Look Alike, Sound Alike: Phenocopies in Steroid-Resistant Nephrotic Syndrome

 , , , and
, , , and
Abstract
1. Introduction
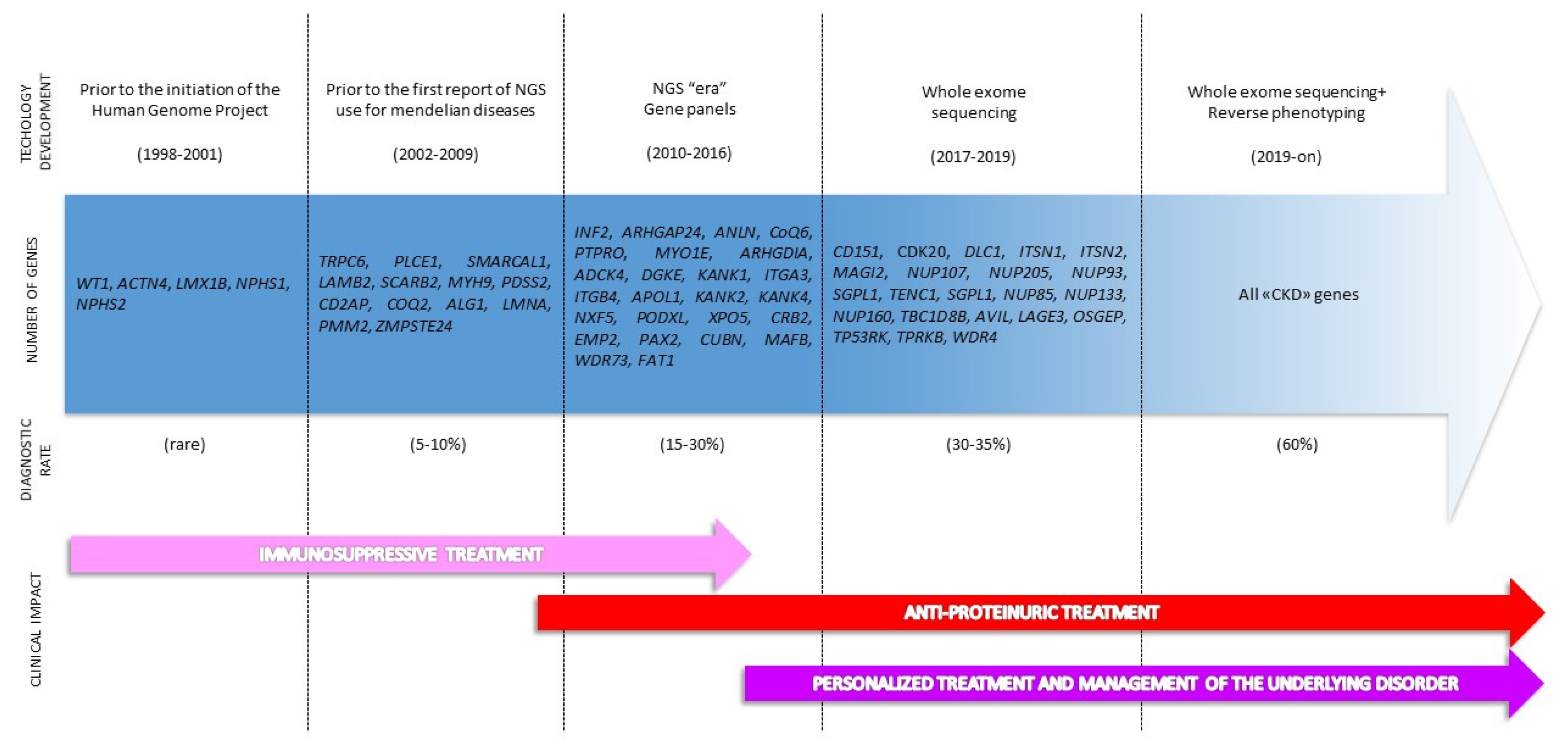
2. The Genetic Revolution: From Single Gene Analysis to Extended Genotyping
