancer Development and Progression
4.1. DNA Methylation and Cancer
4.2. Histone Modifications and Cancers
4.3. MicroRNAs and Cancers
5. Epigenetic Effects Associated with Carcinogenic Chemicals from Gasoline
5.1. Benzene Induces Epigenetic Changes
5.2. The Impacts of Toluene, Xylene, 1,3–Butadiene, 1,2,4–Trimethylbenzene and 2–Methylnaphthalene on Epigenetics in Cancers
5.3. Polycyclic Aromatic Hydrocarbons (PAHs)
6. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Rossner, P., Jr.; Cervena, T.; Vojtisek–Lom, M.; Vrbova, K.; Ambroz, A.; Novakova, Z.; Elzeinova, F.; Margaryan, H.; Beranek, V.; Pechout, M.; et al. The Biological Effects of Complete Gasoline Engine Emissions Exposure in a 3D Human Airway Model (MucilAir(TM)) and in Human Bronchial Epithelial Cells (BEAS–2B). Int. J. Mol. Sci. 2019, 20, 5710. [Google Scholar] [CrossRef]
- Lewtas, J. Air pollution combustion emissions: Characterization of causative agents and mechanisms associated with cancer, reproductive, and cardiovascular effects. Mutat. Res. Mutat. Res. 2007, 636, 95–133. [Google Scholar] [CrossRef]
- Ribeiro, C.B.; Martins, K.G.; Gueri, M.V.D.; Pavanello, G.P.; Schirmer, W.N. Effect of anhydrous ethanol/gasoline blends on performance and exhaust emissions of spark-ignited non-road engines. Environ. Sci. Pollut. Res. 2018, 25, 24192–24200. [Google Scholar] [CrossRef]
- DeMarini, D.M. Genotoxicity biomarkers associated with exposure to traffic and near-road atmospheres: A review. Mutagenesis 2013, 28, 485–505. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Wang, G.; Guo, S.; Zamora, M.L.; Ying, Q.; Lin, Y.; Wang, W.; Hu, M.; Wang, Y. Formation of Urban Fine Particulate Matter. Chem. Rev. 2015, 115, 3803–3855. [Google Scholar] [CrossRef] [PubMed]
- Lelieveld, J.; Evans, J.S.; Fnais, M.; Giannadaki, D.; Pozzer, A. The contribution of outdoor air pollution sources to premature mortality on a global scale. Nat. Cell Biol. 2015, 525, 367–371. [Google Scholar] [CrossRef]
- Su, J.G.; Apte, J.S.; Lipsitt, J.; Garcia–Gonzales, D.A.; Beckerman, B.S.; de Nazelle, A.; Texcalac–Sangrador, J.L.; Jerrett, M. Populations potentially exposed to traffic–related air pollution in seven world cities. Environ. Int. 2015, 78, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Sanchez–Guerra, M.; Zhang, Z.; Joyce, B.T.; Zhong, J.; Kresovich, J.K.; Liu, L.; Zhang, W.; Gao, T.; Chang, D.; et al. Traffic–derived particulate matter exposure and histone H3 modification: A repeated measures study. Environ. Res. 2017, 153, 112–119. [Google Scholar] [CrossRef]
- Künzli, N.; Kaiser, R.; Medina, S.; Studnicka, M.; Chanel, O.; Filliger, P.; Herry, M.; Horak, F.; Puybonnieux-Texier, V.; Quénel, P.; et al. Public-health impact of outdoor and traffic-related air pollution: A European assessment. Lancet 2000, 356, 795–801. [Google Scholar] [CrossRef]
- Raaschou-Nielsen, O.; Andersen, Z.J.; Hvidberg, M.; Jensen, S.S.; Ketzel, M.; Sørensen, M.; Loft, S.; Overvad, K.; Tjønneland, A. Lung Cancer Incidence and Long-Term Exposure to Air Pollution from Traffic. Environ. Heal. Perspect. 2011, 119, 860–865. [Google Scholar] [CrossRef]
- Tonne, C.; Melly, S.; Mittleman, M.; Coull, B.; Goldberg, R.; Schwartz, J. A Case–Control Analysis of Exposure to Traffic and Acute Myocardial Infarction. Environ. Heal. Perspect. 2007, 115, 53–57. [Google Scholar] [CrossRef]
- Weuve, J.; Kaufman, J.; Szpiro, A.A.; Curl, C.; Puett, R.C.; Beck, T.; Evans, D.A.; De Leon, C.F.M. Exposure to Traffic-Related Air Pollution in Relation to Progression in Physical Disability among Older Adults. Environ. Heal. Perspect. 2016, 124, 1000–1008. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.; Slezakova, K.; Delerue–Matos, C.; Pereira, M.C.; Morais, S. Children environmental exposure to particulate matter and polycyclic aromatic hydrocarbons and biomonitoring in school environments: A review on indoor and outdoor exposure levels, major sources and health impacts. Environ. Int. 2019, 124, 180–204. [Google Scholar] [CrossRef] [PubMed]
- Shiraiwa, M.; Selzle, K.; Poschl, U. Hazardous components and health effects of atmospheric aerosol particles: Reactive oxygen species, soot, polycyclic aromatic compounds and allergenic proteins. Free Radic Res. 2012, 46, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Lewis, L.; Borowa–Mazgaj, B.; de Conti, A.; Chappell, G.A.; Luo, Y.S.; Bodnar, W.; Konganti, K.; Wright, F.A.; Threadgill, D.W.; Chiu, W.A.; et al. Population–Based Analysis of DNA Damage and Epigenetic Effects of 1,3–Butadiene in the Mouse. Chem. Res. Toxicol. 2019, 32, 887–898. [Google Scholar] [CrossRef]
- Warden, H.; Richardson, H.; Richardson, L.; Siemiatycki, J.; Ho, V. Associations between occupational exposure to benzene, toluene and xylene and risk of lung cancer in Montreal. Occup. Environ. Med. 2018, 75, 696–702. [Google Scholar] [CrossRef]
- Loomis, D.; Guyton, K.Z.; Grosse, Y.; El Ghissassi, F.; Bouvard, V.; Benbrahim–Tallaa, L.; Guha, N.; Vilahur, N.; Mattock, H.; Straif, K.; et al. Carcinogenicity of benzene. Lancet Oncol. 2017, 18, 1574–1575. [Google Scholar] [CrossRef]
- Blanc-Lapierre, A.; Sauvé, J.-F.; Parent, M.-E. Occupational exposure to benzene, toluene, xylene and styrene and risk of prostate cancer in a population-based study. Occup. Environ. Med. 2018, 75, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Sha, Y.; Zhou, W.; Yang, Z.; Zhu, X.; Xiang, Y.; Li, T.; Zhu, D.; Yang, X. Changes in Poly(ADP-Ribosyl)ation Patterns in Workers Exposed to BTX. PLoS One 2014, 9, e106146. [Google Scholar] [CrossRef]
- Guyton, K.Z.; Kyle, A.D.; Aubrecht, J.; Cogliano, V.J.; Eastmond, D.A.; Jackson, M.; Keshava, N.; Sandy, M.S.; Sonawane, B.; Zhang, L.; et al. Improving prediction of chemical carcinogenicity by considering multiple mechanisms and applying toxicogenomic approaches. Mutat. Res. Mutat. Res. 2009, 681, 230–240. [Google Scholar] [CrossRef]
- Moorthy, B.; Chu, C.; Carlin, D.J. Polycyclic Aromatic Hydrocarbons: From Metabolism to Lung Cancer. Toxicol. Sci. 2015, 145, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Mizumoto, A.; Ohashi, S.; Hirohashi, K.; Amanuma, Y.; Matsuda, T.; Muto, M. Molecular Mechanisms of Acetaldehyde-Mediated Carcinogenesis in Squamous Epithelium. Int. J. Mol. Sci. 2017, 18, 1943. [Google Scholar] [CrossRef] [PubMed]
- Chappell, G.; Kobets, T.; O’Brien, B.; Tretyakova, N.; Sangaraju, D.; Kosyk, O.; Sexton, K.G.; Bodnar, W.; Pogribny, I.P.; Rusyn, I. Epigenetic events determine tissue-specific toxicity of inhalational exposure to the genotoxic chemical 1,3-butadiene in male C57BL/6J mice. Toxicol. Sci. 2014, 142, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Fenga, C.; Gangemi, S.; Costa, C. Benzene exposure is associated with epigenetic changes. Mol. Med. Rep. 2016, 13, 3401–3405. [Google Scholar] [CrossRef]
- Blair, A.; Marrett, L.; Freeman, L.B. Occupational cancer in developed countries. Environ. Health 2011, 10, S9. [Google Scholar] [CrossRef] [PubMed]
- Infante, P.F.; Rinsky, R.A.; Wagoner, J.K.; Young, R.J. Leukaemia in benzene workers. Lancet 1977, 2, 76–78. [Google Scholar] [CrossRef]
- Infante, P.F. Benzene exposure and multiple myeloma: A detailed meta–analysis of benzene cohort studies. Ann. N. Y. Acad. Sci. 2006, 1076, 90–109. [Google Scholar] [CrossRef]
- Duval, R.; Bui, L.C.; Mathieu, C.; Nian, Q.; Berthelet, J.; Xu, X.; Haddad, I.; Vinh, J.; Dupret, J.M.; Busi, F.; et al. Benzoquinone, a leukemogenic metabolite of benzene, catalytically inhibits the protein tyrosine phosphatase PTPN2 and alters STAT1 signaling. J. Biol. Chem. 2019, 294, 12483–12494. [Google Scholar] [CrossRef] [PubMed]
- Teras, L.R.; Diver, W.R.; Deubler, E.L.; Krewski, D.; Flowers, C.R.; Switchenko, J.M.; Gapstur, S.M. Residential ambient benzene exposure in the United States and subsequent risk of hematologic malignancies. Int. J. Cancer 2019, 145, 2647–2660. [Google Scholar] [CrossRef]
- Grigoryan, H.; Edmands, W.M.B.; Lan, Q.; Carlsson, H.; Vermeulen, R.; Zhang, L.; Yin, S.N.; Li, G.L.; Smith, M.T.; Rothman, N.; et al. Adductomic signatures of benzene exposure provide insights into cancer induction. Carcinogenesis 2018, 39, 661–668. [Google Scholar] [CrossRef]
- Chen, J.; Zheng, Z.; Chen, Y.; Li, J.; Qian, S.; Shi, Y.; Sun, L.; Han, Y.; Zhang, S.; Yu, K. Histone Deacetylase Inhibitors Trichostatin A and MCP30 Relieve Benzene–Induced Hematotoxicity via Restoring Topoisomerase IIalpha. PLoS One 2016, 11, e0153330. [Google Scholar] [CrossRef]
