Rhizosphere Bacterial Community Structure and Predicted Functional Analysis in the Water-Level Fluctuation Zone of the Danjiangkou Reservoir in China During the Dry Period

 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Site, Sampling, and Soil Physicochemical Properties
2.2. DNA Extraction and Sequencing
2.3. Bioinformatics Analysis
2.4. Statistical Analyses
3. Results
3.1. Soil Physicochemical Properties in Rhizosphere of Plants in the WLFZ of the Danjiangkou Reservoir
3.2. Rhizosphere Bacterial Community Structure Analysis in Different Samples
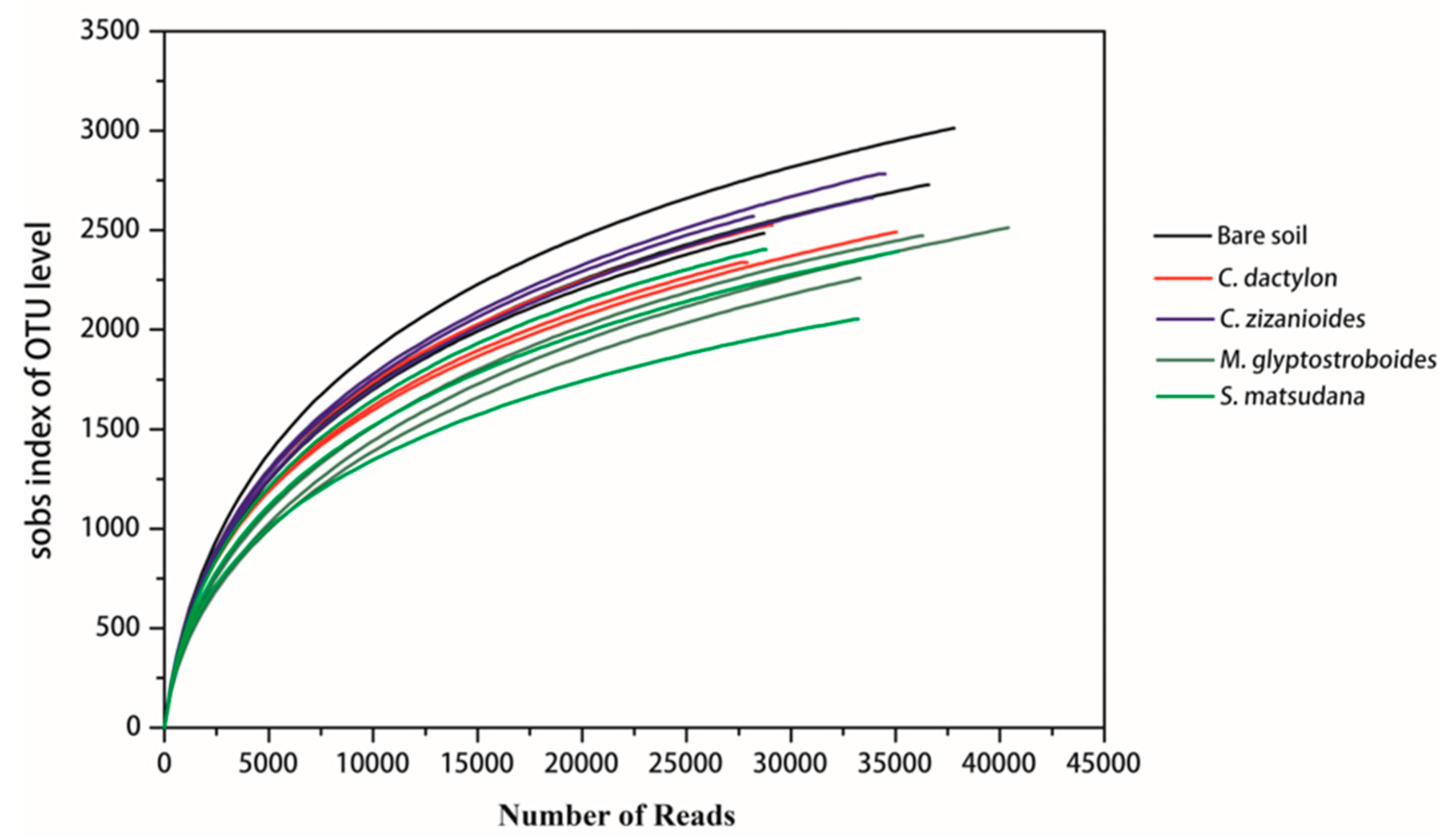
3.2.1. High-Throughput Sequencing Data and Diversity Assessment
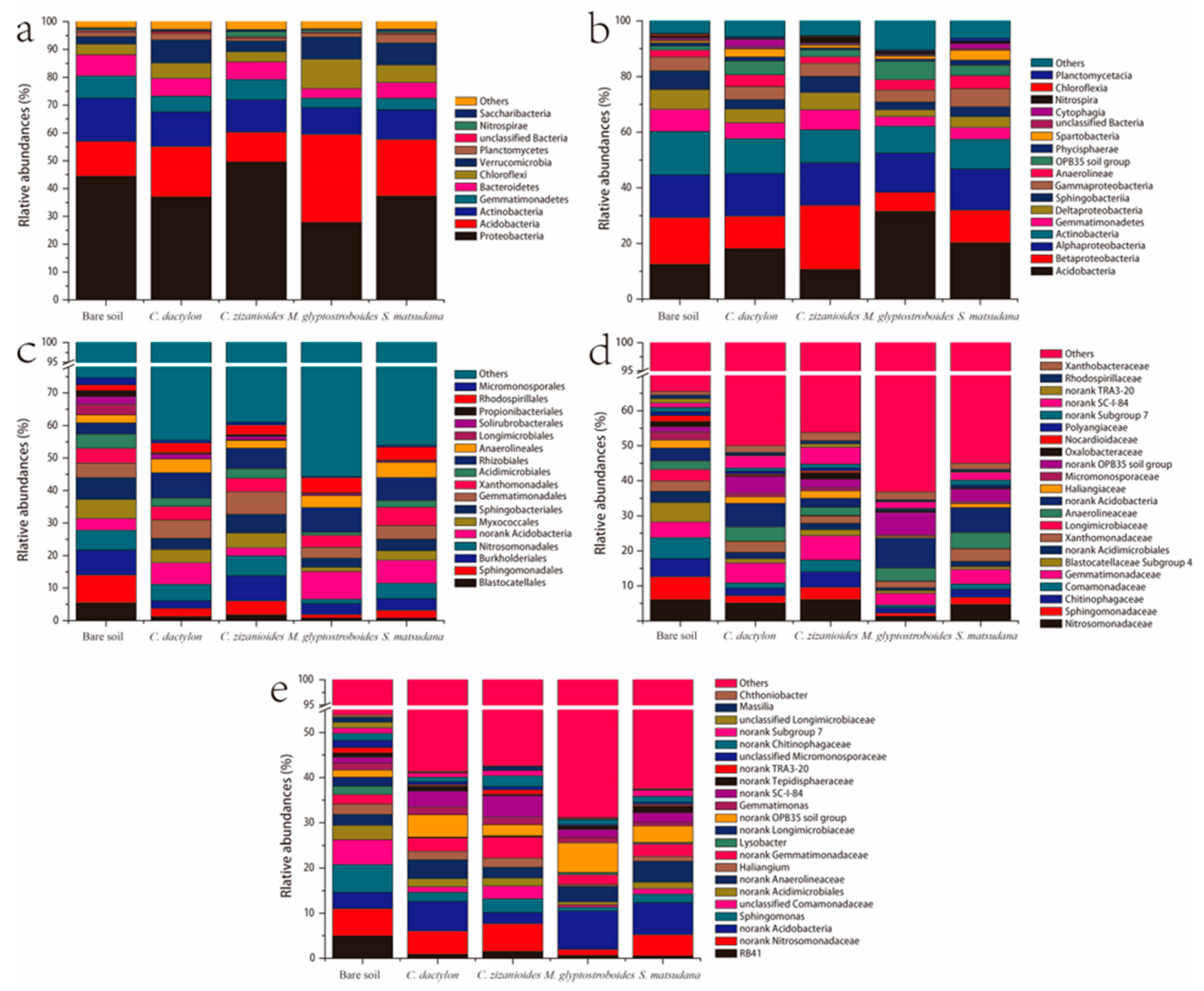
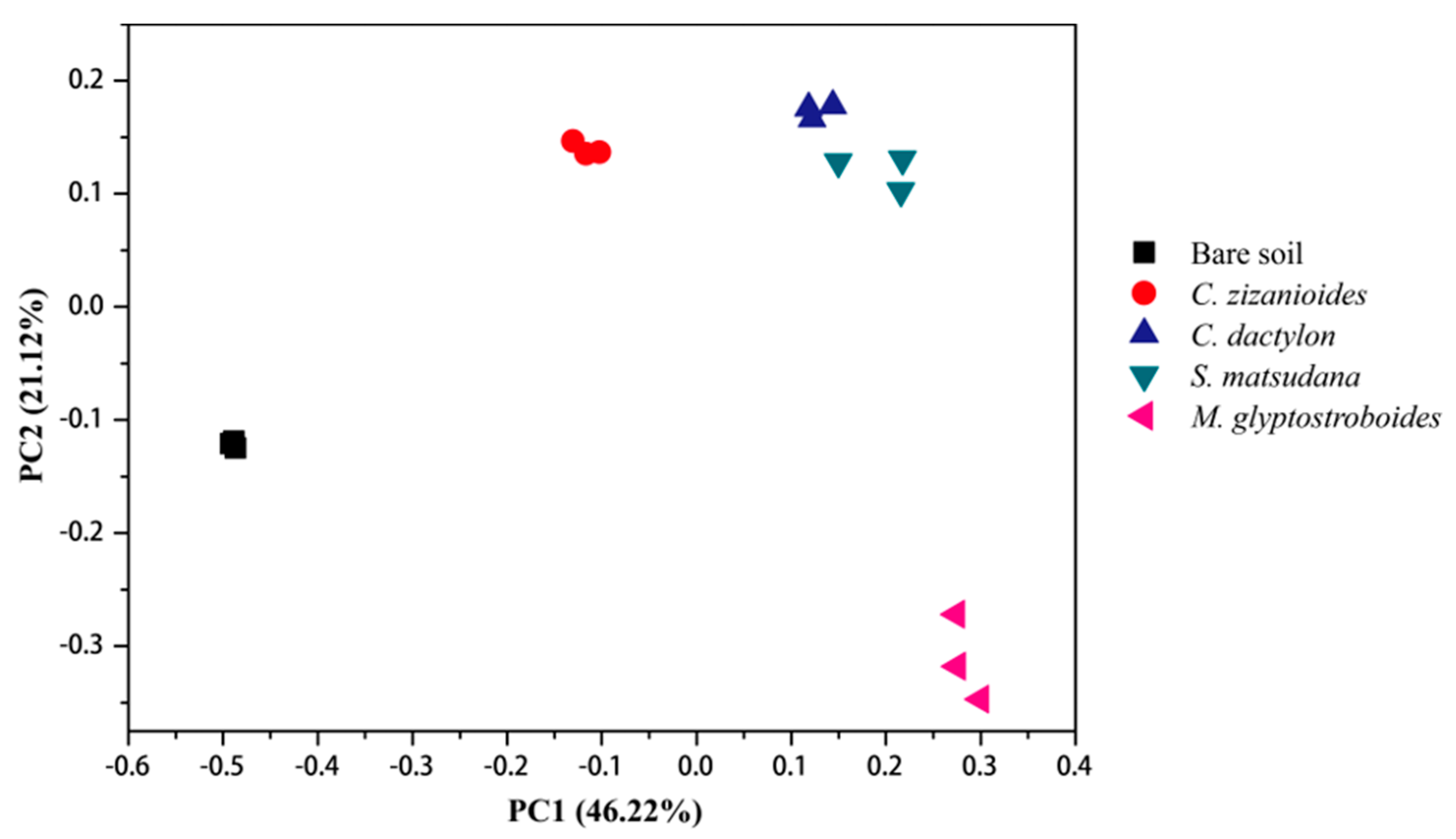
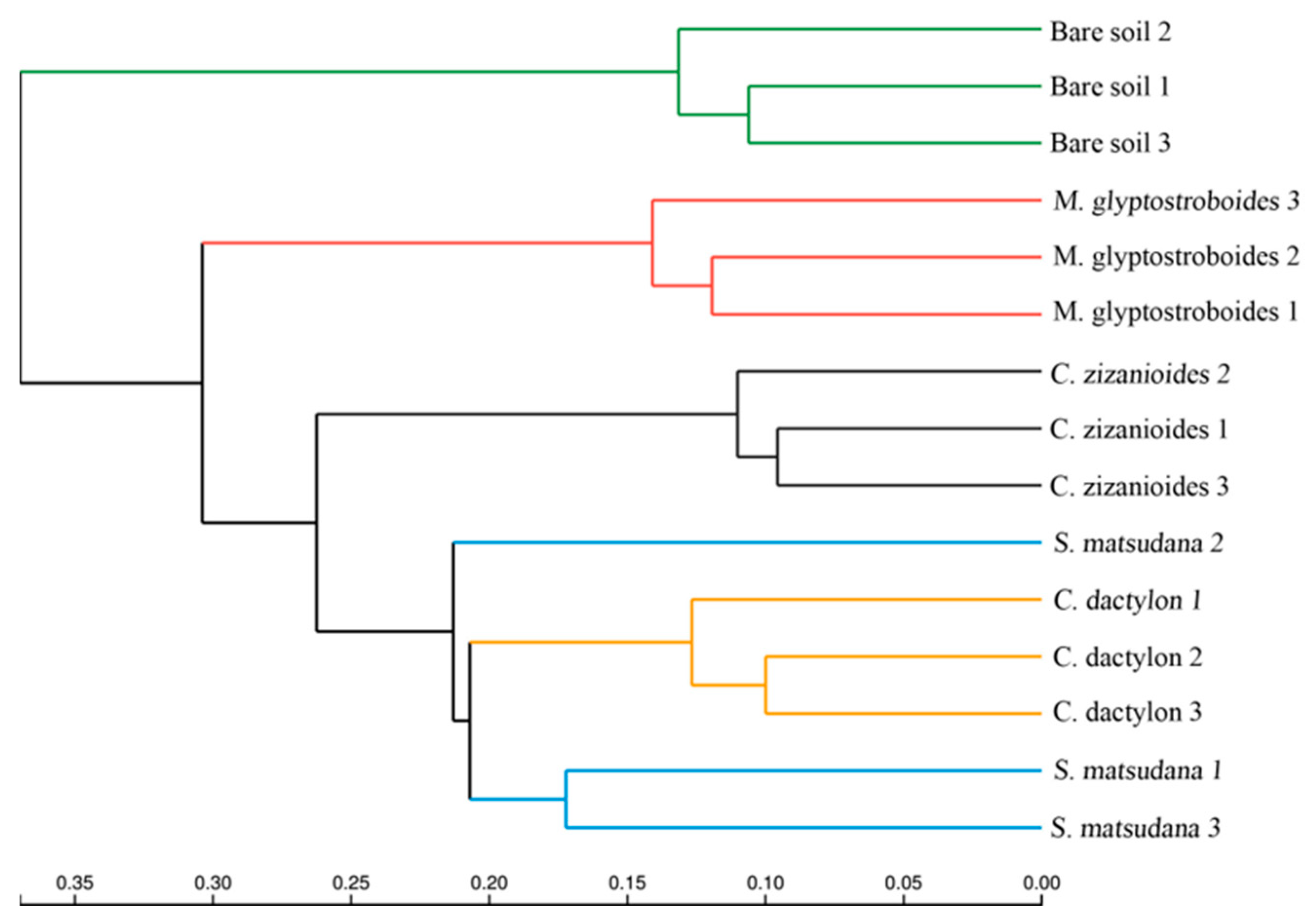
3.2.2. Bacterial Community Composition and Beta Diversity Analysis
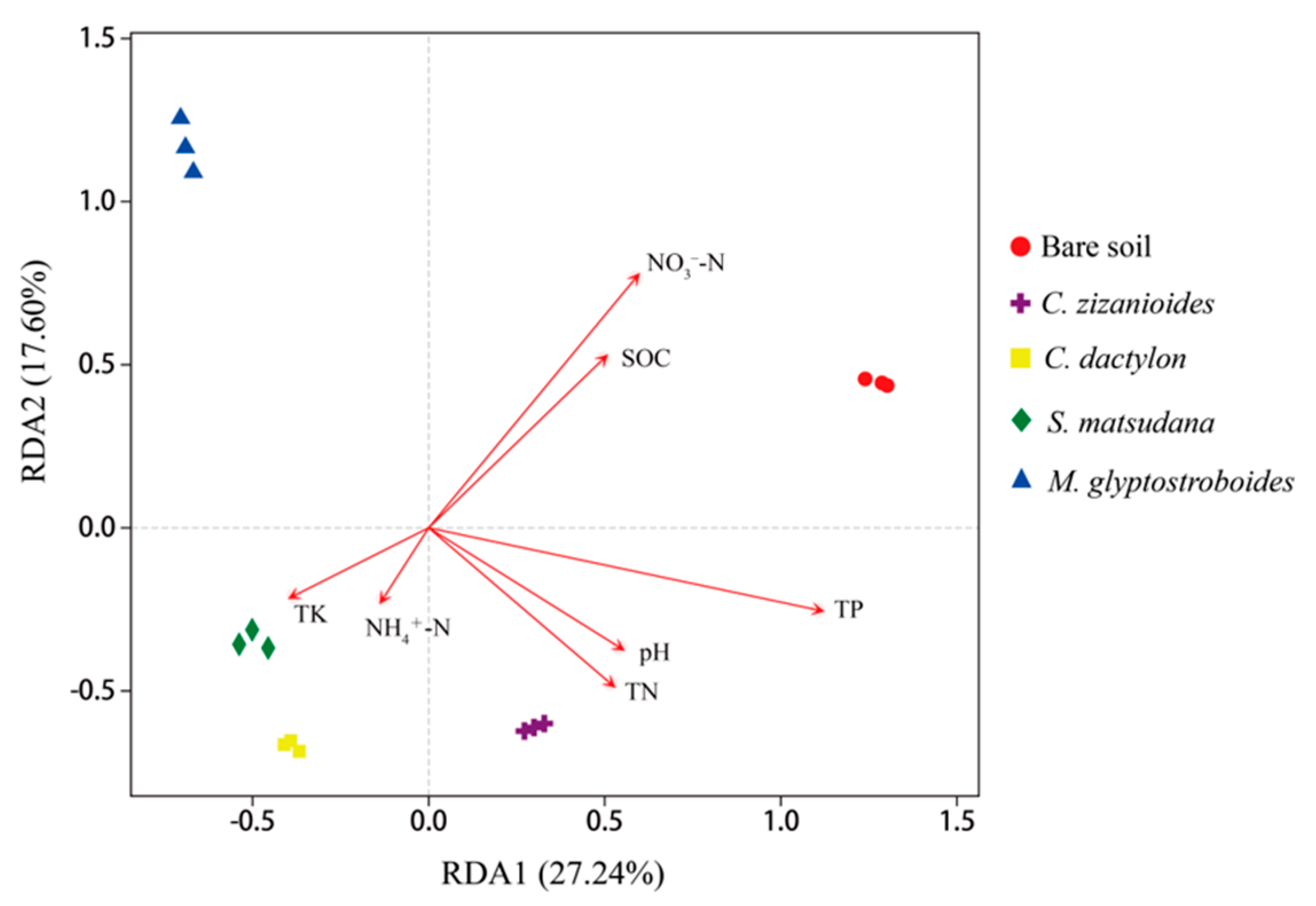
3.2.3. Correlation Analysis of Bacterial Community and Environmental Factors
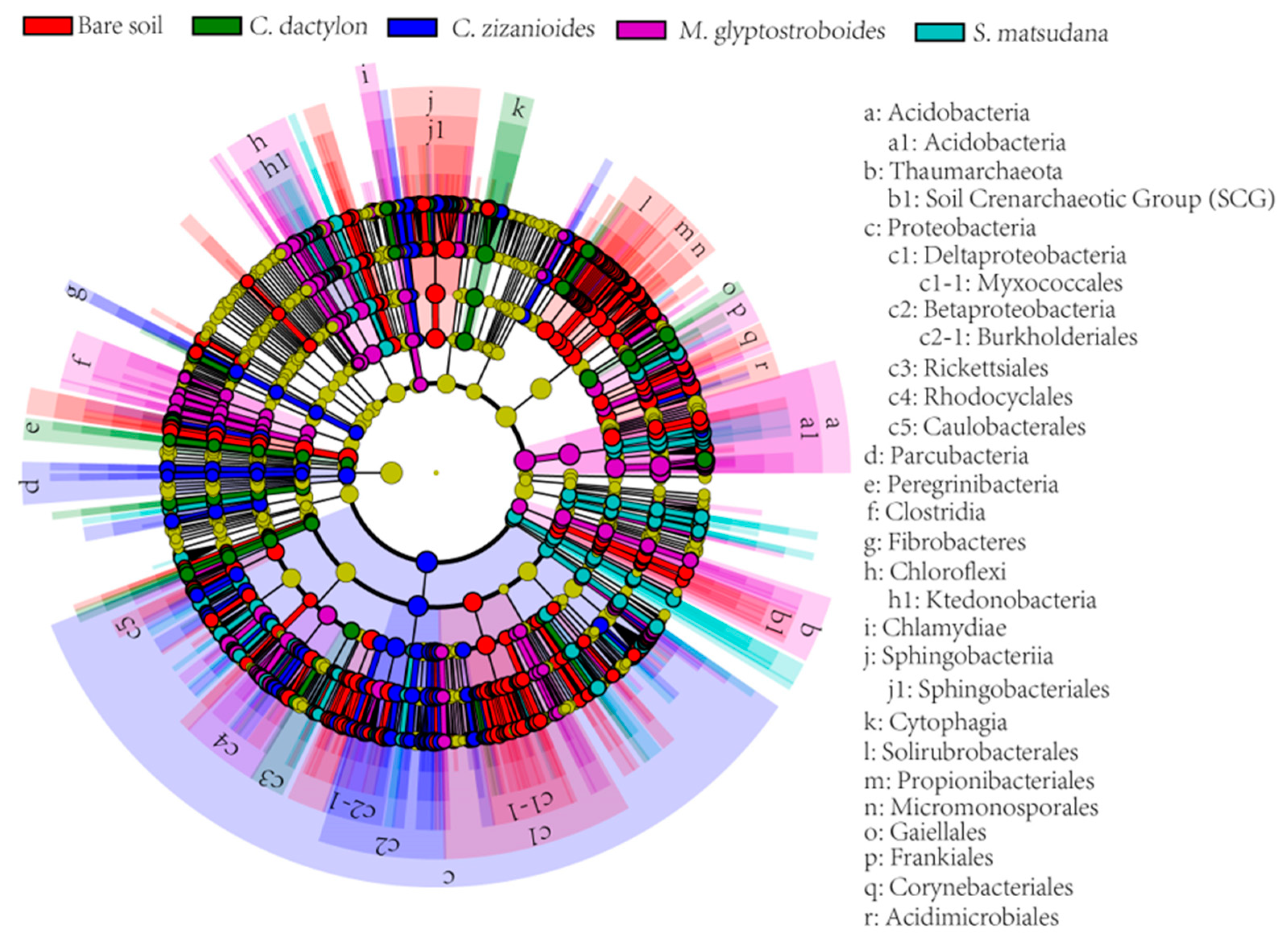
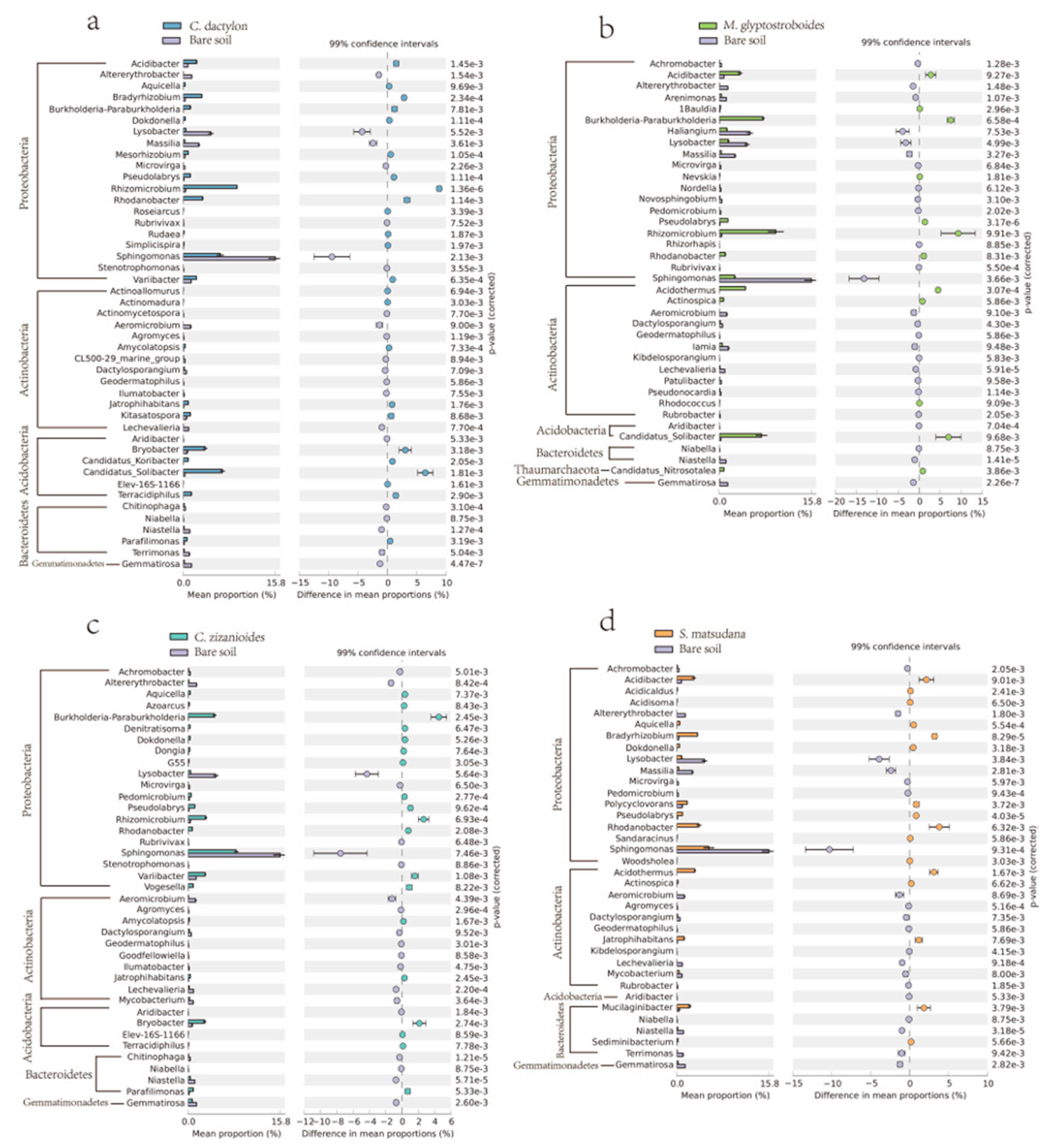
3.2.4. Analysis of Different Bacteria From Different Samples
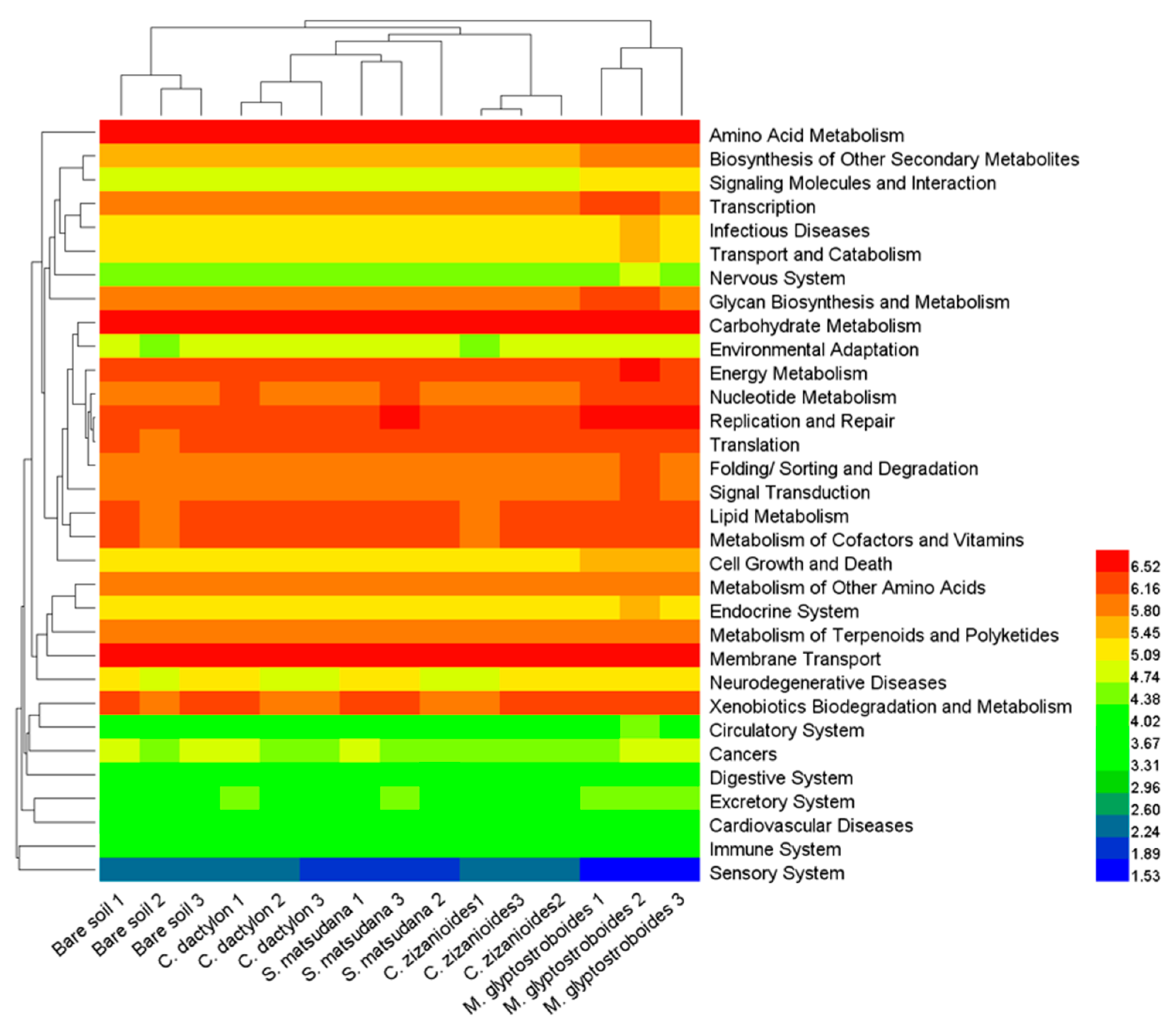
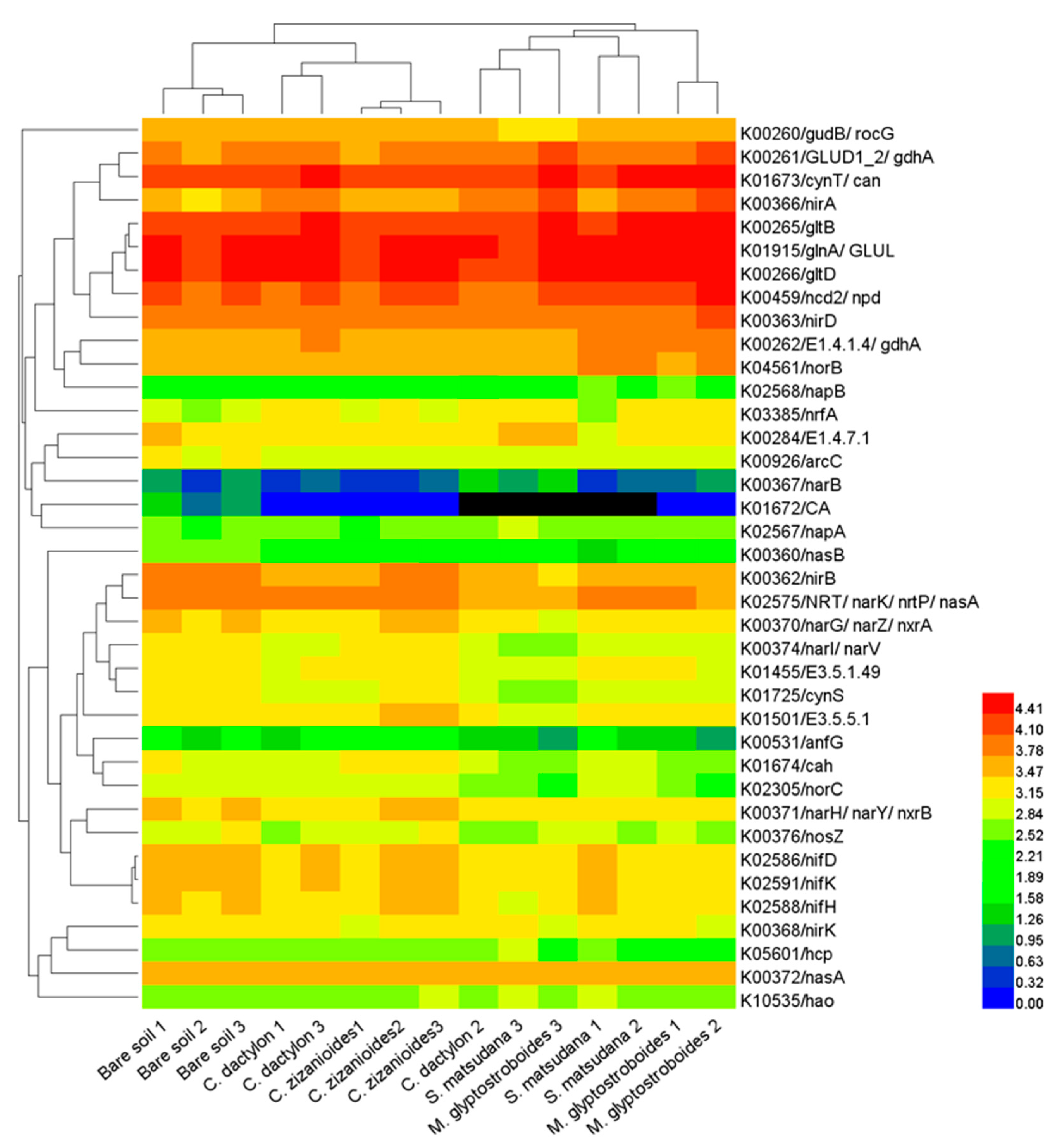
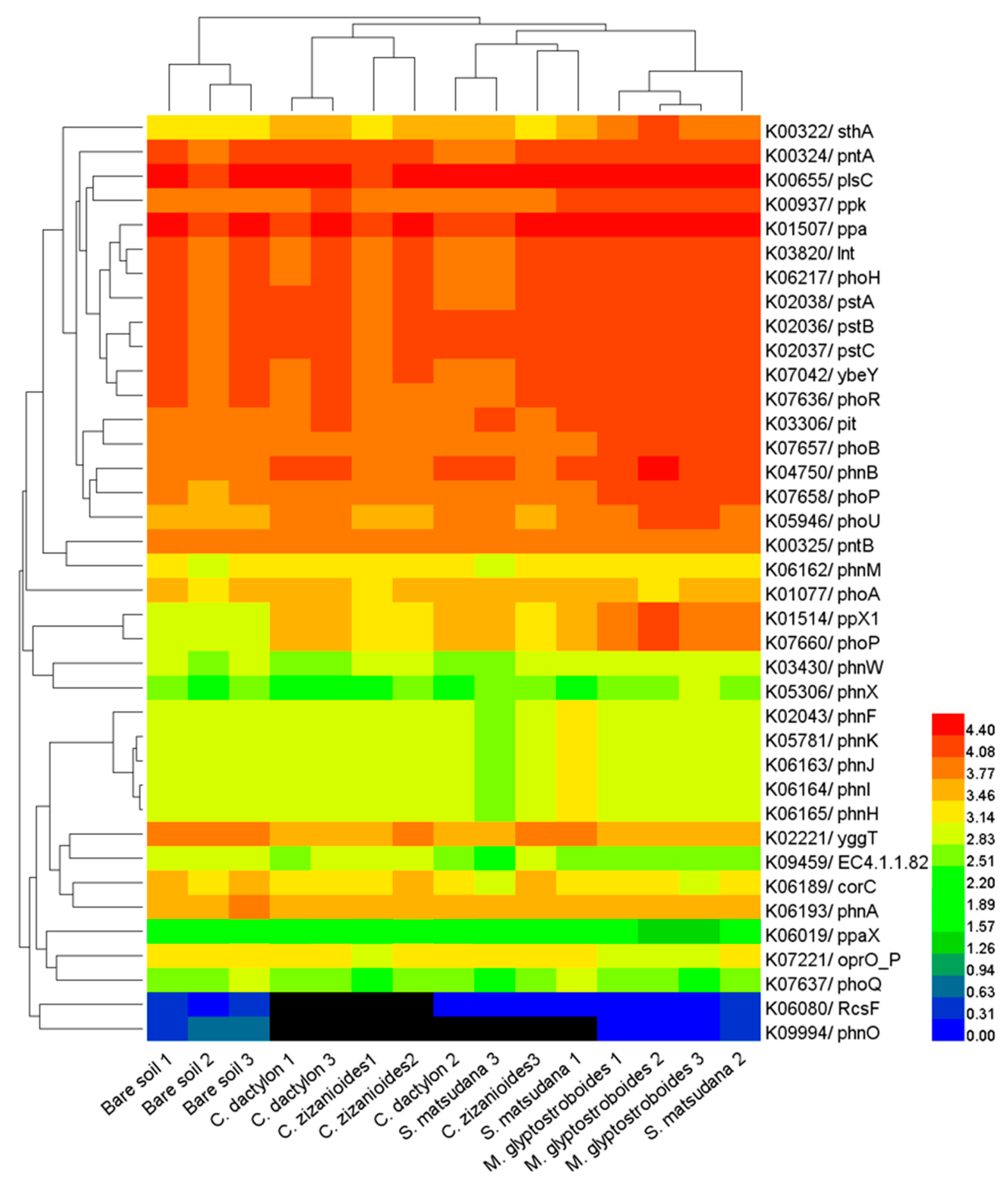
3.3. Functional Prediction by PICRUSt
4. Discussion
4.1. Bacterial Community Composition and Its Influence in the WLFZ of the Danjiangkou Reservoir
4.2. Soil Bacterial Function and Influencing Factors in the WLFZ of the Danjiangkou Reservoir
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- White, M.S.; Xenopoulos, M.A.; Hogsden, K.; Metcalfe, R.A.; Dillon, P.J. Natural lake level fluctuation and associated concordance with water quality and aquatic communities within small lakes of the Laurentian Great Lakes region. Hydrobiologia 2008, 613, 21–31. [Google Scholar] [CrossRef]