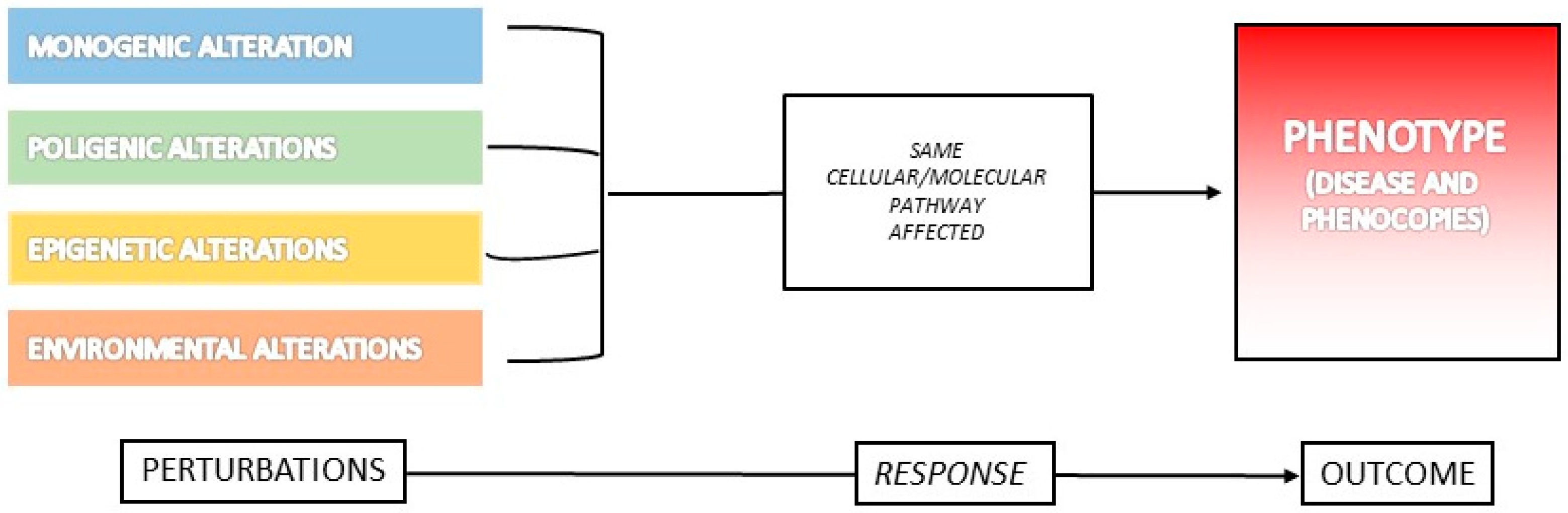
3. What Is a Phenocopy?
Examples of Phenocopies in Human Diseases
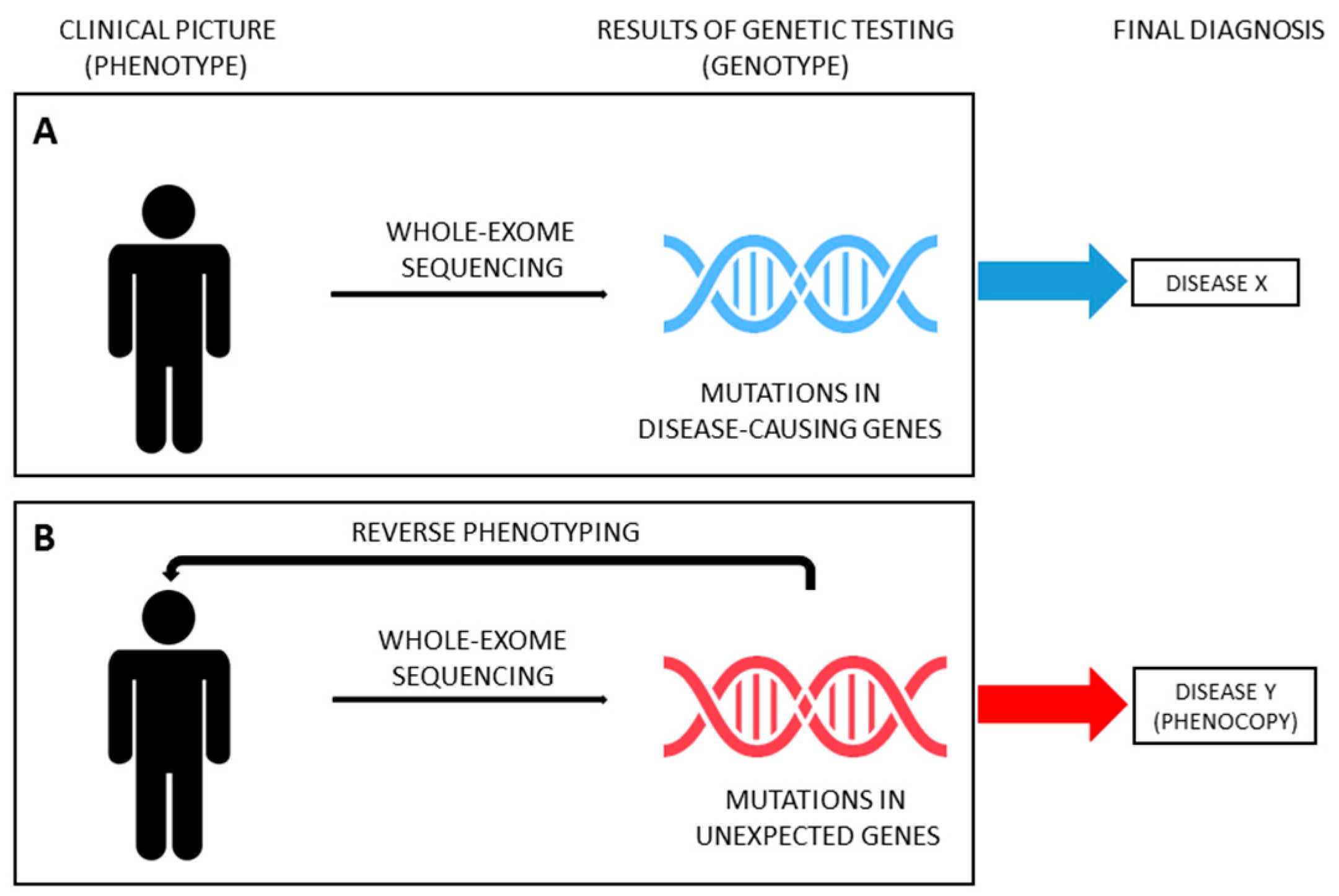
4. From Phenotype to Genotype and Backwards in SRNS
5. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Report, North American Pediatric Renal Trials and Collaborative Studies: NAPRTCS Annual Transplant. Available online: http://we.emmes.com/study/ped/annlrept/annualrept2014.pdf (accessed on 1 September 2020).