- Zhang, H.; Yuan, Q.; Pan, Z.; Ling, X.; Tan, Q.; Wu, M.; Zheng, D.; Xie, P.; Xie, D.; Liu, L. Up–regulation of DNMT3b contributes to HOTAIRM1 silencing via DNA hypermethylation in cells transformed by long–term exposure to hydroquinone and workers exposed to benzene. Toxicol. Lett. 2020, 322, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.H.; Li, Y.; Zhao, X.; Yang, S.Q.; Li, L.; Cui, N.X.; Rong, L.; Yi, Z.C. Benzene metabolite 1,2,4–benzenetriol changes DNA methylation and histone acetylation of erythroid–specific genes in K562 cells. Arch. Toxicol. 2019, 93, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Thys, R.G.; Lehman, C.E.; Pierce, L.C.; Wang, Y.H. Environmental and chemotherapeutic agents induce breakage at genes involved in leukemia–causing gene rearrangements in human hematopoietic stem/progenitor cells. Mutat. Res. 2015, 779, 86–95. [Google Scholar] [CrossRef]
- McHale, C.M.; Zhang, L.; Smith, M.T. Current understanding of the mechanism of benzene–induced leukemia in humans: Implications for risk assessment. Carcinogenesis 2012, 33, 240–252. [Google Scholar] [CrossRef]
- Bollati, V.; Baccarelli, A.; Hou, L.; Bonzini, M.; Fustinoni, S.; Cavallo, D.; Byun, H.M.; Jiang, J.; Marinelli, B.; Pesatori, A.C.; et al. Changes in DNA methylation patterns in subjects exposed to low–dose benzene. Cancer Res. 2007, 67, 876–880. [Google Scholar] [CrossRef]
- Smith, M.T.; Jones, R.M.; Smith, A.H. Benzene exposure and risk of non–Hodgkin lymphoma. Cancer Epidemiol. Biomark. Prev. 2007, 16, 385–391. [Google Scholar] [CrossRef]
- Shu, X.O.; Gao, Y.T.; Brinton, L.A.; Linet, M.S.; Tu, J.T.; Zheng, W.; Fraumeni, J.F., Jr. A population–based case–control study of childhood leukemia in Shanghai. Cancer 1988, 62, 635–644. [Google Scholar] [CrossRef]
- Adegoke, O.J.; Blair, A.; Shu, X.O.; Sanderson, M.; Jin, F.; Dosemeci, M.; Addy, C.L.; Zheng, W. Occupational history and exposure and the risk of adult leukemia in Shanghai. Ann. Epidemiol. 2003, 13, 485–494. [Google Scholar] [CrossRef]
- Glass, D.C.; Gray, C.N.; Jolley, D.J.; Gibbons, C.; Sim, M.R.; Fritschi, L.; Adams, G.G.; Bisby, J.A.; Manuell, R. Leukemia risk associated with low–level benzene exposure. Epidemiology 2003, 14, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Glass, D.C.; Gray, C.N.; Jolley, D.J.; Gibbons, C.; Sim, M.R. Health Watch exposure estimates: Do they underestimate benzene exposure? Chem. Biol. Interact. 2005, 153–154, 23–32. [Google Scholar] [CrossRef]
- Lan, Q.; Zhang, L.; Li, G.; Vermeulen, R.; Weinberg, R.S.; Dosemeci, M.; Rappaport, S.M.; Shen, M.; Alter, B.P.; Wu, Y.; et al. Hematotoxicity in workers exposed to low levels of benzene. Science 2004, 306, 1774–1776. [Google Scholar] [CrossRef]
- Snyder, R. Leukemia and benzene. Int. J. Environ. Res. Public Health 2012, 9, 2875–2893. [Google Scholar] [CrossRef]
- Yin, S.N.; Hayes, R.B.; Linet, M.S.; Li, G.L.; Dosemeci, M.; Travis, L.B.; Li, C.Y.; Zhang, Z.N.; Li, D.G.; Chow, W.H.; et al. A cohort study of cancer among benzene–exposed workers in China: Overall results. Am. J. Ind. Med. 1996, 29, 227–235. [Google Scholar] [CrossRef]
- Hayes, R.B.; Yin, S.N.; Dosemeci, M.; Li, G.L.; Wacholder, S.; Chow, W.H.; Rothman, N.; Wang, Y.Z.; Dai, T.R.; Chao, X.J.; et al. Mortality among benzene–exposed workers in China. Environ. Health Perspect. 1996, 104 (Suppl. S6), 1349–1352. [Google Scholar] [CrossRef]
- Yin, S.N.; Hayes, R.B.; Linet, M.S.; Li, G.L.; Dosemeci, M.; Travis, L.B.; Zhang, Z.N.; Li, D.G.; Chow, W.H.; Wacholder, S.; et al. An expanded cohort study of cancer among benzene–exposed workers in China. Benzene Study Group. Environ. Health Perspect. 1996, 104 (Suppl. S6), 1339–1341. [Google Scholar] [CrossRef]
- Wolff, M.S.; Collman, G.W.; Barrett, J.C.; Huff, J. Breast cancer and environmental risk factors: Epidemiological and experimental findings. Annu. Rev. Pharmacol. Toxicol. 1996, 36, 573–596. [Google Scholar] [CrossRef]
- Bennett, L.M.; Davis, B.J. Identification of mammary carcinogens in rodent bioassays. Environ. Mol. Mutagen. 2002, 39, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Rudel, R.A.; Attfield, K.R.; Schifano, J.N.; Brody, J.G. Chemicals causing mammary gland tumors in animals signal new directions for epidemiology, chemicals testing, and risk assessment for breast cancer prevention. Cancer 2007, 109 (Suppl. S12), 2635–2666. [Google Scholar] [CrossRef]
- Falzone, L.; Marconi, A.; Loreto, C.; Franco, S.; Spandidos, D.A.; Libra, M. Occupational exposure to carcinogens: Benzene, pesticides and fibers. Mol. Med. Rep. 2016, 14, 4467–4474. [Google Scholar] [CrossRef]
- Chappell, G.; Pogribny, I.P.; Guyton, K.Z.; Rusyn, I. Epigenetic alterations induced by genotoxic occupational and environmental human chemical carcinogens: A systematic literature review. Mutat. Res. Rev. Mutat. Res. 2016, 768, 27–45. [Google Scholar] [CrossRef]
- Hadkhale, K.; Martinsen, J.I.; Weiderpass, E.; Kjaerheim, K.; Sparen, P.; Tryggvadottir, L.; Lynge, E.; Pukkala, E. Occupational exposure to solvents and bladder cancer: A population–based case control study in Nordic countries. Int. J. Cancer 2017, 140, 1736–1746. [Google Scholar] [CrossRef]
- Petralia, S.A.; Vena, J.E.; Freudenheim, J.L.; Dosemeci, M.; Michalek, A.; Goldberg, M.S.; Brasure, J.; Graham, S. Risk of premenopausal breast cancer in association with occupational exposure to polycyclic aromatic hydrocarbons and benzene. Scand. J. Work Environ. Health 1999, 25, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Costantini, A.S.; Gorini, G.; Consonni, D.; Miligi, L.; Giovannetti, L.; Quinn, M. Exposure to benzene and risk of breast cancer among shoe factory workers in Italy. Tumori 2009, 95, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Langman, J.M. Xylene: Its toxicity, measurement of exposure levels, absorption, metabolism and clearance. Pathology 1994, 26, 301–309. [Google Scholar] [CrossRef]
- Huy, L.N.; Lee, S.C.; Zhang, Z. Human cancer risk estimation for 1,3–butadiene: An assessment of personal exposure and different microenvironments. Sci. Total Environ. 2018, 616–617, 1599–1611. [Google Scholar] [CrossRef] [PubMed]
- Cote, I.L.; Bayard, S.P. Cancer risk assessment of 1,3–butadiene. Environ. Health Perspect. 1990, 86, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Sielken, R.L., Jr.; Valdez–Flores, C. A comprehensive review of occupational and general population cancer risk: 1,3–Butadiene exposure–response modeling for all leukemia, acute myelogenous leukemia, chronic lymphocytic leukemia, chronic myelogenous leukemia, myeloid neoplasm and lymphoid neoplasm. Chem. Biol. Interact. 2015, 241, 50–58. [Google Scholar] [CrossRef]
- Melnick, R.L.; Kohn, M.C. Mechanistic data indicate that 1,3–butadiene is a human carcinogen. Carcinogenesis 1995, 16, 157–163. [Google Scholar] [CrossRef][Green Version]
- Koturbash, I.; Scherhag, A.; Sorrentino, J.; Sexton, K.; Bodnar, W.; Tryndyak, V.; Latendresse, J.R.; Swenberg, J.A.; Beland, F.A.; Pogribny, I.P.; et al. Epigenetic alterations in liver of C57BL/6J mice after short–term inhalational exposure to 1,3–butadiene. Environ. Health Perspect. 2011, 119, 635–640. [Google Scholar] [CrossRef][Green Version]
- Stec, A.A.; Dickens, K.E.; Salden, M.; Hewitt, F.E.; Watts, D.P.; Houldsworth, P.E.; Martin, F.L. Occupational Exposure to Polycyclic Aromatic Hydrocarbons and Elevated Cancer Incidence in Firefighters. Sci. Rep. 2018, 8, 2476. [Google Scholar] [CrossRef]
- Mastrangelo, G.; Fadda, E.; Marzia, V. Polycyclic aromatic hydrocarbons and cancer in man. Environ. Health Perspect. 1996, 104, 1166–1170. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Luo, Y.; Zhong, R.; Law, P.T.Y.; Boon, S.S.; Chen, Z.; Wong, C.H.; Chan, P.K.S. Role of polycyclic aromatic hydrocarbons as a co–factor in human papillomavirus–mediated carcinogenesis. BMC Cancer 2019, 19, 138. [Google Scholar] [CrossRef]
- Chen, D.; Fang, L.; Li, H.; Jin, C. The effects of acetaldehyde exposure on histone modifications and chromatin structure in human lung bronchial epithelial cells. Environ. Mol. Mutagen. 2018, 59, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Stickel, F. Acetaldehyde as an underestimated risk factor for cancer development: Role of genetics in ethanol metabolism. Genes Nutr. 2010, 5, 121–128. [Google Scholar] [CrossRef]