- Naiman, R.J.; Décamps, H. The Ecology of Interfaces: Riparian Zones. Annu. Rev. Ecol. Syst. 1997, 28, 621–658. [Google Scholar] [CrossRef]
- Yin, J.; Liu, H.; Chen, X. Dynamic Change in the Water-Level Fluctuation Zone of the Danjiangkou Reservoir and Its Influence on Water Quality. Sustainability 2018, 10, 1025. [Google Scholar] [CrossRef]
- Webber, M.; Crow-Miller, B.; Rogers, S. The South–North Water Transfer Project: Remaking the geography of China. Reg. Stud. 2017, 51, 370–382. [Google Scholar] [CrossRef]
- Gao, W.; Chen, Z.; Li, Y.; Pan, Y.; Zhu, J.; Guo, S.; Hu, L.; Huang, J. Bioassessment of a Drinking Water Reservoir Using Plankton: High Throughput Sequencing vs. Traditional Morphological Method. Water 2018, 10, 82. [Google Scholar] [CrossRef]
- Shu, X.; Zhang, K.; Zhang, Q.; Wang, W. Response of soil physico-chemical properties to restoration approaches and submergence in the water level fluctuation zone of the Danjiangkou Reservoir, China. Ecotoxicol. Environ. Saf. 2017, 145, 119–125. [Google Scholar] [CrossRef]
- Chen, Z.-J.; Xu, G.; Ding, C.-Y.; Zheng, B.-H.; Chen, Y.; Han, H.; Li, Y.-Y.; Shi, J.-W.; Hu, L.-Q. Illumina MiSeq sequencing and network analysis the distribution and co-occurrence of bacterioplankton in Danjiangkou Reservoir, China. Arch. Microbiol. 2020. [Google Scholar] [CrossRef]
- Chen, Z.; Yuan, J.; Sun, F.; Zhang, F.; Chen, Y.; Ding, C.; Shi, J.; Li, Y.; Yao, L. Planktonic fungal community structures and their relationship to water quality in the Danjiangkou Reservoir, China. Sci. Rep. 2018, 8, 10596. [Google Scholar] [CrossRef]