- Becherucci, F.; Roperto, R.M.; Materassi, M.; Romagnani, P. Chronic kidney disease in children. Clin. Kidney J. 2016, 9, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Warejko, J.K.; Tan, W.; Daga, A.; Schapiro, D.; Lawson, J.A.; Shril, S.; Lovric, S.; Ashraf, S.; Rao, J.; Hermle, T.; et al. Whole Exome Sequencing of Patients with Steroid-Resistant Nephrotic Syndrome. Clin. J. Am. Soc. Nephrol. 2018, 13, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Landini, S.; Mazzinghi, B.; Becherucci, F.; Allinovi, M.; Provenzano, A.; Palazzo, V.; Ravaglia, F.; Artuso, R.; Bosi, E.; Stagi, S.; et al. Reverse Phenotyping after Whole-Exome Sequencing in Steroid-Resistant Nephrotic Syndrome. Clin. J. Am. Soc. Nephrol. 2020, 15, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef]
- Frank, M.; Prenzler, A.; Eils, R.; von der Schulenburg, J.M. Genome sequencing: A systematic review of health economic evidence. Health Econ. Rev. 2013, 3, 29. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.T.; Campeau, P.M.; Lee, B.H. Genotype-phenotype correlation—Promiscuity in the era of next-generation sequencing. N. Engl. J. Med. 2014, 371, 593–596. [Google Scholar] [CrossRef]
- Boycott, K.M.; Vanstone, M.R.; Bulman, D.E.; MacKenzie, A.E. Rare-disease genetics in the era of next-generation sequencing: Discovery to translation. Nat. Rev. Genet. 2013, 14, 681–691. [Google Scholar] [CrossRef]
- Posey, J.E.; Genomics, C.F.M.; Odonnell-Luria, A.H.; Chong, J.X.; Harel, T.; Jhangiani, S.N.; Akdemir, Z.H.C.; Buyske, S.; Pehlivan, D.; Carvalho, C.M.B.; et al. Insights into genetics, human biology and disease gleaned from family based genomic studies. Genet. Med. 2019, 21, 798–812. [Google Scholar] [CrossRef]
- Córdoba, M.; Rodriguez-Quiroga, S.A.; Vega, P.A.; Salinas, V.; Perez-Maturo, J.; Amartino, H.; Vásquez-Dusefante, C.; Medina, N.; González-Morón, D.; Kauffman, M.A. Whole exome sequencing in neurogenetic odysseys: An effective, cost- and time-saving diagnostic approach. PLoS ONE 2018, 13, e0191228. [Google Scholar] [CrossRef]
- Baker, S.W.; Murrell, J.R.; Nesbitt, A.I.; Pechter, K.B.; Balciuniene, J.; Zhao, X.; Yu, Z.; Denenberg, E.H.; DeChene, E.T.; Wilkens, A.; et al. Automated Clinical Exome Reanalysis Reveals Novel Diagnoses. J. Mol. Diagn. 2019, 21, 38–48. [Google Scholar] [CrossRef]
- Groopman, E.E.; Rasouly, H.M.; Gharavi, A.G. Genomic medicine for kidney diseases. Nat. Rev. Nephrol. 2018, 14, 83–104. [Google Scholar] [CrossRef]
- Groopman, E.E.; Povysil, G.; Goldstein, D.B.; Gharavi, A.G. Rare genetic causes of complex kidney and urological diseases. Nat. Rev. Nephrol. 2020. [Google Scholar] [CrossRef]
- Chong, J.X.; Buckingham, K.J.; Jhangiani, S.N.; Boehm, C.D.; Sobreira, N.; Smith, J.D.; Harrell, T.M.; McMillin, M.J.; Wiszniewski, W.; Gambin, T.; et al. The Genetic Basis of Mendelian Phenotypes: Discoveries, Challenges, and Opportunities. Am. J. Hum. Genet. 2015, 97, 199–215. [Google Scholar] [CrossRef] [PubMed]
- Jha, V.; Garcia-Garcia, G.; Iseki, K.; Li, Z.; Naicker, S.; Plattner, B.; Saran, R.; Wang, A.Y.-M.; Yang, C.-W. Chronic kidney disease: Global dimension and perspectives. Lancet 2013, 82, 260–272. [Google Scholar] [CrossRef]