- Lachenmeier, D.W.; Kanteres, F.; Rehm, J. Carcinogenicity of acetaldehyde in alcoholic beverages: Risk assessment outside ethanol metabolism. Addiction 2009, 104, 533–550. [Google Scholar] [CrossRef] [PubMed]
- Murata, Y.; Denda, A.; Maruyama, H.; Nakae, D.; Tsutsumi, M.; Tsujiuchi, T.; Konishi, Y. Chronic toxicity and carcinogenicity studies of 2–methylnaphthalene in B6C3F1 mice. Fundam. Appl. Toxicol. 1997, 36, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Stein, R.; Anderson, J.; Wallington, T. An Overview of the Effects of Ethanol–Gasoline Blends on SI Engine Performance, Fuel Efficiency, and Emissions. SAE Int. J. Engines 2013, 6, 470–487. [Google Scholar] [CrossRef]
- Durbin, T.D.; Miller, J.W.; Younglove, T.; Huai, T.; Cockert, K. Effects of fuel ethanol content and volatility on regulated and unregulated exhaust emissions for the latest technology gasoline vehicles. Environ. Sci. Technol. 2007, 41, 4059–4064. [Google Scholar] [CrossRef]
- Munoz, M.; Heeb, N.V.; Haag, R.; Honegger, P.; Zeyer, K.; Mohn, J.; Comte, P.; Czerwinski, J. Bioethanol Blending Reduces Nanoparticle, PAH, and Alkyl– and Nitro–PAH Emissions and the Genotoxic Potential of Exhaust from a Gasoline Direct Injection Flex–Fuel Vehicle. Environ. Sci. Technol. 2016, 50, 11853–11861. [Google Scholar] [CrossRef]
- Comunian, S.; Dongo, D.; Milani, C.; Palestini, P. Air Pollution and Covid–19: The Role of Particulate Matter in the Spread and Increase of Covid–19’s Morbidity and Mortality. Int. J. Environ. Res. Public Health 2020, 17, 4487. [Google Scholar] [CrossRef]
- Lolli, S.; Chen, Y.C.; Wang, S.H.; Vivone, G. Impact of meteorological conditions and air pollution on COVID–19 pandemic transmission in Italy. Sci. Rep. 2020, 10, 16213. [Google Scholar] [CrossRef]
- Barakat, T.; Muylkens, B.; Su, B.L. Is Particulate Matter of Air Pollution a Vector of Covid–19 Pandemic? Matter 2020, 3, 977–980. [Google Scholar] [CrossRef]
- Conticini, E.; Frediani, B.; Caro, D. Can atmospheric pollution be considered a co–factor in extremely high level of SARS–CoV–2 lethality in Northern Italy? Environ. Pollut. 2020, 261, 114465. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Nethery, R.C.; Sabath, B.M.; Braun, D.; Dominici, F. Exposure to air pollution and COVID–19 mortality in the United States: A nationwide cross–sectional study. MedRxiv 2020. [Google Scholar] [CrossRef]
- Baylin, S.B. DNA methylation and gene silencing in cancer. Nat. Clin. Pract. Oncol. 2005, 2 (Suppl. S1), S4–S11. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Ohm, J.E. Epigenetic gene silencing in cancer—A mechanism for early oncogenic pathway addiction? Nat. Rev. Cancer 2006, 6, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Wen, H.; Li, Y.; Lyu, J.; Xi, Y.; Hoshii, T.; Joseph, J.K.; Wang, X.; Loh, Y.E.; Erb, M.A.; et al. ENL links histone acetylation to oncogenic gene expression in acute myeloid leukaemia. Nature 2017, 543, 265–269. [Google Scholar] [CrossRef]
- Bueno, M.J.; Perez de Castro, I.; Gomez de Cedron, M.; Santos, J.; Calin, G.A.; Cigudosa, J.C.; Croce, C.M.; Fernandez–Piqueras, J.; Malumbres, M. Genetic and epigenetic silencing of microRNA–203 enhances ABL1 and BCR–ABL1 oncogene expression. Cancer Cell 2008, 13, 496–506. [Google Scholar] [CrossRef]
- Pinweha, P.; Rattanapornsompong, K.; Charoensawan, V.; Jitrapakdee, S. MicroRNAs and oncogenic transcriptional regulatory networks controlling metabolic reprogramming in cancers. Comput. Struct. Biotechnol. J. 2016, 14, 223–233. [Google Scholar] [CrossRef]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Gregory, R.I. MicroRNA biogenesis pathways in cancer. Nat. Rev. Cancer 2015, 15, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Bracken, C.P.; Scott, H.S.; Goodall, G.J. A network–biology perspective of microRNA function and dysfunction in cancer. Nat. Rev. Genet. 2016, 17, 719–732. [Google Scholar] [CrossRef] [PubMed]
- Michalak, E.M.; Burr, M.L.; Bannister, A.J.; Dawson, M.A. The roles of DNA, RNA and histone methylation in ageing and cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 573–589. [Google Scholar] [CrossRef] [PubMed]
- Fardi, M.; Solali, S.; Farshdousti Hagh, M. Epigenetic mechanisms as a new approach in cancer treatment: An updated review. Genes Dis. 2018, 5, 304–311. [Google Scholar] [CrossRef]
- Chi, P.; Allis, C.D.; Wang, G.G. Covalent histone modifications––miswritten, misinterpreted and mis–erased in human cancers. Nat. Rev. Cancer 2010, 10, 457–469. [Google Scholar] [CrossRef]
- Esteller, M. Cancer epigenomics: DNA methylomes and histone–modification maps. Nat. Rev. Genet. 2007, 8, 286–298. [Google Scholar] [CrossRef]
- Zhao, Z.; Shilatifard, A. Epigenetic modifications of histones in cancer. Genome Biol. 2019, 20, 245. [Google Scholar] [CrossRef]
- Kinnaird, A.; Zhao, S.; Wellen, K.E.; Michelakis, E.D. Metabolic control of epigenetics in cancer. Nat. Rev. Cancer 2016, 16, 694–707. [Google Scholar] [CrossRef]
- Dart, A. Tumour metabolism: When metabolic and epigenetic states converge. Nat. Rev. Cancer 2016, 16, 757. [Google Scholar] [CrossRef]
- Arand, J.; Spieler, D.; Karius, T.; Branco, M.R.; Meilinger, D.; Meissner, A.; Jenuwein, T.; Xu, G.; Leonhardt, H.; Wolf, V.; et al. In vivo control of CpG and non–CpG DNA methylation by DNA methyltransferases. PLoS Genet. 2012, 8, e1002750. [Google Scholar] [CrossRef]
- Gowher, H.; Stockdale, C.J.; Goyal, R.; Ferreira, H.; Owen–Hughes, T.; Jeltsch, A. De novo methylation of nucleosomal DNA by the mammalian Dnmt1 and Dnmt3A DNA methyltransferases. Biochemistry 2005, 44, 9899–9904. [Google Scholar] [CrossRef]
- Jeltsch, A.; Jurkowska, R.Z. New concepts in DNA methylation. Trends Biochem. Sci. 2014, 39, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, S.; Chijiwa, T.; Okamura, T.; Akashi, K.; Fukumaki, Y.; Niho, Y.; Sasaki, H. Expression of DNA methyltransferases DNMT1, 3A, and 3B in normal hematopoiesis and in acute and chronic myelogenous leukemia. Blood 2001, 97, 1172–1179. [Google Scholar] [CrossRef]
- Yan, F.; Shen, N.; Pang, J.; Zhao, N.; Deng, B.; Li, B.; Yang, Y.; Yang, P.; Molina, J.R.; Liu, S. A regulatory circuit composed of DNA methyltransferases and receptor tyrosine kinases controls lung cancer cell aggressiveness. Oncogene 2017, 36, 6919–6928. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Shen, N.; Pang, J.X.; Zhang, Y.W.; Rao, E.Y.; Bode, A.M.; Al–Kali, A.; Zhang, D.E.; Litzow, M.R.; Li, B.; et al. Fatty acid–binding protein FABP4 mechanistically links obesity with aggressive AML by enhancing aberrant DNA methylation in AML cells. Leukemia 2017, 31, 1434–1442. [Google Scholar] [CrossRef]
- Shen, N.; Yan, F.; Pang, J.; Wu, L.C.; Al–Kali, A.; Litzow, M.R.; Liu, S. A nucleolin–DNMT1 regulatory axis in acute myeloid leukemogenesis. Oncotarget 2014, 5, 5494–5509. [Google Scholar] [CrossRef]
- Liu, S. Epigenetics advancing personalized nanomedicine in cancer therapy. Adv. Drug Deliv. Rev. 2012, 64, 1532–1543. [Google Scholar] [CrossRef] [PubMed]
- Garzon, R.; Liu, S.; Fabbri, M.; Liu, Z.; Heaphy, C.E.; Callegari, E.; Schwind, S.; Pang, J.; Yu, J.; Muthusamy, N.; et al. MicroRNA–29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood 2009, 113, 6411–6418. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, N.; Rabbani, S.A. DNA Methylation Readers and Cancer: Mechanistic and Therapeutic Applications. Front. Oncol. 2019, 9, 489. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Ni, M.; Chen, Z.; Zheng, Z. Expression and clinical significance of methyl–CpG binding domain protein 2 in high–grade serous ovarian cancer. Oncol. Lett. 2020, 20, 2749–2756. [Google Scholar] [CrossRef] [PubMed]
- Ballestar, E.; Esteller, M. Methyl–CpG–binding proteins in cancer: Blaming the DNA methylation messenger. Biochem. Cell Biol. 2005, 83, 374–384. [Google Scholar] [CrossRef]
- Mishra, A.; Liu, S.; Sams, G.H.; Curphey, D.P.; Santhanam, R.; Rush, L.J.; Schaefer, D.; Falkenberg, L.G.; Sullivan, L.; Jaroncyk, L.; et al. Aberrant overexpression of IL–15 initiates large granular lymphocyte leukemia through chromosomal instability and DNA hypermethylation. Cancer Cell 2012, 22, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Ley, T.J.; Ding, L.; Walter, M.J.; McLellan, M.D.; Lamprecht, T.; Larson, D.E.; Kandoth, C.; Payton, J.E.; Baty, J.; Welch, J.; et al. DNMT3A mutations in acute myeloid leukemia. New Engl. J. Med. 2010, 363, 2424–2433. [Google Scholar] [CrossRef]