- Ye, C.; Cheng, X.; Liu, W.; Zhang, Q. Revegetation impacts soil nitrogen dynamics in the water level fluctuation zone of the Three Gorges Reservoir, China. Sci. Total. Environ 2015, 517, 76–85. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, P.; Ye, F.; Jiang, Y.; Song, L.; Op den Camp, H.J.M.; Zhu, G.; Wu, S. Nitrite-dependent anaerobic methane oxidizing bacteria along the water level fluctuation zone of the Three Gorges Reservoir. Appl. Microbiol. Biotechnol. 2016, 100, 1977–1986. [Google Scholar] [CrossRef]
- Peng, C.; Zhang, L.; Qin, H.; Li, D. Revegetation in the water level fluctuation zone of a reservoir: An ideal measure to reduce the input of nutrients and sediment. Ecol. Eng. 2014, 71, 574–577. [Google Scholar] [CrossRef]
- Kuypers, M.M.M.; Marchant, H.K.; Kartal, B. The microbial nitrogen-cycling network. Nat. Rev. Microbiol. 2018, 16, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Li, S.; Zhang, Y.; Tong, X.; Zhang, Q. Assessing heavy metal pollution in the water level fluctuation zone of China’s Three Gorges Reservoir using geochemical and soil microbial approaches. Environ. Monit. Assess. 2013, 185, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, E.L.; Auger, H.; Jaszczyszyn, Y.; Thermes, C. Ten years of next-generation sequencing technology. Trends Genet. 2014, 30, 418–426. [Google Scholar] [CrossRef]
- Chen, Z.; Zheng, Y.; Ding, C.; Ren, X.; Yuan, J.; Sun, F.; Li, Y. Integrated metagenomics and molecular ecological network analysis of bacterial community composition during the phytoremediation of cadmium-contaminated soils by bioenergy crops. Ecotoxicol. Environ. Saf. 2017, 145, 111–118. [Google Scholar] [CrossRef]