- Wühl, E.; Van Stralen, K.J.; Wanner, C.; Ariceta, G.; Heaf, J.G.; Bjerre, A.K.; Palsson, R.; Duneau, G.; Hoitsma, A.J.; Ravani, P.; et al. Renal replacement therapy for rare diseases affecting the kidney: An analysis of the ERA-EDTA Registry. Nephrol. Dial. Transplant. 2014, 29 (Suppl. 4), v1–v8. [Google Scholar] [CrossRef] [PubMed]
- Vivante, A.; Hildebrandt, F. Exploring the genetic basis of early-onset chronic kidney disease. Nat. Rev. Nephrol. 2016, 12, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Ingelfinger, J.R.; Kalantar-Zadeh, K.; Schaefer, F. World Kidney Day Steering Committee. Averting the legacy of kidney disease-focus on childhood. Pediatr. Nephrol. 2016, 31, 343–348. [Google Scholar] [CrossRef]
- Kestilä, M.; Lenkkeri, U.; Männikkö, M.; Lamerdin, J.; McCready, P.; Putaala, H.; Ruotsalainen, V.; Morita, T.; Nissinen, M.; Herva, R.; et al. Positionally cloned gene for a novel glomerular protein— nephrin—is mutated in congenital nephrotic syndrome. Mol. Cell 1998, 1, 575–582. [Google Scholar] [CrossRef]
- Lovric, S.; Ashraf, S.; Tan, W.; Hildebrandt, F. Genetic testing in steroid-resistant nephrotic syndrome: When and how? Nephrol. Dial. Transpl. 2016, 31, 1802–1813. [Google Scholar] [CrossRef]
- Online Mendelian Inheritance in Man Database (OMIM). Available online: https://www.ncbi.nlm.nih.gov/omim (accessed on 1 September 2020).
- Kopp, J.B.; Anders, H.-J.; Susztak, K.; Podestà, M.A.; Remuzzi, G.; Hildebrandt, F.; Romagnani, P. Podocytopathies. Nat. Rev. Dis. Primers 2020, 6, 68. [Google Scholar] [CrossRef]
- Lipska-Ziętkiewicz, B.S.; Iatropoulos, P.; Maranta, R.; Caridi, G.; Ozaltin, F.; Anarat, A.; Balat, A.; Gellermann, J.; Trautmann, A.; Erdogan, O.; et al. Genetic screening in adolescents with steroid-resistant nephrotic syndrome. Kidney Int. 2013, 84, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Trautmann, A.; Bodria, M.; Ozaltin, F.; Gheisari, A.; Melk, A.; Azocar, M.; Anarat, A.; Caliskan, S.; Emma, F.; Gellermann, J.; et al. Spectrum of steroid-resistant and congenital nephrotic syndrome in children: The PodoNet registry cohort. Clin. J. Am. Soc. Nephrol. 2015, 10, 592–600. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, H.J.; Bierzynska, A.; Wherlock, M.; Ognjanovic, M.; Kerecuk, L.; Hegde, S.; Feather, S.; Gilbert, R.D.; Krischock, L.; Jones, C.; et al. Simultaneous sequencing of 24 genes associated with steroid-resistant nephrotic syndrome. Clin. J. Am. Soc. Nephrol. 2013, 8, 637–648. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sadowski, C.E.; Lovric, S.; Ashraf, S.; Pabst, W.L.; Gee, H.Y.; Kohl, S.; Engelmann, S.; Vega-Warner, V.; Fang, H.; Halbritter, J.; et al. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J. Am. Soc. Nephrol. 2015, 26, 1279–1289. [Google Scholar] [CrossRef] [PubMed]
- Santín, S.; Bullich, G.; Tazón-Vega, B.; García-Maset, R.; Giménez, I.; Silva, I.; Ruíz, P.; Ballarín, J.; Torra, R.; Ars, E. Clinical utility of genetic testing in children and adults with steroid-resistant nephrotic syndrome. Clin. J. Am. Soc. Nephrol. 2011, 6, 1139–1148. [Google Scholar] [CrossRef]