- Yan, F.; Shen, N.; Pang, J.; Molina, J.R.; Yang, P.; Liu, S. The DNA Methyltransferase DNMT1 and Tyrosine–Protein Kinase KIT Cooperatively Promote Resistance to 5–Aza–2’–deoxycytidine (Decitabine) and Midostaurin (PKC412) in Lung Cancer Cells. J. Biol. Chem. 2015, 290, 18480–18494. [Google Scholar] [CrossRef]
- Liu, S.; Liu, Z.; Xie, Z.; Pang, J.; Yu, J.; Lehmann, E.; Huynh, L.; Vukosavljevic, T.; Takeki, M.; Klisovic, R.B.; et al. Bortezomib induces DNA hypomethylation and silenced gene transcription by interfering with Sp1/NF–kappaB–dependent DNA methyltransferase activity in acute myeloid leukemia. Blood 2008, 111, 2364–2373. [Google Scholar] [CrossRef]
- Yan, F.; Pang, J.; Peng, Y.; Molina, J.R.; Yang, P.; Liu, S. Elevated Cellular PD1/PD–L1 Expression Confers Acquired Resistance to Cisplatin in Small Cell Lung Cancer Cells. PLoS One 2016, 11, e0162925. [Google Scholar] [CrossRef]
- Yan, F.; Shen, N.; Pang, J.X.; Zhao, N.; Zhang, Y.W.; Bode, A.M.; Al–Kali, A.; Litzow, M.R.; Li, B.; Liu, S.J. A vicious loop of fatty acid–binding protein 4 and DNA methyltransferase 1 promotes acute myeloid leukemia and acts as a therapeutic target. Leukemia 2018, 32, 865–873. [Google Scholar] [CrossRef]
- Shen, N.; Yan, F.; Pang, J.; Zhao, N.; Gangat, N.; Wu, L.; Bode, A.M.; Al–Kali, A.; Litzow, M.R.; Liu, S. Inactivation of Receptor Tyrosine Kinases Reverts Aberrant DNA Methylation in Acute Myeloid Leukemia. Clin. Cancer Res. 2017, 23, 6254–6266. [Google Scholar] [CrossRef]
- Gao, X.N.; Yan, F.; Lin, J.; Gao, L.; Lu, X.L.; Wei, S.C.; Shen, N.; Pang, J.X.; Ning, Q.Y.; Komeno, Y.; et al. AML1/ETO cooperates with HIF1alpha to promote leukemogenesis through DNMT3a transactivation. Leukemia 2015, 29, 1730–1740. [Google Scholar] [CrossRef]
- Joo, J.E.; Dowty, J.G.; Milne, R.L.; Wong, E.M.; Dugue, P.A.; English, D.; Hopper, J.L.; Goldgar, D.E.; Giles, G.G.; Southey, M.C.; et al. Heritable DNA methylation marks associated with susceptibility to breast cancer. Nature Commun. 2018, 9, 867. [Google Scholar] [CrossRef]
- Szyf, M. DNA methylation signatures for breast cancer classification and prognosis. Genome Med. 2012, 4, 26. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, J.; Ronneberg, J.A.; Tost, J.; Kristensen, V. The epigenetics of breast cancer. Mol. Oncol. 2010, 4, 242–254. [Google Scholar] [CrossRef] [PubMed]
- Fleischer, T.; Tekpli, X.; Mathelier, A.; Wang, S.; Nebdal, D.; Dhakal, H.P.; Sahlberg, K.K.; Schlichting, E.; Oslo Breast Cancer Research Consortium; Borresen–Dale, A.L.; et al. DNA methylation at enhancers identifies distinct breast cancer lineages. Nat. Commun. 2017, 8, 1379. [Google Scholar] [CrossRef]
- Temian, D.C.; Pop, L.A.; Irimie, A.I.; Berindan–Neagoe, I. The Epigenetics of Triple–Negative and Basal–Like Breast Cancer: Current Knowledge. J. Breast Cancer 2018, 21, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kwon, Y.M.; Kim, J.S.; Han, J.; Shim, Y.M.; Park, J.; Kim, D.H. Elevated mRNA levels of DNA methyltransferase–1 as an independent prognostic factor in primary nonsmall cell lung cancer. Cancer 2006, 107, 1042–1049. [Google Scholar] [CrossRef]
- Lin, R.K.; Hsu, H.S.; Chang, J.W.; Chen, C.Y.; Chen, J.T.; Wang, Y.C. Alteration of DNA methyltransferases contributes to 5’CpG methylation and poor prognosis in lung cancer. Lung Cancer 2007, 55, 205–213. [Google Scholar] [CrossRef]
- Lin, R.K.; Wu, C.Y.; Chang, J.W.; Juan, L.J.; Hsu, H.S.; Chen, C.Y.; Lu, Y.Y.; Tang, Y.A.; Yang, Y.C.; Yang, P.C.; et al. Dysregulation of p53/Sp1 control leads to DNA methyltransferase–1 overexpression in lung cancer. Cancer Res. 2010, 70, 5807–5817. [Google Scholar] [CrossRef]
- Hua, X.; Zhao, W.; Pesatori, A.C.; Consonni, D.; Caporaso, N.E.; Zhang, T.; Zhu, B.; Wang, M.; Jones, K.; Hicks, B.; et al. Genetic and epigenetic intratumor heterogeneity impacts prognosis of lung adenocarcinoma. Nat. Commun. 2020, 11, 2459. [Google Scholar] [CrossRef]
- Ferreira, H.J.; Heyn, H.; Vizoso, M.; Moutinho, C.; Vidal, E.; Gomez, A.; Martinez–Cardus, A.; Simo–Riudalbas, L.; Moran, S.; Jost, E.; et al. DNMT3A mutations mediate the epigenetic reactivation of the leukemogenic factor MEIS1 in acute myeloid leukemia. Oncogene 2016, 35, 3079–3082. [Google Scholar] [CrossRef]
- Ribeiro, A.F.; Pratcorona, M.; Erpelinck–Verschueren, C.; Rockova, V.; Sanders, M.; Abbas, S.; Figueroa, M.E.; Zeilemaker, A.; Melnick, A.; Lowenberg, B.; et al. Mutant DNMT3A: A marker of poor prognosis in acute myeloid leukemia. Blood 2012, 119, 5824–5831. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.J.; Ding, L.; Shen, D.; Shao, J.; Grillot, M.; McLellan, M.; Fulton, R.; Schmidt, H.; Kalicki–Veizer, J.; O’Laughlin, M.; et al. Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia 2011, 25, 1153–1158. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Rau, R.; Goodell, M.A. DNMT3A in haematological malignancies. Nat. Rev. Cancer 2015, 15, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Im, A.P.; Sehgal, A.R.; Carroll, M.P.; Smith, B.D.; Tefferi, A.; Johnson, D.E.; Boyiadzis, M. DNMT3A and IDH mutations in acute myeloid leukemia and other myeloid malignancies: Associations with prognosis and potential treatment strategies. Leukemia 2014, 28, 1774–1783. [Google Scholar] [CrossRef]
- Hajkova, H.; Markova, J.; Haskovec, C.; Sarova, I.; Fuchs, O.; Kostecka, A.; Cetkovsky, P.; Michalova, K.; Schwarz, J. Decreased DNA methylation in acute myeloid leukemia patients with DNMT3A mutations and prognostic implications of DNA methylation. Leuk. Res. 2012, 36, 1128–1133. [Google Scholar] [CrossRef]
- Zhang, Z.M.; Lu, R.; Wang, P.; Yu, Y.; Chen, D.; Gao, L.; Liu, S.; Ji, D.; Rothbart, S.B.; Wang, Y.; et al. Structural basis for DNMT3A–mediated de novo DNA methylation. Nature 2018, 554, 387–391. [Google Scholar] [CrossRef]
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5–methylcytosine to 5–formylcytosine and 5–carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef]
- Inoue, A.; Shen, L.; Dai, Q.; He, C.; Zhang, Y. Generation and replication–dependent dilution of 5fC and 5caC during mouse preimplantation development. Cell Res. 2011, 21, 1670–1676. [Google Scholar] [CrossRef]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet–mediated formation of 5–carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef]
- Guo, J.U.; Su, Y.; Zhong, C.; Ming, G.L.; Song, H. Hydroxylation of 5–methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 2011, 145, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Kohli, R.M.; Zhang, Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013, 502, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.U.; Su, Y.; Zhong, C.; Ming, G.L.; Song, H. Emerging roles of TET proteins and 5–hydroxymethylcytosines in active DNA demethylation and beyond. Cell Cycle 2011, 10, 2662–2668. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, Y. TET–mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef] [PubMed]
- Scourzic, L.; Mouly, E.; Bernard, O.A. TET proteins and the control of cytosine demethylation in cancer. Genome Med. 2015, 7, 9. [Google Scholar] [CrossRef]
- Chou, W.C.; Chou, S.C.; Liu, C.Y.; Chen, C.Y.; Hou, H.A.; Kuo, Y.Y.; Lee, M.C.; Ko, B.S.; Tang, J.L.; Yao, M.; et al. TET2 mutation is an unfavorable prognostic factor in acute myeloid leukemia patients with intermediate–risk cytogenetics. Blood 2011, 118, 3803–3810. [Google Scholar] [CrossRef]
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Masse, A.; Kosmider, O.; Le Couedic, J.P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in myeloid cancers. New Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Zahid, M.F.; Lasho, T.L.; Finke, C.; Ketterling, R.L.; Gangat, N.; Robertson, K.D.; Hanson, C.A.; Tefferi, A. Number and type of TET2 mutations in chronic myelomonocytic leukemia and their clinical relevance. Blood Cancer J. 2016, 6, e472. [Google Scholar] [CrossRef]
- Huang, Y.; Rao, A. Connections between TET proteins and aberrant DNA modification in cancer. Trends Genet. 2014, 30, 464–474. [Google Scholar] [CrossRef]
- Huang, H.; Jiang, X.; Li, Z.; Li, Y.; Song, C.X.; He, C.; Sun, M.; Chen, P.; Gurbuxani, S.; Wang, J.; et al. TET1 plays an essential oncogenic role in MLL–rearranged leukemia. Proc. Natl. Acad. Sci. USA 2013, 110, 11994–11999. [Google Scholar] [CrossRef]
