The Effects of Sulglycotide on the Adhesion and the Inflammation of Helicobacter Pylori

 , ,
, , 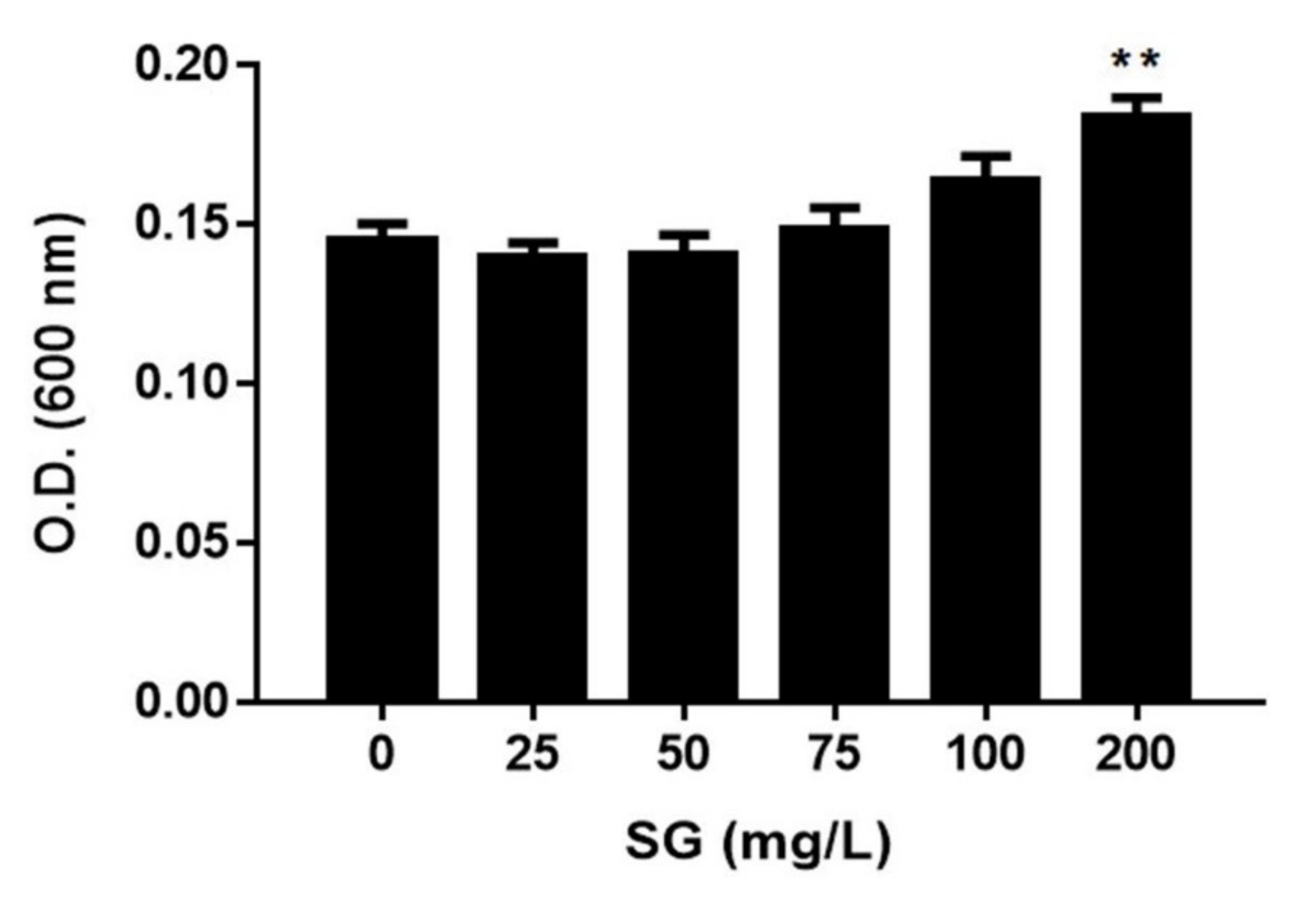{kind=link}
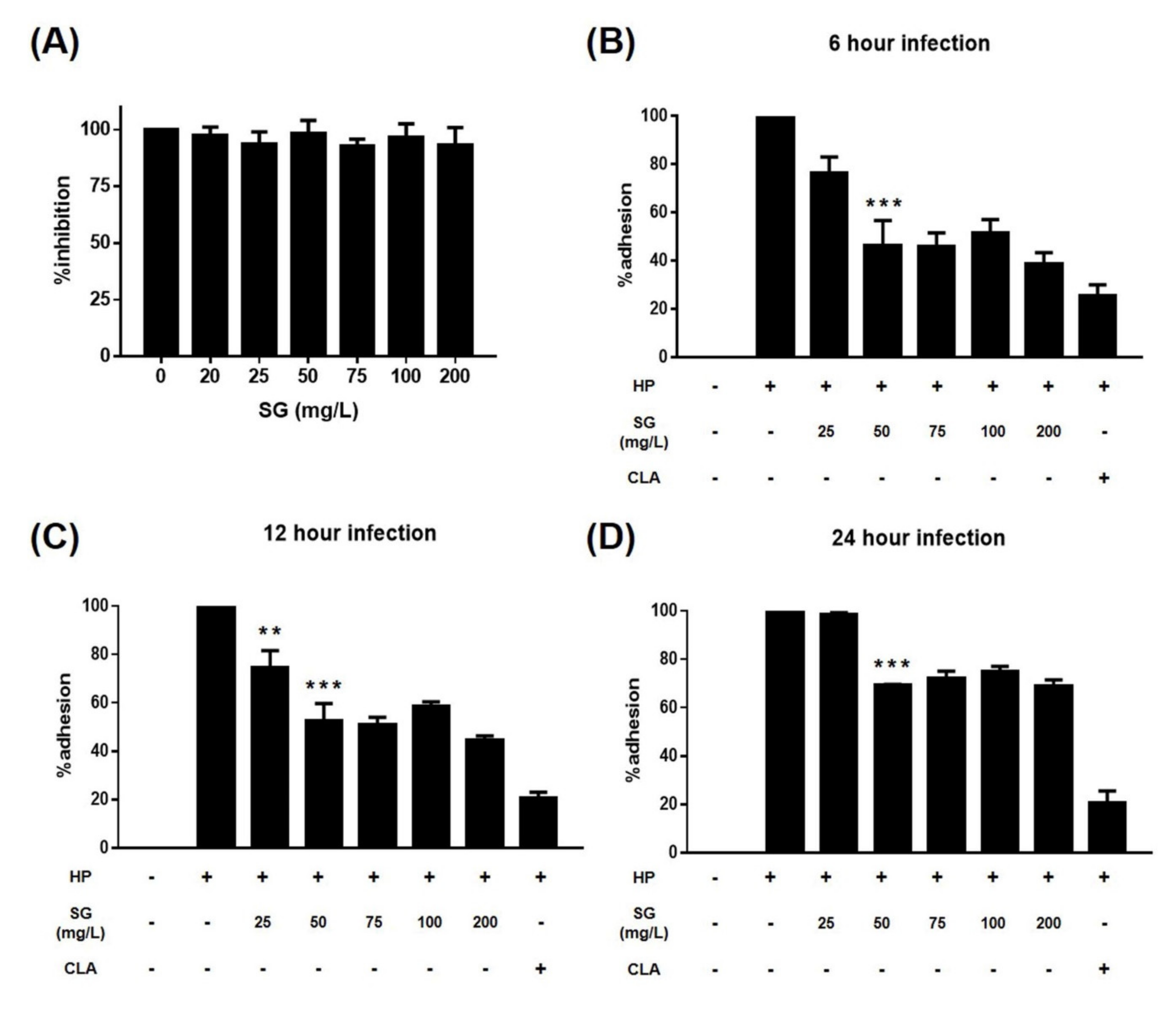{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
3. Discussion
4. Methods
4.1. Materials
4.2. Bacterial and Mammalian Cell Culture
4.3. Broth Dilution Method
4.4. Urease Activity Test
4.5. Adhesion Test
4.6. Western Blotting
4.7. Enzyme-Linked Immunosorbent Assay (ELISA)
4.8. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Schistosomes, Liver Flukes and Helicobacter pylori. IARC Monogr. Eval. Carcinog. Risks Hum. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Available online: https://www.ncbi.nlm.nih.gov/books/NBK487782/pdf/Bookshelf_NBK487782.pdf (accessed on 28 March 2011).