- Bullich, G.; Trujillano, D.; Santín, S.; Ossowski, S.; Mendizábal, S.; Fraga, G.; Madrid, Á.; Ariceta, G.; Ballarín, J.; Torra, R.; et al. Targeted next-generation sequencing in steroid-resistant nephrotic syndrome: Mutations in multiple glomerular genes may influence disease severity. Eur. J. Hum. Genet. 2015, 23, 1192–1199. [Google Scholar] [CrossRef]
- Büscher, A.K.; Beck, B.B.; Melk, A.; Hoefele, J.; Kranz, B.; Bamborschke, D.; Baig, S.; Lange-Sperandio, B.; Jungraithmayr, T.; Weber, L.T.; et al. Rapid response to cyclosporin A and favorable renal outcome in non- genetic versus genetic steroid-resistant nephrotic syndrome. J. Am. Soc. Nephrol. 2016, 11, 245–253. [Google Scholar] [CrossRef]
- Lovric, S.; Fang, H.; Vega-Warner, V.; Sadowski, C.E.; Gee, H.Y.; Halbritter, J.; Ashraf, S.; Saisawat, P.; Soliman, N.A.; Kari, J.A.; et al. Rapid detection of monogenic causes of childhood-onset steroid-resistant nephrotic syndrome. Clin. J. Am. Soc. Nephrol. 2014, 9, 1109–1116. [Google Scholar] [CrossRef]
- Giglio, S.; Provenzano, A.; Mazzinghi, B.; Becherucci, F.; Giunti, L.; Sansavini, G.; Ravaglia, F.; Roperto, R.M.; Farsetti, S.; Benetti, E.; et al. Heterogeneous genetic alterations in sporadic nephrotic syndrome associate with resistance to immunosuppression. J. Am. Soc. Nephrol. 2015, 26, 230–236. [Google Scholar] [CrossRef]
- Gipson, D.S.; Troost, J.P.; Lafayette, R.A.; Hladunewich, M.A.; Trachtman, H.; Gadegbeku, C.A.; Sedor, J.R.; Holzman, L.B.; Moxey-Mims, M.M.; Perumal, K.; et al. Complete remission in the nephrotic syndrome study network. Clin. J. Am. Soc. Nephrol. 2016, 11, 81–89. [Google Scholar] [CrossRef]
- Bergmann, C. Advances in renal genetic diagnosis. Cell Tissue Res. 2017, 369, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Lenz, W. Phenocopies. J. Med Genet. 1973, 10, 34–49. [Google Scholar] [CrossRef] [PubMed]
- Baum, P.; Schmid, R.; Ittrich, C.; Rust, W.; Fundel-Clemens, K.; Siewert, S.; Baur, M.; Mara, L.; Gruenbaum, L.; Heckel, A.; et al. Phenocopy—A Strategy to Qualify Chemical Compounds during it-to-Lead and/or Lead Optimization. PLoS ONE 2010, 5, e14272. [Google Scholar] [CrossRef] [PubMed]
- Grebe, H. Gene und phaenokopien in der aetiologie menschlicher missbildungen. Acta Genet. Med. Gemellogiae 1954, 3, 197–209. [Google Scholar] [CrossRef]
- Rimoin, D.L.; Schimke, R.N. Genetic Disorders of the Endocrine Glands; Mosby: St. Louis, MO, USA, 1971. [Google Scholar]
- Cassina, M.; Cagnoli, G.A.; Zuccarello, D.; Di Gianantonio, E.; Clementi, M. Human teratogens and genetic phenocopies. Understanding pathogenesis through human genes mutation. Eur. J. Med. Genet. 2017, 60, 22–31. [Google Scholar] [CrossRef]
- Berman, J.J. Disease convergence. In Precision Medicine and the Reinvention of Human Diseases; Berman, J.J., Ed.; Elzevier: Amsterdam, The Netherlands, 2018; pp. 117–151. [Google Scholar] [CrossRef]
- NIH-National Cancer Insitute. Available online: https://www.cancer.gov/publications/dictionaries/genetics-dictionary/def/phenocopy (accessed on 1 September 2020).