- Yang, H.; Liu, Y.; Bai, F.; Zhang, J.Y.; Ma, S.H.; Liu, J.; Xu, Z.D.; Zhu, H.G.; Ling, Z.Q.; Ye, D.; et al. Tumor development is associated with decrease of TET gene expression and 5–methylcytosine hydroxylation. Oncogene 2013, 32, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Weissmann, S.; Alpermann, T.; Grossmann, V.; Kowarsch, A.; Nadarajah, N.; Eder, C.; Dicker, F.; Fasan, A.; Haferlach, C.; Haferlach, T.; et al. Landscape of TET2 mutations in acute myeloid leukemia. Leukemia 2012, 26, 934–942. [Google Scholar] [CrossRef]
- Xu, Y.P.; Lv, L.; Liu, Y.; Smith, M.D.; Li, W.C.; Tan, X.M.; Cheng, M.; Li, Z.; Bovino, M.; Aube, J.; et al. Tumor suppressor TET2 promotes cancer immunity and immunotherapy efficacy. J. Clin. Invest. 2019, 129, 4316–4331. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Yin, Y.; Hong, S.; Cao, S.; Huang, Y.; Chen, S.; Liu, Y.; Guan, H.; Zhang, Q.; Li, Y.; et al. TET1 is a Tumor Suppressor That Inhibits Papillary Thyroid Carcinoma Cell Migration and Invasion. Int. J. Endocrinol. 2020, 2020, 3909610. [Google Scholar] [CrossRef] [PubMed]
- Filipczak, P.T.; Leng, S.; Tellez, C.S.; Do, K.C.; Grimes, M.J.; Thomas, C.L.; Walton–Filipczak, S.R.; Picchi, M.A.; Belinsky, S.A. p53–Suppressed Oncogene TET1 Prevents Cellular Aging in Lung Cancer. Cancer Res. 2019, 79, 1758–1768. [Google Scholar] [CrossRef]
- Rivas, M.P.; Aguiar, T.F.M.; Fernandes, G.R.; Caires–Junior, L.C.; Goulart, E.; Telles–Silva, K.A.; Cypriano, M.; de Toledo, S.R.C.; Rosenberg, C.; Carraro, D.M.; et al. TET Upregulation Leads to 5–Hydroxymethylation Enrichment in Hepatoblastoma. Front. Genet. 2019, 10, 553. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Gao, X.; Yu, L. The prognostic impact of tet oncogene family member 2 mutations in patients with acute myeloid leukemia: A systematic–review and meta–analysis. BMC Cancer 2019, 19, 389. [Google Scholar] [CrossRef]
- Gaidzik, V.I.; Paschka, P.; Spath, D.; Habdank, M.; Kohne, C.H.; Germing, U.; von Lilienfeld–Toal, M.; Held, G.; Horst, H.A.; Haase, D.; et al. TET2 mutations in acute myeloid leukemia (AML): Results from a comprehensive genetic and clinical analysis of the AML study group. J. Clin. Oncol. 2012, 30, 1350–1357. [Google Scholar] [CrossRef]
- Yang, L.; Yu, S.J.; Hong, Q.; Yang, Y.; Shao, Z.M. Reduced Expression of TET1, TET2, TET3 and TDG mRNAs Are Associated with Poor Prognosis of Patients with Early Breast Cancer. Plos One 2015, 10, e0133896. [Google Scholar] [CrossRef]
- Rider, C.F.; Carlsten, C. Air pollution and DNA methylation: Effects of exposure in humans. Clin. Epigenetics 2019, 11, 131. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.L.; He, S.W.; Zhang, Y.D.; Duan, H.X.; Huang, T.; Huang, Y.C.; Li, G.F.; Wang, P.; Ma, L.J.; Zhou, G.B.; et al. Air pollution and DNA methylation alterations in lung cancer: A systematic and comparative study. Oncotarget 2017, 8, 1369–1391. [Google Scholar] [CrossRef]
- Soberanes, S.; Gonzalez, A.; Urich, D.; Chiarella, S.E.; Radigan, K.A.; Osornio–Vargas, A.; Joseph, J.; Kalyanaraman, B.; Ridge, K.M.; Chandel, N.S.; et al. Particulate matter Air Pollution induces hypermethylation of the p16 promoter Via a mitochondrial ROS–JNK–DNMT1 pathway. Sci. Rep. 2012, 2, 275. [Google Scholar] [CrossRef]
- Cheung, P.; Allis, C.D.; Sassone–Corsi, P. Signaling to chromatin through histone modifications. Cell 2000, 103, 263–271. [Google Scholar] [CrossRef]
- Rossetto, D.; Avvakumov, N.; Cote, J. Histone phosphorylation: A chromatin modification involved in diverse nuclear events. Epigenetics 2012, 7, 1098–1108. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, T.; Chakravarti, D. A peek into the complex realm of histone phosphorylation. Mol. Cell Biol. 2011, 31, 4858–4873. [Google Scholar] [CrossRef] [PubMed]
- Weake, V.M.; Workman, J.L. Histone ubiquitination: Triggering gene activity. Mol. Cell 2008, 29, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, F.; Dann, G.P.; Beh, L.Y.; Debelouchina, G.T.; Hofmann, R.; Muir, T.W. Functional crosstalk between histone H2B ubiquitylation and H2A modifications and variants. Nat. Commun. 2018, 9, 1394. [Google Scholar] [CrossRef]
- Bannister, A.J.; Schneider, R.; Kouzarides, T. Histone methylation: Dynamic or static? Cell 2002, 109, 801–806. [Google Scholar] [CrossRef]
- Schneider, R.; Bannister, A.J.; Myers, F.A.; Thorne, A.W.; Crane–Robinson, C.; Kouzarides, T. Histone H3 lysine 4 methylation patterns in higher eukaryotic genes. Nat. Cell Biol. 2004, 6, 73–77. [Google Scholar] [CrossRef]
- Shi, X.; Hong, T.; Walter, K.L.; Ewalt, M.; Michishita, E.; Hung, T.; Carney, D.; Pena, P.; Lan, F.; Kaadige, M.R.; et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature 2006, 442, 96–99. [Google Scholar] [CrossRef]
- Volpe, T.A.; Kidner, C.; Hall, I.M.; Teng, G.; Grewal, S.I.; Martienssen, R.A. Regulation of heterochromatic silencing and histone H3 lysine–9 methylation by RNAi. Science 2002, 297, 1833–1837. [Google Scholar] [CrossRef]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument–Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H3 lysine 27 methylation in Polycomb–group silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef]
- Graff, J.; Tsai, L.H. Histone acetylation: Molecular mnemonics on the chromatin. Nat. Rev. Neurosci. 2013, 14, 97–111. [Google Scholar] [CrossRef]
- Wade, P.A.; Pruss, D.; Wolffe, A.P. Histone acetylation: Chromatin in action. Trends Biochem. Sci. 1997, 22, 128–132. [Google Scholar] [CrossRef]
- Peleg, S.; Feller, C.; Ladurner, A.G.; Imhof, A. The Metabolic Impact on Histone Acetylation and Transcription in Ageing. Trends Biochem. Sci. 2016, 41, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Yan, Q. Histone ubiquitination and deubiquitination in transcription, DNA damage response, and cancer. Front. Oncol 2012, 2, 26. [Google Scholar] [CrossRef]
- Shanmugam, M.K.; Arfuso, F.; Arumugam, S.; Chinnathambi, A.; Jinsong, B.; Warrier, S.; Wang, L.Z.; Kumar, A.P.; Ahn, K.S.; Sethi, G.; et al. Role of novel histone modifications in cancer. Oncotarget 2018, 9, 11414–11426. [Google Scholar] [CrossRef]
- Li, J.; Zhu, S.; Ke, X.X.; Cui, H. Role of several histone lysine methyltransferases in tumor development. Biomed. Rep. 2016, 4, 293–299. [Google Scholar] [CrossRef]
- Di Cerbo, V.; Schneider, R. Cancers with wrong HATs: The impact of acetylation. Brief. Funct. Genom. 2013, 12, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Glozak, M.A.; Seto, E. Histone deacetylases and cancer. Oncogene 2007, 26, 5420–5432. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.J.; Byun, D.S.; Nasser, S.; Murray, L.B.; Ayyanar, K.; Arango, D.; Figueroa, M.; Melnick, A.; Kao, G.D.; Augenlicht, L.H.; et al. HDAC4 promotes growth of colon cancer cells via repression of p21. Mol. Biol. Cell 2008, 19, 4062–4075. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Fang, D. The Roles of SIRT1 in Cancer. Genes Cancer 2013, 4, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Witt, O.; Deubzer, H.E.; Milde, T.; Oehme, I. HDAC family: What are the cancer relevant targets? Cancer Lett. 2009, 277, 8–21. [Google Scholar] [CrossRef]
- Johnstone, R.W.; Licht, J.D. Histone deacetylase inhibitors in cancer therapy: Is transcription the primary target? Cancer Cell 2003, 4, 13–18. [Google Scholar] [CrossRef]
- Leslie, P.L.; Chao, Y.L.; Tsai, Y.H.; Ghosh, S.K.; Porrello, A.; Van Swearingen, A.E.D.; Harrison, E.B.; Cooley, B.C.; Parker, J.S.; Carey, L.A.; et al. Histone deacetylase 11 inhibition promotes breast cancer metastasis from lymph nodes. Nat. Commun. 2019, 10, 4192. [Google Scholar] [CrossRef]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Invest. 2014, 124, 30–39. [Google Scholar] [CrossRef]
- Thurn, K.T.; Thomas, S.; Moore, A.; Munster, P.N. Rational therapeutic combinations with histone deacetylase inhibitors for the treatment of cancer. Future Oncol. 2011, 7, 263–283. [Google Scholar] [CrossRef]
- Zhu, W.G.; Otterson, G.A. The interaction of histone deacetylase inhibitors and DNA methyltransferase inhibitors in the treatment of human cancer cells. Curr. Med. Chem. Anticancer. Agents 2003, 3, 187–199. [Google Scholar] [CrossRef]
- Wang, L.; Gural, A.; Sun, X.J.; Zhao, X.; Perna, F.; Huang, G.; Hatlen, M.A.; Vu, L.; Liu, F.; Xu, H.; et al. The leukemogenicity of AML1–ETO is dependent on site–specific lysine acetylation. Science 2011, 333, 765–769. [Google Scholar] [CrossRef]