- El-Omar, E.M.; Oien, K.; El-Nujumi, A.; Gillen, D.; Wirz, A.; Dahill, S.; Williams, C.; Ardill, J.E.; McColl, K.E. Helicobacter pylori infection and chronic gastric acid hyposecretion. Gastroenterology 1997, 113, 15–24. [Google Scholar] [CrossRef]
- Hamlet, A.; Thoreson, A.C.; Nilsson, O.; Svennerholm, A.M.; Olbe, L. Duodenal Helicobacter pylori infection differs in cagA genotype between asymptomatic subjects and patients with duodenal ulcers. Gastroenterology 1999, 116, 259–268. [Google Scholar] [CrossRef]
- Amieva, M.; Peek, R.M., Jr. Pathobiology of Helicobacter pylori-Induced Gastric Cancer. Gastroenterology 2016, 150, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Suerbaum, S.; Michetti, P. Helicobacter pylori infection. N. Engl. J. Med. 2002, 347, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Uemura, N.; Okamoto, S.; Yamamoto, S.; Matsumura, N.; Yamaguchi, S.; Yamakido, M.; Taniyama, K.; Sasaki, N.; Schlemper, R.J. Helicobacter pylori infection and the development of gastric cancer. N. Engl. J. Med. 2001, 345, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Yim, J.Y.; Kim, N.; Choi, S.H.; Kim, Y.S.; Cho, K.R.; Kim, S.S.; Seo, G.S.; Kim, H.U.; Baik, G.H.; Sin, C.S.; et al. Seroprevalence of Helicobacter pylori in South Korea. Helicobacter 2007, 12, 333–340. [Google Scholar] [CrossRef]
- Slomiany, B.L.; Piotrowski, J.; Slomiany, A. Induction of caspase-3 and nitric oxide synthase-2 during gastric mucosal inflammatory reaction to Helicobacter pylori lipopolysaccharide. Biochem. Mol. Biol. Int. 1998, 46, 1063–1070. [Google Scholar] [CrossRef]
- Piotrowski, J. Lipopolysaccharide a virulence factor of Helicobacter pylori: Effect of antiulcer agents. J. Physiol. Pharmacol. 1998, 49, 3–24. [Google Scholar]
- Slomiany, B.L.; Piotrowski, J.; Slomiany, A. Anti-Helicobacter pylori activities of ebrotidine. A review of biochemical and animal experimental studies and data. Arzneimittelforschung 1997, 47, 475–482. [Google Scholar]
- Stein, M.; Bagnoli, F.; Halenbeck, R.; Rappuoli, R.; Fantl, W.J.; Covacci, A. c-Src/Lyn kinases activate Helicobacter pylori CagA through tyrosine phosphorylation of the EPIYA motifs. Mol. Microbiol. 2002, 43, 971–980. [Google Scholar] [CrossRef]
- Higashi, H.; Tsutsumi, R.; Muto, S.; Sugiyama, T.; Azuma, T.; Asaka, M.; Hatakeyama, M. SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science 2002, 295, 683–686. [Google Scholar] [CrossRef] [PubMed]
- Backert, S.; Ziska, E.; Brinkmann, V.; Zimny-Arndt, U.; Fauconnier, A.; Jungblut, P.R.; Naumann, M.; Meyer, T.F. Translocation of the Helicobacter pylori CagA protein in gastric epithelial cells by a type IV secretion apparatus. Cell. Microbiol. 2000, 2, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Saadat, I.; Higashi, H.; Obuse, C.; Umeda, M.; Murata-Kamiya, N.; Saito, Y.; Lu, H.; Ohnishi, N.; Azuma, T.; Suzuki, A.; et al. Helicobacter pylori CagA targets PAR1/MARK kinase to disrupt epithelial cell polarity. Nature 2007, 447, 330–333. [Google Scholar] [CrossRef]
- Hatakeyama, M. Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat. Rev. Cancer 2004, 4, 688–694. [Google Scholar] [CrossRef]
- Cover, T.L.; Krishna, U.S.; Israel, D.A.; Peek, R.M., Jr. Induction of gastric epithelial cell apoptosis by Helicobacter pylori vacuolating cytotoxin. Cancer Res. 2003, 63, 951–957. [Google Scholar] [PubMed]