- Small, K.W.; DeLuca, A.P.; Whitmore, S.S. North Carolina macular dystrophy is caused by dysregulation of the retinal transcription factor PRDM13. Ophthalmology 2016, 123, 9–18. [Google Scholar] [CrossRef]
- Limongelli, G.; Masarone, D.; Verrengia, M.; Gravino, R.; Salerno, G.; Castelletti, S.; Rubino, M.; Marrazzo, T.; Pisani, A.; Cecchi, F.; et al. Diagnostic Clues for the Diagnosis of Nonsarcomeric Hypertrophic Cardiomyopathy (Phenocopies): Amyloidosis, Fabry Disease, and Mitochondrial Disease. J. Cardiovasc. Echogr. 2018, 28, 120–123. [Google Scholar] [CrossRef]
- Akhtar, M.; Elliott, P. The genetics of hypertrophic cardiomyopathy. Glob. Cardiol. Sci. Pract. 2018, 2018, 36. [Google Scholar] [CrossRef]
- Sankaranarayanan, R.; Fleming, E.J.; Garratt, C.J. Mimics of Hypertrophic Cardiomyopathy—Diagnostic Clues to Aid Early Identification of Phenocopies. Arrhythm. Electrophysiol. Rev. 2013, 2, 36–40. [Google Scholar] [CrossRef]
- Yu, J.E.; Orange, J.S.; Demirdag, Y.Y. New primary immunodeficiency diseases: Context and future. Curr. Opin. Pediatr. 2018, 30, 806–820. [Google Scholar] [CrossRef]
- Bousfiha, A.; Jeddane, L.; Picard, C. The 2017 IUIS phenotypic classification for primary immunodeficiencies. J. Clin. Immunol. 2018, 38, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Jindal, A.K.; Joshi, V.; Anjani, G.; Rawat, A. An updated review on phenocopies of primary immunodeficiency diseases. Genes Dis. 2019, 7, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Neven, B.; Magerus-Chatinet, A.; Florkin, B.; Gobert, D.; Lambotte, O.; De Somer, L.; Lanzarotti, N.; Stolzenberg, M.-C.; Bader-Meunier, B.; Aladjidi, N.; et al. A survey of 90 patients with autoimmune lymphoproliferative syndrome related to TNFRSF6 mutation. Blood 2011, 118, 4798–4807. [Google Scholar] [CrossRef] [PubMed]
- Rieux-Laucat, F. What’s up in the ALPS. Curr. Opin. Immunol. 2017, 49, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Alsenz, J.; Bork, K.; Loos, M. Autoantibody-mediated acquired deficiency of C1 inhibitor. N. Engl. J. Med. 1987, 316, 1360–1366. [Google Scholar] [CrossRef] [PubMed]
- Peterson, P.; Pitkänen, J.; Sillanpää, N.; Krohn, K. Autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED): A model disease to study molecular aspects of endocrine autoimmunity. Clin. Exp. Immunol. 2004, 135, 348–357. [Google Scholar] [CrossRef]
- Karimi-Moghadam, A.; Charsouei, S.; Bell, B.; Jabalameli, M.R. Parkinson Disease from Mendelian Forms to Genetic Susceptibility: New Molecular Insights into the Neurodegeneration Process. Cell Mol. Neurobiol. 2018, 38, 1153–1178. [Google Scholar] [CrossRef]
- Klein, C.; Chuang, R.; Marras, C.; Lang, A.E. The curious case of phenocopies in families with genetic Parkinson’s disease. Mov. Disord. 2011, 26, 1793–1802. [Google Scholar] [CrossRef]
- Illés, A.; Csabán, D.; Grosz, Z.; Balicza, P.; Gézsi, A.; Molnár, V.; Bencsik, R.; Gál, A.; Klivényi, P.; Molnar, M.J. The Role of Genetic Testing in the Clinical Practice and Research of Early-Onset Parkinsonian Disorders in a Hungarian Cohort: Increasing Challenge in Genetic Counselling, Improving Chances in Stratification for Clinical Trials. Front. Genet. 2019, 10, 1061. [Google Scholar] [CrossRef]