- Ogryzko, V.V.; Schiltz, R.L.; Russanova, V.; Howard, B.H.; Nakatani, Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 1996, 87, 953–959. [Google Scholar] [CrossRef]
- Johnstone, R.W. Histone–deacetylase inhibitors: Novel drugs for the treatment of cancer. Nat. Rev. Drug Discov. 2002, 1, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Dekker, F.J.; Haisma, H.J. Histone acetyl transferases as emerging drug targets. Drug Discov. Today 2009, 14, 19–20. [Google Scholar] [CrossRef]
- Eliseeva, E.D.; Valkov, V.; Jung, M.; Jung, M.O. Characterization of novel inhibitors of histone acetyltransferases. Mol. Cancer Ther 2007, 6, 2391–2398. [Google Scholar] [CrossRef]
- Wapenaar, H.; Dekker, F.J. Histone acetyltransferases: Challenges in targeting bi–substrate enzymes. Clin. Epigenetics 2016, 8, 59. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin–4 encodes small RNAs with antisense complementarity to lin–14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Esquela–Kerscher, A.; Slack, F.J. Oncomirs–microRNAs with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, A.; Scott, J.G.; Harris, A.L.; Buffa, F.M. Pan–cancer characterisation of microRNA across cancer hallmarks reveals microRNA–mediated downregulation of tumour suppressors. Nat. Commun. 2018, 9, 5228. [Google Scholar] [CrossRef] [PubMed]
- Fazi, F.; Racanicchi, S.; Zardo, G.; Starnes, L.M.; Mancini, M.; Travaglini, L.; Diverio, D.; Ammatuna, E.; Cimino, G.; Lo–Coco, F.; et al. Epigenetic silencing of the myelopoiesis regulator microRNA–223 by the AML1/ETO oncoprotein. Cancer Cell 2007, 12, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gao, L.; Luo, X.; Wang, L.; Gao, X.; Wang, W.; Sun, J.; Dou, L.; Li, J.; Xu, C.; et al. Epigenetic silencing of microRNA–193a contributes to leukemogenesis in t(8;21) acute myeloid leukemia by activating the PTEN/PI3K signal pathway. Blood 2013, 121, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, S.K.; Perez, A.W.; White, E.S.; Lian, J.B.; Stein, J.L.; Stein, G.S. An AML1–ETO/miR–29b–1 regulatory circuit modulates phenotypic properties of acute myeloid leukemia cells. Oncotarget 2017, 8, 39994–40005. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wu, L.C.; Pang, J.; Santhanam, R.; Schwind, S.; Wu, Y.Z.; Hickey, C.J.; Yu, J.; Becker, H.; Maharry, K.; et al. Sp1/NFkappaB/HDAC/miR–29b regulatory network in KIT–driven myeloid leukemia. Cancer Cell 2010, 17, 333–347. [Google Scholar] [CrossRef]
- Kumar, V.; Palermo, R.; Talora, C.; Campese, A.F.; Checquolo, S.; Bellavia, D.; Tottone, L.; Testa, G.; Miele, E.; Indraccolo, S.; et al. Notch and NF–kB signaling pathways regulate miR–223/FBXW7 axis in T–cell acute lymphoblastic leukemia. Leukemia 2014, 28, 2324–2335. [Google Scholar] [CrossRef] [PubMed]
- Scott, G.K.; Mattie, M.D.; Berger, C.E.; Benz, S.C.; Benz, C.C. Rapid alteration of microRNA levels by histone deacetylase inhibition. Cancer Res. 2006, 66, 1277–1281. [Google Scholar] [CrossRef]
- Sato, F.; Tsuchiya, S.; Meltzer, S.J.; Shimizu, K. MicroRNAs and epigenetics. FEBS J. 2011, 278, 1598–1609. [Google Scholar] [CrossRef]
- Han, L.; Witmer, P.D.; Casey, E.; Valle, D.; Sukumar, S. DNA methylation regulates MicroRNA expression. Cancer Biol. Ther. 2007, 6, 1284–1288. [Google Scholar] [CrossRef]
- Chuang, J.C.; Jones, P.A. Epigenetics and microRNAs. Pediatr. Res. 2007, 61, 24R–29R. [Google Scholar] [CrossRef]
- Palmer, J.D.; Soule, B.P.; Simone, B.A.; Zaorsky, N.G.; Jin, L.; Simone, N.L. MicroRNA expression altered by diet: Can food be medicinal? Ageing Res. Rev. 2014, 17, 16–24. [Google Scholar] [CrossRef]
- Slattery, M.L.; Herrick, J.S.; Mullany, L.E.; Stevens, J.R.; Wolff, R.K. Diet and lifestyle factors associated with miRNA expression in colorectal tissue. Pharmgenomics Pers. Med. 2017, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Landrier, J.F.; Derghal, A.; Mounien, L. MicroRNAs in Obesity and Related Metabolic Disorders. Cells 2019, 8, 859. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Li, H.; Cai, J.; Wang, C.; Lin, Z.; Liu, C.; Niu, Y.; Zhao, Z.; Li, W.; Kan, H. Fine Particulate Air Pollution and the Expression of microRNAs and Circulating Cytokines Relevant to Inflammation, Coagulation, and Vasoconstriction. Environ. Health Perspect. 2018, 126, 017007. [Google Scholar] [CrossRef]
- Tanaka, N.; Toyooka, S.; Soh, J.; Tsukuda, K.; Shien, K.; Furukawa, M.; Muraoka, T.; Maki, Y.; Ueno, T.; Yamamoto, H.; et al. Downregulation of microRNA–34 induces cell proliferation and invasion of human mesothelial cells. Oncol. Rep. 2013, 29, 2169–2174. [Google Scholar] [CrossRef] [PubMed]
- Kubo, T.; Toyooka, S.; Tsukuda, K.; Sakaguchi, M.; Fukazawa, T.; Soh, J.; Asano, H.; Ueno, T.; Muraoka, T.; Yamamoto, H.; et al. Epigenetic silencing of microRNA–34b/c plays an important role in the pathogenesis of malignant pleural mesothelioma. Clin. Cancer Res. 2011, 17, 4965–4974. [Google Scholar] [CrossRef] [PubMed]
- Misso, G.; Di Martino, M.T.; De Rosa, G.; Farooqi, A.A.; Lombardi, A.; Campani, V.; Zarone, M.R.; Gulla, A.; Tagliaferri, P.; Tassone, P.; et al. Mir–34: A new weapon against cancer? Mol. Ther. Nucleic Acids 2014, 3, e194. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Croce, C.M. MicroRNA–cancer connection: The beginning of a new tale. Cancer Res. 2006, 66, 7390–7394. [Google Scholar] [CrossRef]
- Garzon, R.; Calin, G.A.; Croce, C.M. MicroRNAs in Cancer. Annu. Rev. Med. 2009, 60, 167–179. [Google Scholar] [CrossRef]
- Manikandan, J.; Aarthi, J.J.; Kumar, S.D.; Pushparaj, P.N. Oncomirs: The potential role of non–coding microRNAs in understanding cancer. Bioinformation 2008, 2, 330–334. [Google Scholar] [CrossRef]
- Bhere, D.; Arghiani, N.; Lechtich, E.R.; Yao, Y.; Alsaab, S.; Bei, F.; Matin, M.M.; Shah, K. Simultaneous downregulation of miR–21 and upregulation of miR–7 has anti–tumor efficacy. Sci. Rep. 2020, 10, 1779. [Google Scholar] [CrossRef]
- Sochor, M.; Basova, P.; Pesta, M.; Dusilkova, N.; Bartos, J.; Burda, P.; Pospisil, V.; Stopka, T. Oncogenic microRNAs: miR–155, miR–19a, miR–181b, and miR–24 enable monitoring of early breast cancer in serum. BMC Cancer 2014, 14, 448. [Google Scholar] [CrossRef]
- Cimmino, A.; Calin, G.A.; Fabbri, M.; Iorio, M.V.; Ferracin, M.; Shimizu, M.; Wojcik, S.E.; Aqeilan, R.I.; Zupo, S.; Dono, M.; et al. miR–15 and miR–16 induce apoptosis by targeting BCL2. Proc. Natl. Acad. Sci. USA 2005, 102, 13944–13949. [Google Scholar] [CrossRef]
- Wang, J.; Li, Y.; Ding, M.; Zhang, H.; Xu, X.; Tang, J. Molecular mechanisms and clinical applications of miR–22 in regulating malignant progression in human cancer. Int. J. Oncol. 2017, 50, 345–355. [Google Scholar] [CrossRef]
- Bader, A.G.; Brown, D.; Stoudemire, J.; Lammers, P. Developing therapeutic microRNAs for cancer. Gene Ther. 2011, 18, 1121–1126. [Google Scholar] [CrossRef]
- Zealy, R.W.; Wrenn, S.P.; Davila, S.; Min, K.W.; Yoon, J.H. MicroRNA–binding proteins: Specificity and function. Wiley Interdiscip. Rev. RNA 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Hoareau–Aveilla, C.; Meggetto, F. Crosstalk between microRNA and DNA Methylation Offers Potential Biomarkers and Targeted Therapies in ALK–Positive Lymphomas. Cancers 2017, 9, 100. [Google Scholar] [CrossRef] [PubMed]
- Anwar, S.L.; Lehmann, U. DNA methylation, microRNAs, and their crosstalk as potential biomarkers in hepatocellular carcinoma. World J. Gastroenterol. 2014, 20, 7894–7913. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Ma, Y.; Wang, H.; Qin, H. Reciprocal regulation between microRNAs and epigenetic machinery in colorectal cancer. Oncol. Lett. 2017, 13, 1048–1057. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wu, W.; Claret, F.X. Mutual regulation of microRNAs and DNA methylation in human cancers. Epigenetics 2017, 12, 187–197. [Google Scholar] [CrossRef]
- Sun, X.; He, Y.; Huang, C.; Ma, T.T.; Li, J. The epigenetic feedback loop between DNA methylation and microRNAs in fibrotic disease with an emphasis on DNA methyltransferases. Cell Signal. 2013, 25, 1870–1876. [Google Scholar] [CrossRef] [PubMed]