- Kuck, D.; Kolmerer, B.; Iking-Konert, C.; Krammer, P.H.; Stremmel, W.; Rudi, J. Vacuolating cytotoxin of Helicobacter pylori induces apoptosis in the human gastric epithelial cell line AGS. Infect. Immun. 2001, 69, 5080–5087. [Google Scholar] [CrossRef] [PubMed]
- Galmiche, A.; Rassow, J.; Doye, A.; Cagnol, S.; Chambard, J.C.; Contamin, S.; de Thillot, V.; Just, I.; Ricci, V.; Solcia, E.; et al. The N-terminal 34 kDa fragment of Helicobacter pylori vacuolating cytotoxin targets mitochondria and induces cytochrome c release. EMBO J. 2000, 19, 6361–6370. [Google Scholar] [CrossRef]
- McClain, M.S.; Schraw, W.; Ricci, V.; Boquet, P.; Cover, T.L. Acid activation of Helicobacter pylori vacuolating cytotoxin (VacA) results in toxin internalization by eukaryotic cells. Mol. Microbiol. 2000, 37, 433–442. [Google Scholar] [CrossRef]
- Isomoto, H.; Matsushima, K.; Inoue, N.; Hayashi, T.; Nakayama, T.; Kunizaki, M.; Hidaka, S.; Nakayama, M.; Hisatsune, J.; Nakashima, M.; et al. Interweaving microRNAs and proinflammatory cytokines in gastric mucosa with reference to H. pylori infection. J. Clin. Immunol. 2012, 32, 290–299. [Google Scholar] [CrossRef]
- Fox, J.G.; Wang, T.C. Inflammation, atrophy, and gastric cancer. J. Clin. Investig. 2007, 117, 60–69. [Google Scholar] [CrossRef]
- Slomiany, B.L.; Piotrowski, J.; Czajkowski, A.; Murty, V.L.; Slomiany, A. Characterization of gastric mucosal mucin receptor. Biochem. Mol. Biol. Int. 1993, 31, 745–753. [Google Scholar]
- Lord, D.A.; McLaren, J.W.; Wheeler, R.C. Determination of trace metals in fresh water mussels by atomic absorption spectrometry with direct solid sample injection. Anal. Chem. 1977, 49, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Piotrowski, J.; Majka, J.; Murty, V.L.; Czajkowski, A.; Slomiany, A.; Slomiany, B.L. Inhibition of gastric mucosal mucin receptor by Helicobacter pylori lipopolysaccharide: Effect of sulglycotide. Gen. Pharmacol. 1994, 25, 969–976. [Google Scholar] [CrossRef]
- Slomiany, B.L.; Piotrowski, J.; Slomiany, A. Suppression of gastric mucosal inflammatory responses to Helicobacter pylori lipopolysaccharide by sulglycotide. Gen. Pharmacol. 1999, 32, 251–257. [Google Scholar] [CrossRef]
- Slomiany, B.L.; Piotrowski, J.; Slomiany, A. Gastric mucosal inflammatory responses to Helicobacter pylori lipopolysaccharide: Down-regulation of nitric oxide synthase-2 and caspase-3 by sulglycotide. Biochem. Biophys. Res. Commun. 1999, 261, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Camargo, M.C.; Piazuelo, M.B.; Mera, R.M.; Fontham, E.T.; Delgado, A.G.; Yepez, M.C.; Ceron, C.; Bravo, L.E.; Bravo, J.C.; Correa, P. Effect of smoking on failure of H. pylori therapy and gastric histology in a high gastric cancer risk area of Colombia. Acta Gastroenterol. Latinoam. 2007, 37, 238–245. [Google Scholar]
- Suzuki, T.; Matsuo, K.; Ito, H.; Sawaki, A.; Hirose, K.; Wakai, K.; Sato, S.; Nakamura, T.; Yamao, K.; Ueda, R.; et al. Smoking increases the treatment failure for Helicobacter pylori eradication. Am. J. Med. 2006, 119, 217–224. [Google Scholar] [CrossRef]
- Ghotaslou, R.; Leylabadlo, H.E.; Asl, Y.M. Prevalence of antibiotic resistance in Helicobacter pylori: A recent literature review. World J. Methodol. 2015, 5, 164–174. [Google Scholar] [CrossRef]