- Roesch, L.F.W.; Fulthorpe, R.R.; Riva, A.; Casella, G.; Hadwin, A.K.M.; Kent, A.D.; Daroub, S.H.; Camargo, F.A.O.; Farmerie, W.G.; Triplett, E.W. Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J. 2007, 1, 283–290. [Google Scholar] [CrossRef]
- Buée, M.; Reich, M.; Murat, C.; Morin, E.; Nilsson, R.H.; Uroz, S.; Martin, F. 454 Pyrosequencing analyses of forest soils reveal an unexpectedly high fungal diversity. New Phytol. 2009, 184, 449–456. [Google Scholar] [CrossRef]
- Smets, W.; Leff, J.W.; Bradford, M.A.; McCulley, R.L.; Lebeer, S.; Fierer, N. A method for simultaneous measurement of soil bacterial abundances and community composition via 16S rRNA gene sequencing. Soil Biol. Biochem. 2016, 96, 145–151. [Google Scholar] [CrossRef]
- Ye, F.; Wu, S.; Jiang, Y.; Op den Camp, H.J.M.; Li, Z.; Zhu, G.; Zheng, J.; Wang, Y. Shifts of archaeal community structure in soil along an elevation gradient in a reservoir water level fluctuation zone. J. Soils Sediments 2016, 16, 2728–2739. [Google Scholar] [CrossRef]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Hartman, W.H.; Ye, R.; Horwath, W.R.; Tringe, S.G. A genomic perspective on stoichiometric regulation of soil carbon cycling. ISME J. 2017, 11, 2652–2665. [Google Scholar] [CrossRef] [PubMed]
- LeBrun, E.; Kang, S. A comparison of computationally predicted functional metagenomes and microarray analysis for microbial P cycle genes in a unique basalt-soil forest. F1000Research 2018, 7, 179. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, H.; de Sousa, T.; Santos, J.P.; Sousa, A.G.G.; Teixeira, C.; Monteiro, M.R.; Salgado, P.; Mucha, A.P.; Almeida, C.M.R.; Torgo, L.; et al. Potential of dissimilatory nitrate reduction pathways in polycyclic aromatic hydrocarbon degradation. Chemosphere 2018, 199, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Pii, Y.; Borruso, L.; Brusetti, L.; Crecchio, C.; Cesco, S.; Mimmo, T. The interaction between iron nutrition, plant species and soil type shapes the rhizosphere microbiome. Plant Physiol. Biochem. 2016, 99, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Druzhinina, I.S.; Labbé, J.; Redman, R.; Qin, Y.; Rodriguez, R.; Zhang, C.; Tuskan, G.A.; Lin, F. Specialized Microbiome of a Halophyte and its Role in Helping Non-Host Plants to Withstand Salinity. Sci. Rep. 2016, 6, 32467. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Tao, Q.; Wu, K.; Li, J.; Qian, J.; Liang, Y.; Yang, X.; Li, T. Structural and functional variability in root-associated bacterial microbiomes of Cd/Zn hyperaccumulator Sedum alfredii. Appl. Microbiol. Biotechnol. 2017, 101, 7961–7976. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Song, M.; Yang, L.; Zhang, D.; Sun, Y.; Shen, Z.; Luo, C.; Zhang, G. Exploring the Influence of Environmental Factors on Bacterial Communities within the Rhizosphere of the Cu-tolerant plant, Elsholtzia splendens. Sci. Rep. 2016, 6, 36302. [Google Scholar] [CrossRef]
- Feng, M.-H.; Shan, X.-Q.; Zhang, S.; Wen, B. A comparison of the rhizosphere-based method with DTPA, EDTA, CaCl2, and NaNO3 extraction methods for prediction of bioavailability of metals in soil to barley. Environ. Pollut. 2005, 137, 231–240. [Google Scholar] [CrossRef]