- Bourassa, M.W.; Ratan, R.R. The interplay between microRNAs and histone deacetylases in neurological diseases. Neurochem. Int. 2014, 77, 33–39. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhao, K.; Shen, Q.; Han, Y.; Gu, Y.; Li, X.; Zhao, D.; Liu, Y.; Wang, C.; Zhang, X.; et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL–6. Nature 2015, 525, 389–393. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Wang, Z.; Xie, W.; Cai, Y.; Xia, L.; Easwaran, H.; Luo, J.; Yen, R.C.; Li, Y.; Baylin, S.B. Acetylation Enhances TET2 Function in Protecting against Abnormal DNA Methylation during Oxidative Stress. Mol. Cell 2017, 65, 323–335. [Google Scholar] [CrossRef]
- Robertson, K.D.; Ait–Si–Ali, S.; Yokochi, T.; Wade, P.A.; Jones, P.L.; Wolffe, A.P. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F–responsive promoters. Nat. Genet. 2000, 25, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Shen, T.; Huynh, L.; Klisovic, M.I.; Rush, L.J.; Ford, J.L.; Yu, J.; Becknell, B.; Li, Y.; Liu, C.; et al. Interplay of RUNX1/MTG8 and DNA methyltransferase 1 in acute myeloid leukemia. Cancer Res. 2005, 65, 1277–1284. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Klisovic, R.B.; Vukosavljevic, T.; Yu, J.; Paschka, P.; Huynh, L.; Pang, J.; Neviani, P.; Liu, Z.; Blum, W.; et al. Targeting AML1/ETO–histone deacetylase repressor complex: A novel mechanism for valproic acid–mediated gene expression and cellular differentiation in AML1/ETO–positive acute myeloid leukemia cells. J. Pharmacol. Exp. Ther. 2007, 321, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by beta–hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Shyh–Chang, N.; Locasale, J.W.; Lyssiotis, C.A.; Zheng, Y.; Teo, R.Y.; Ratanasirintrawoot, S.; Zhang, J.; Onder, T.; Unternaehrer, J.J.; Zhu, H.; et al. Influence of threonine metabolism on S–adenosylmethionine and histone methylation. Science 2013, 339, 222–226. [Google Scholar] [CrossRef]
- Vogelstein, B.; Fearon, E.R.; Hamilton, S.R.; Kern, S.E.; Preisinger, A.C.; Leppert, M.; Nakamura, Y.; White, R.; Smits, A.M.; Bos, J.L. Genetic alterations during colorectal–tumor development. New Engl. J. Med. 1988, 319, 525–532. [Google Scholar] [CrossRef]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef]
- Jones, P.A. Epigenetics in carcinogenesis and cancer prevention. Ann. N. Y. Acad. Sci. 2003, 983, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Widschwendter, M.; Jones, P.A. DNA methylation and breast carcinogenesis. Oncogene 2002, 21, 5462–5482. [Google Scholar] [CrossRef]
- Jones, P.A.; Laird, P.W. Cancer epigenetics comes of age. Nature Genet. 1999, 21, 163–167. [Google Scholar] [CrossRef]
- Schoofs, T.; Berdel, W.E.; Muller–Tidow, C. Origins of aberrant DNA methylation in acute myeloid leukemia. Leukemia 2014, 28, 1–14. [Google Scholar] [CrossRef]
- Fustinoni, S.; Rossella, F.; Polledri, E.; Bollati, V.; Campo, L.; Byun, H.M.; Agnello, L.; Consonni, D.; Pesatori, A.C.; Baccarelli, A.; et al. Global DNA methylation and low–level exposure to benzene. Med. Lav. 2012, 103, 84–95. [Google Scholar]
- Salemi, R.; Marconi, A.; Di Salvatore, V.; Franco, S.; Rapisarda, V.; Libra, M. Epigenetic alterations and occupational exposure to benzene, fibers, and heavy metals associated with tumor development. Mol. Med. Rep. 2017, 15, 3366–3371. [Google Scholar] [CrossRef] [PubMed]
- Baccarelli, A.; Bollati, V. Epigenetics and environmental chemicals. Curr. Opin. Pediatr. 2009, 21, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Jimenez–Garza, O.; Guo, L.; Byun, H.M.; Carrieri, M.; Bartolucci, G.B.; Barron–Vivanco, B.S.; Baccarelli, A.A. Aberrant promoter methylation in genes related to hematopoietic malignancy in workers exposed to a VOC mixture. Toxicol. Appl. Pharmacol. 2018, 339, 65–72. [Google Scholar] [CrossRef]
- Xing, C.; Wang, Q.F.; Li, B.; Tian, H.; Ni, Y.; Yin, S.; Li, G. Methylation and expression analysis of tumor suppressor genes p15 and p16 in benzene poisoning. Chem. Biol. Interact. 2010, 184, 306–309. [Google Scholar] [CrossRef]
- Zheng, M.; Lin, F.; Hou, F.; Li, G.; Zhu, C.; Xu, P.; Xing, C.; Wang, Q. Association between Promoter Methylation of Gene ERCC3 and Benzene Hematotoxicity. Int. J. Environ. Res. Public Health 2017, 14, 921. [Google Scholar] [CrossRef] [PubMed]
- Gao, A.; Zuo, X.; Liu, Q.; Lu, X.; Guo, W.; Tian, L. Methylation of PARP–1 promoter involved in the regulation of benzene–induced decrease of PARP–1 mRNA expression. Toxicol. Lett. 2010, 195, 114–118. [Google Scholar] [CrossRef]
- Yu, K.; Shi, Y.F.; Yang, K.Y.; Zhuang, Y.; Zhu, R.H.; Xu, X.; Cai, G. Decreased topoisomerase IIalpha expression and altered histone and regulatory factors of topoisomerase IIalpha promoter in patients with chronic benzene poisoning. Toxicol. Lett. 2011, 203, 111–117. [Google Scholar] [CrossRef]
- Li, J.; Xing, X.; Zhang, X.; Liang, B.; He, Z.; Gao, C.; Wang, S.; Wang, F.; Zhang, H.; Zeng, S.; et al. Enhanced H3K4me3 modifications are involved in the transactivation of DNA damage responsive genes in workers exposed to low–level benzene. Environ. Pollut. 2018, 234, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Zhang, J.; Tan, K.; Sun, R.; Yin, L.; Pu, Y. Benzene–Induced Aberrant miRNA Expression Profile in Hematopoietic Progenitor Cells in C57BL/6 Mice. Int. J. Mol. Sci. 2015, 16, 27058–27071. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, X.; Bian, Q.; Shi, Y.; Liu, Q.; Ding, L.; Zhang, H.; Zhu, B. Analysis of plasma microRNA expression profiles in a Chinese population occupationally exposed to benzene and in a population with chronic benzene poisoning. J. Thorac. Dis. 2016, 8, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Kotsyfakis, M.; Patelarou, E. MicroRNAs as biomarkers of harmful environmental and occupational exposures: A systematic review. Biomarkers 2019, 24, 623–630. [Google Scholar] [CrossRef]
- Liang, B.; Chen, Y.; Yuan, W.; Qin, F.; Zhang, Q.; Deng, N.; Liu, X.; Ma, X.; Zhang, X.; Zhang, B.; et al. Down–regulation of miRNA–451a and miRNA–486–5p involved in benzene–induced inhibition on erythroid cell differentiation in vitro and in vivo. Arch. Toxicol. 2018, 92, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Habieb, M.S.E.; Younis, F.E.; Safan, M.; Allam, H.K. PARP1–DNMT1–CTCF complex and the apoptotic–induced factor mRNA expressions in workers occupationally exposed to benzene. Environ. Sci. Pollut. Res. Int. 2020, 27, 22648–22657. [Google Scholar] [CrossRef] [PubMed]
- Rothman, N.; Smith, M.T.; Hayes, R.B.; Li, G.L.; Irons, R.D.; Dosemeci, M.; Haas, R.; Stillman, W.S.; Linet, M.; Xi, L.Q.; et al. An epidemiologic study of early biologic effects of benzene in Chinese workers. Environ. Health Perspect. 1996, 104 (Suppl. S6), 1365–1370. [Google Scholar] [CrossRef]
- Gillis, B.; Gavin, I.M.; Arbieva, Z.; King, S.T.; Jayaraman, S.; Prabhakar, B.S. Identification of human cell responses to benzene and benzene metabolites. Genomics 2007, 90, 324–333. [Google Scholar] [CrossRef]
- Wang, F.; Li, C.; Liu, W.; Jin, Y. Modulation of microRNA expression by volatile organic compounds in mouse lung. Environ. Toxicol. 2014, 29, 679–689. [Google Scholar] [CrossRef]
- Lim, J.H.; Song, M.K.; Cho, Y.; Kim, W.; Han, S.O.; Ryu, J.C. Comparative analysis of microRNA and mRNA expression profiles in cells and exosomes under toluene exposure. Toxicol. In Vitro 2017, 41, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Silvestre, R.T.; Bravo, M.; Santiago, F.; Delmonico, L.; Scherrer, L.; Otero, U.B.; Liehr, T.; Alves, G.; Chantre–Justino, M.; Ornellas, M.H. Hypermethylation in Gene Promoters Are Induced by Chronic Exposure to Benzene, Toluene, Ethylbenzene and Xylenes. Pak. J. Biol. Sci. 2020, 23, 518–525. [Google Scholar] [CrossRef]
- Hong, J.Y.; Yu, S.Y.; Kim, S.Y.; Ahn, J.J.; Kim, Y.; Kim, G.W.; Son, S.W.; Park, J.T.; Hwang, S.Y. Association analysis of toluene exposure time with high–throughput mRNA expressions and methylation patterns using in vivo samples. Environ. Res. 2016, 146, 59–64. [Google Scholar] [CrossRef]
- Jimenez–Garza, O.; Baccarelli, A.A.; Byun, H.M.; Marquez–Gamino, S.; Barron–Vivanco, B.S.; Albores, A. CYP2E1 epigenetic regulation in chronic, low–level toluene exposure: Relationship with oxidative stress and smoking habit. Toxicol. Appl. Pharmacol. 2015, 286, 207–215. [Google Scholar] [CrossRef]
- Dick, A.L.W.; Zhao, Q.; Crossin, R.; Baker–Andresen, D.; Li, X.; Edson, J.; Roeh, S.; Marshall, V.; Bredy, T.W.; Lawrence, A.J.; et al. Adolescent chronic intermittent toluene inhalation dynamically regulates the transcriptome and neuronal methylome within the rat medial prefrontal cortex. Addict. Biol. 2021, 26, e12937. [Google Scholar] [CrossRef]