- De Francesco, V.; Giorgio, F.; Hassan, C.; Manes, G.; Vannella, L.; Panella, C.; Ierardi, E.; Zullo, A. Worldwide H. pylori antibiotic resistance: A systematic review. J. Gastrointest. Liver Dis. 2010, 19, 409–414. [Google Scholar]
- Bruchard, M.; Rebe, C.; Derangere, V.; Togbe, D.; Ryffel, B.; Boidot, R.; Humblin, E.; Hamman, A.; Chalmin, F.; Berger, H.; et al. The receptor NLRP3 is a transcriptional regulator of TH2 differentiation. Nat. Immunol. 2015, 16, 859–870. [Google Scholar] [CrossRef]
- Lu, H.; Ouyang, W.; Huang, C. Inflammation, a key event in cancer development. Mol. Cancer Res. 2006, 4, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Lamb, A.; Chen, L.F. Role of the Helicobacter pylori-induced inflammatory response in the development of gastric cancer. J. Cell. Biochem. 2013, 114, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.; Bhagat, G.; Cui, G.; Takaishi, S.; Kurt-Jones, E.A.; Rickman, B.; Betz, K.S.; Penz-Oesterreicher, M.; Bjorkdahl, O.; Fox, J.G.; et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell 2008, 14, 408–419. [Google Scholar] [CrossRef] [PubMed]
- El-Omar, E.M.; Carrington, M.; Chow, W.H.; McColl, K.E.; Bream, J.H.; Young, H.A.; Herrera, J.; Lissowska, J.; Yuan, C.C.; Rothman, N.; et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature 2000, 404, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Piotrowski, J.; Slomiany, A.; Slomiany, B.L. Suppression of Helicobacter pylori protease activity towards growth factors by sulglycotide. J. Physiol. Pharmacol. 1997, 48, 345–351. [Google Scholar] [PubMed]
- Cover, T.L.; Blaser, M.J. Helicobacter pylori in health and disease. Gastroenterology 2009, 136, 1863–1873. [Google Scholar] [CrossRef]
- Tharmalingam, N.; Kim, S.H.; Park, M.; Woo, H.J.; Kim, H.W.; Yang, J.Y.; Rhee, K.J.; Kim, J.B. Inhibitory effect of piperine on Helicobacter pylori growth and adhesion to gastric adenocarcinoma cells. Infect. Agents Cancer 2014, 9, 43. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.Y.; Kim, P.; Jeong, S.-H.; Lee, S.W.; Myung, Y.S.; Baeg, M.K.; Kim, J.-B. The Effects of Sulglycotide on the Adhesion and the Inflammation of Helicobacter Pylori. Int. J. Environ. Res. Public Health 2020, 17, 2918. https://doi.org/10.3390/ijerph17082918
Yang JY, Kim P, Jeong S-H, Lee SW, Myung YS, Baeg MK, Kim J-B. The Effects of Sulglycotide on the Adhesion and the Inflammation of Helicobacter Pylori. International Journal of Environmental Research and Public Health. 2020; 17(8):2918. https://doi.org/10.3390/ijerph17082918
Chicago/Turabian StyleYang, Ji Yeong, Pumsoo Kim, Seok-Hoo Jeong, Seong Woong Lee, Yu Sik Myung, Myong Ki Baeg, and Jong-Bae Kim. 2020. "The Effects of Sulglycotide on the Adhesion and the Inflammation of Helicobacter Pylori" International Journal of Environmental Research and Public Health 17, no. 8: 2918. https://doi.org/10.3390/ijerph17082918
APA StyleYang, J. Y., Kim, P., Jeong, S.-H., Lee, S. W., Myung, Y. S., Baeg, M. K., & Kim, J.-B. (2020). The Effects of Sulglycotide on the Adhesion and the Inflammation of Helicobacter Pylori. International Journal of Environmental Research and Public Health, 17(8), 2918. https://doi.org/10.3390/ijerph17082918