- Caporaso, J.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.; Costello, E. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a Chimera-Checked 16S rRNA Gene Database and Workbench Compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Wang, Y.; Liu, Z.; Cheng, H.; Xue, Y. HemI: A Toolkit for Illustrating Heatmaps. PLoS ONE 2014, 9, e111988. [Google Scholar] [CrossRef]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [PubMed]
- Bais, H.P.; Weir, T.L.; Perry, L.G.; Gilroy, S.; Vivanco, J.M. The role of root exudates in rhizosphere interactions with plants and other organisms. Annu. Rev. Plant Biol. 2006, 57, 233–266. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Cañizares, C.; Jorrín, B.; Poole, P.S.; Tkacz, A. Understanding the holobiont: The interdependence of plants and their microbiome. Curr. Opin. Microbiol. 2017, 38, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Widder, S.; Allen, R.J.; Pfeiffer, T.; Curtis, T.P.; Wiuf, C.; Sloan, W.T.; Cordero, O.X.; Brown, S.P.; Momeni, B.; Shou, W.; et al. Challenges in microbial ecology: Building predictive understanding of community function and dynamics. ISME J. 2016, 10, 2557–2568. [Google Scholar] [CrossRef] [PubMed]
- Franzosa, E.A.; Hsu, T.; Sirota-Madi, A.; Shafquat, A.; Abu-Ali, G.; Morgan, X.C.; Huttenhower, C. Sequencing and beyond: Integrating molecular ‘omics’ for microbial community profiling. Nat. Rev. Microbiol. 2015, 13, 360–372. [Google Scholar] [CrossRef]
- Sun, D.; Meng, J.; Xu, E.G.; Chen, W. Microbial community structure and predicted bacterial metabolic functions in biochar pellets aged in soil after 34 months. Appl. Soil Ecol. 2016, 100, 135–143. [Google Scholar] [CrossRef]
- Chen, L.; Luo, Y.; Xu, J.; Yu, Z.; Zhang, K.; Brookes, P.C. Assessment of Bacterial Communities and Predictive Functional Profiling in Soils Subjected to Short-Term Fumigation-Incubation. Microb. Ecol. 2016, 72, 240–251. [Google Scholar] [CrossRef]