- Sanchez–Serrano, S.L.; Cruz, S.L.; Lamas, M. Repeated toluene exposure modifies the acetylation pattern of histones H3 and H4 in the rat brain. Neurosci. Lett. 2011, 489, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Huerta–Rivas, A.; Lopez–Rubalcava, C.; Sanchez–Serrano, S.L.; Valdez–Tapia, M.; Lamas, M.; Cruz, S.L. Toluene impairs learning and memory, has antinociceptive effects, and modifies histone acetylation in the dentate gyrus of adolescent and adult rats. Pharmacol. Biochem. Behav. 2012, 102, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Sisto, R.; Capone, P.; Cerini, L.; Sanjust, F.; Paci, E.; Pigini, D.; Gatto, M.P.; Gherardi, M.; Gordiani, A.; L’Episcopo, N.; et al. Circulating microRNAs as potential biomarkers of occupational exposure to low dose organic solvents. Toxicol. Rep. 2019, 6, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Sisto, R.; Capone, P.; Cerini, L.; Paci, E.; Pigini, D.; Gherardi, M.; Gordiani, A.; L’Episcopo, N.; Tranfo, G.; Chiarella, P. Occupational exposure to volatile organic compounds affects microRNA profiling: Towards the identification of novel biomarkers. Toxicol. Rep. 2020, 7, 700–710. [Google Scholar] [CrossRef]
- Herbstman, J.B.; Tang, D.; Zhu, D.; Qu, L.; Sjodin, A.; Li, Z.; Camann, D.; Perera, F.P. Prenatal exposure to polycyclic aromatic hydrocarbons, benzo[a]pyrene–DNA adducts, and genomic DNA methylation in cord blood. Environ. Health Perspect. 2012, 120, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kalia, V.; Perera, F.; Herbstman, J.; Li, T.; Nie, J.; Qu, L.R.; Yu, J.; Tang, D. Prenatal airborne polycyclic aromatic hydrocarbon exposure, LINE1 methylation and child development in a Chinese cohort. Environ. Int. 2017, 99, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhu, X.; Yu, K.; Jiang, H.; Zhang, Y.; Wang, B.; Liu, X.; Deng, S.; Hu, J.; Deng, Q.; et al. Exposure to Polycyclic Aromatic Hydrocarbons and Accelerated DNA Methylation Aging. Environ. Health Perspect. 2018, 126, 067005. [Google Scholar] [CrossRef]
- Ma, Y.; Lu, Z.; Wang, L.; Qiang, M. Correlation of Internal Exposure Levels of Polycyclic Aromatic Hydrocarbons to Methylation of Imprinting Genes of Sperm DNA. Int. J. Environ. Res. Public Health 2019, 16, 2606. [Google Scholar] [CrossRef]
- Alegria–Torres, J.A.; Barretta, F.; Batres–Esquivel, L.E.; Carrizales–Yanez, L.; Perez–Maldonado, I.N.; Baccarelli, A.; Bertazzi, P.A. Epigenetic markers of exposure to polycyclic aromatic hydrocarbons in Mexican brickmakers: A pilot study. Chemosphere 2013, 91, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Pavanello, S.; Bollati, V.; Pesatori, A.C.; Kapka, L.; Bolognesi, C.; Bertazzi, P.A.; Baccarelli, A. Global and gene–specific promoter methylation changes are related to anti–B[a]PDE–DNA adduct levels and influence micronuclei levels in polycyclic aromatic hydrocarbon–exposed individuals. Int. J. Cancer 2009, 125, 1692–1697. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Ma, J.; Zhang, B.; Duan, H.; He, Z.; Zeng, J.; Zeng, X.; Li, D.; Wang, Q.; Xiao, Y.; et al. CpG site–specific hypermethylation of p16INK4alpha in peripheral blood lymphocytes of PAH–exposed workers. Cancer Epidemiol. Biomarkers Prev. 2012, 21, 182–190. [Google Scholar] [CrossRef]
- White, A.J.; Chen, J.; McCullough, L.E.; Xu, X.; Cho, Y.H.; Teitelbaum, S.L.; Neugut, A.I.; Terry, M.B.; Hibshoosh, H.; Santella, R.M.; et al. Polycyclic aromatic hydrocarbon (PAH)–DNA adducts and breast cancer: Modification by gene promoter methylation in a population–based study. Cancer Causes Control. 2015, 26, 1791–1802. [Google Scholar] [CrossRef] [PubMed]
- White, A.J.; Chen, J.; Teitelbaum, S.L.; McCullough, L.E.; Xu, X.; Hee Cho, Y.; Conway, K.; Beyea, J.; Stellman, S.D.; Steck, S.E.; et al. Sources of polycyclic aromatic hydrocarbons are associated with gene–specific promoter methylation in women with breast cancer. Environ. Res. 2016, 145, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Lee, Y.S.; Lee, D.H.; Kim, D.S. Polycyclic aromatic hydrocarbons are associated with insulin receptor substrate 2 methylation in adipose tissues of Korean women. Environ. Res. 2016, 150, 47–51. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, L.; Xing, X.; Li, D.; Gao, C.; He, Z.; Li, J.; Zhu, X.; Xiao, X.; Wang, S.; et al. Specific histone modifications were associated with the PAH–induced DNA damage response in coke oven workers. Toxicol. Res. 2016, 5, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Deng, Q.; Feng, J.; Zhang, X.; Dai, X.; Li, L.; Yang, B.; Wu, T.; Cheng, J. Polycyclic Aromatic Hydrocarbons–Associated MicroRNAs and Heart Rate Variability in Coke Oven Workers. J. Occup. Environ. Med. 2016, 58, e24–e31. [Google Scholar] [CrossRef] [PubMed]
- Gordon, M.W.; Yan, F.; Zhong, X.; Mazumder, P.B.; Xu–Monette, Z.Y.; Zou, D.; Young, K.H.; Ramos, K.S.; Li, Y. Regulation of p53–targeting microRNAs by polycyclic aromatic hydrocarbons: Implications in the etiology of multiple myeloma. Mol. Carcinog. 2015, 54, 1060–1069. [Google Scholar] [CrossRef] [PubMed]
- Roundtree, I.A.; Evans, M.E.; Pan, T.; He, C. Dynamic RNA Modifications in Gene Expression Regulation. Cell 2017, 169, 1187–1200. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Weng, H.; Su, R.; Weng, X.; Zuo, Z.; Li, C.; Huang, H.; Nachtergaele, S.; Dong, L.; Hu, C.; et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N6–Methyladenosine RNA Demethylase. Cancer Cell 2017, 31, 127–141. [Google Scholar] [CrossRef]
- Deng, X.; Su, R.; Weng, H.; Huang, H.; Li, Z.; Chen, J. RNA N(6)–methyladenosine modification in cancers: Current status and perspectives. Cell Res. 2018, 28, 507–517. [Google Scholar] [CrossRef]
- Su, R.; Dong, L.; Li, C.; Nachtergaele, S.; Wunderlich, M.; Qing, Y.; Deng, X.; Wang, Y.; Weng, X.; Hu, C.; et al. R–2HG Exhibits Anti–tumor Activity by Targeting FTO/m(6)A/MYC/CEBPA Signaling. Cell 2018, 172, 90–105. [Google Scholar] [CrossRef]
- Yan, F.; Al–Kali, A.; Zhang, Z.; Liu, J.; Pang, J.; Zhao, N.; He, C.; Litzow, M.R.; Liu, S. A dynamic N(6)–methyladenosine methylome regulates intrinsic and acquired resistance to tyrosine kinase inhibitors. Cell Res. 2018, 28, 1062–1076. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Wei, Q.; Jin, J.; Luo, Q.; Liu, Y.; Yang, Y.; Cheng, C.; Li, L.; Pi, J.; Si, Y.; et al. The m6A reader YTHDF1 promotes ovarian cancer progression via augmenting EIF3C translation. Nucleic Acids Res. 2020, 48, 3816–3831. [Google Scholar] [CrossRef]
- Liu, J.; Eckert, M.A.; Harada, B.T.; Liu, S.M.; Lu, Z.; Yu, K.; Tienda, S.M.; Chryplewicz, A.; Zhu, A.C.; Yang, Y. M(6)A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat. Cell Biol. 2018, 20, 1074–1083. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Acrolein | Inorganic Sulfates and Nitrates |
|---|---|
| Ammonia | Methane |
| Benzene | Methanol |
| 1,3–Butadiene | Nitric acid |
| Carbon monoxide | Metals (e.g., lead and platinum) |
| Formaldehyde/Acetaldehyde | Nitrous acid |
| Formic acid | Nitrogen oxides |
| Heterocyclics and derivatives | Oxides of nitrogen |
| Hydrocarbons (C1–C18) and derivatives | Polycyclic aromatic hydrocarbons and Derivatives |
| Hydrocarbons (C14–C35) and derivatives | Sulfur oxides |
| Hydrogen cyanide | Toluene |
| Hydrogen sulfide | Nitrated hydrocarbons |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mueller, S.; Dennison, G.; Liu, S. An Assessment on Ethanol-Blended Gasoline/Diesel Fuels on Cancer Risk and Mortality. Int. J. Environ. Res. Public Health 2021, 18, 6930. https://doi.org/10.3390/ijerph18136930
Mueller S, Dennison G, Liu S. An Assessment on Ethanol-Blended Gasoline/Diesel Fuels on Cancer Risk and Mortality. International Journal of Environmental Research and Public Health. 2021; 18(13):6930. https://doi.org/10.3390/ijerph18136930
Chicago/Turabian StyleMueller, Steffen, Gail Dennison, and Shujun Liu. 2021. "An Assessment on Ethanol-Blended Gasoline/Diesel Fuels on Cancer Risk and Mortality" International Journal of Environmental Research and Public Health 18, no. 13: 6930. https://doi.org/10.3390/ijerph18136930
APA StyleMueller, S., Dennison, G., & Liu, S. (2021). An Assessment on Ethanol-Blended Gasoline/Diesel Fuels on Cancer Risk and Mortality. International Journal of Environmental Research and Public Health, 18(13), 6930. https://doi.org/10.3390/ijerph18136930