- Mendes, L.W.; Kuramae, E.E.; Navarrete, A.A.; van Veen, J.A.; Tsai, S.M. Taxonomical and functional microbial community selection in soybean rhizosphere. ISME J. 2014, 8, 1577–1587. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | pH | TN (g/kg) | TP (g/kg) | TK (g/kg) | NO3−–N (mg/kg) | NH4+–N (mg/kg) | SOC (g/kg) |
|---|---|---|---|---|---|---|---|
| Bare soil | 8.61 ± 0.20d | 1.34 ± 0.07d | 0.79 ± 0.03c | 4.95 ± 0.22bc | 123.80 ± 4.00c | 79.35 ± 1.96b | 25.60 ± 2.24a |
| C. dactylon | 8.16 ± 0.24c | 0.90 ± 0.04b | 0.82 ± 0.02c | 4.51 ± 0.24a | 133.26 ± 4.40c | 77.93 ± 1.99b | 32.77 ± 2.63b |
| C. zizanioides | 8.37 ± 0.17cd | 0.36 ± 0.02a | 0.71 ± 0.02b | 4.47 ± 0.24a | 56.39 ± 3.19a | 71.60 ± 2.02a | 30.29 ± 2.88b |
| M. glyptostroboides | 7.15 ± 0.14a | 1.09 ± 0.09c | 0.66 ± 0.03ab | 5.15 ± 0.16c | 94.00 ± 3.87b | 92.61 ± 3.48c | 30.92 ± 2.07b |
| S. matsudana | 7.74 ± 0.17b | 0.44 ± 0.01a | 0.65 ± 0.03a | 4.75 ± 0.13ab | 128.70 ± 8.23c | 75.55 ± 4.19ab | 30.77 ± 1.16b |
| Samples | Reads | sobs | Shannon indices | Simpson indices | ACE indices | Chao 1 indices | Coverage (%) |
|---|---|---|---|---|---|---|---|
| Bare soil | 34,367 ± 4943 | 2741 ± 264b | 6.48 ± 0.13bc | 0.0058 ± 0.0011ab | 3489 ± 258a | 3494 ± 259b | 97.64 ± 0.31 |
| C. dactylon | 30,682 ± 3827 | 2451 ± 98ab | 6.46 ± 0.06bc | 0.0047 ± 0.0005a | 3235 ± 108ab | 3246 ± 133ab | 97.48 ± 0.27 |
| C. zizanioides | 32,300 ± 3305 | 2672 ± 107b | 6.52 ± 0.04c | 0.0047 ± 0.0003a | 3525 ± 138a | 3539 ± 236b | 97.37 ± 0.22 |
| M. glyptostroboides | 36,564 ± 3704 | 2414 ± 137ab | 6.10 ± 0.14a | 0.0065 ± 0.0015b | 3274 ± 120ab | 3281 ± 149ab | 97.79 ± 0.19 |
| S. matsudana | 32,369 ± 3233 | 2283 ± 199a | 6.30 ± 0.14ab | 0.0054 ± 0.0005ab | 3005 ± 242a | 3005 ± 294a | 97.80 ± 0.35 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Z.-J.; Shao, Y.; Li, Y.-J.; Lin, L.-A.; Chen, Y.; Tian, W.; Li, B.-L.; Li, Y.-Y. Rhizosphere Bacterial Community Structure and Predicted Functional Analysis in the Water-Level Fluctuation Zone of the Danjiangkou Reservoir in China During the Dry Period. Int. J. Environ. Res. Public Health 2020, 17, 1266. https://doi.org/10.3390/ijerph17041266
Chen Z-J, Shao Y, Li Y-J, Lin L-A, Chen Y, Tian W, Li B-L, Li Y-Y. Rhizosphere Bacterial Community Structure and Predicted Functional Analysis in the Water-Level Fluctuation Zone of the Danjiangkou Reservoir in China During the Dry Period. International Journal of Environmental Research and Public Health. 2020; 17(4):1266. https://doi.org/10.3390/ijerph17041266
Chicago/Turabian StyleChen, Zhao-Jin, Yang Shao, Ying-Jun Li, Li-An Lin, Yan Chen, Wei Tian, Bai-Lian Li, and Yu-Ying Li. 2020. "Rhizosphere Bacterial Community Structure and Predicted Functional Analysis in the Water-Level Fluctuation Zone of the Danjiangkou Reservoir in China During the Dry Period" International Journal of Environmental Research and Public Health 17, no. 4: 1266. https://doi.org/10.3390/ijerph17041266
APA StyleChen, Z.-J., Shao, Y., Li, Y.-J., Lin, L.-A., Chen, Y., Tian, W., Li, B.-L., & Li, Y.-Y. (2020). Rhizosphere Bacterial Community Structure and Predicted Functional Analysis in the Water-Level Fluctuation Zone of the Danjiangkou Reservoir in China During the Dry Period. International Journal of Environmental Research and Public Health, 17(4), 1266. https://doi.org/10.3390/ijerph17041266