- King, M.C.; Marks, J.H.; Mandell, J.B. Breast Cancer Study Group. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science 2003, 302, 643–666. [Google Scholar] [CrossRef]
- Dominguez-Valentin, M.; Evans, D.G.; Nakken, S.; Tubeuf, H.; Vodák, D.; Ekstrøm, P.O.; Nissen, A.M.; Morak, M.; Holinski-Feder, E.; Martins, A.; et al. Genetic variants of prospectively demonstrated phenocopies in BRCA1/2 kindreds. Hered. Cancer Clin. Pract. 2018, 16, 4. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.G.; Ingham, S.L.; Buchan, I.; Woodward, E.R.; Byers, H.; Howell, A.; Maher, E.R.; Newman, W.G.; Lalloo, F. Increased rate of phenocopies in all age groups in BRCA1/BRCA2 mutation kindred, but increased prospective breast cancer risk is confined to BRCA2 mutation carriers. Cancer Epidemiol. Biomark. Prev. 2013, 22, 2269–2276. [Google Scholar] [CrossRef] [PubMed]
- Mariani, L.-L.; Tesson, C.; Charles, P.; Cazeneuve, C.; Hahn, V.; Youssov, K.; Freeman, L.; Grabli, D.; Roze, E.; Noël, S.; et al. Expanding the spectrum of genes involved in Huntington Disease using a combined clinical and genetic approach. JAMA Neurol. 2016, 73, 1105–1114. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.A.; Bird, T. Huntington’s Disease, Huntington’s Disease Look-Alikes, and Benign Hereditary Chorea: What’s New? Mov. Disord. Clin. Pract. 2016, 3, 342–354. [Google Scholar] [CrossRef] [PubMed]
- Baranchuk, A.; Nguyen, T.; Ryu, M.H.; Femenía, F.; Zareba, W.; Wilde, A.A.M.; Shimizu, W.; Brugada, P.; Pérez-Riera, A.R. Brugada phenocopy: New terminology and proposed classification. Ann. Noninvasive Electrocardiol. 2012, 7, 299–314. [Google Scholar] [CrossRef]
- Kocabas, U.; Hasdemir, C.; Kaya, E.; Turkoglu, C.; Baranchuk, A. Brugada syndrome, Brugada phenocopy or none? Ann. Noninvasive Electrocardiol. 2017, 22, e12470. [Google Scholar] [CrossRef]
- Riera, A.R.; Uchida, A.H.; Schapachnik, E.; Dubner, S.; Filho, C.F.; Ferreira, C. Propofol infusion syndrome and Brugada syndrome electrocardiographic phenocopy. Cardiol. J. 2010, 17, 130–135. [Google Scholar]
- Szabó, T.; Orosz, P.; Balogh, E.; Jávorszky, E.; Máttyus, I.; Bereczki, C.; Maróti, Z.; Kalmár, T.; Szabo, A.J.; Reusz, G.; et al. Comprehensive genetic testing in children with a clinical diagnosis of ARPKD identifies phenocopies. Pediatr. Nephrol. 2018, 33, 1713–1721. [Google Scholar] [CrossRef]
- Van Der Ven, A.T.; Connaughton, D.M.; Ityel, H.; Mann, N.; Nakayama, M.; Chen, J.; Vivante, A.; Hwang, D.-Y.; Schulz, J.; Braun, D.A.; et al. Whole-Exome Sequencing Identifies Causative Mutations in Families with Congenital Anomalies of the Kidney and Urinary Tract. J. Am. Soc. Nephrol. 2018, 29, 2348–2361. [Google Scholar] [CrossRef]
- Groopman, E.E.; Marasa, M.; Cameron-Christie, S.; Petrovski, S.; Aggarwal, V.S.; Milo-Rasouly, H.; Li, Y.; Zhang, J.; Nestor, J.; Krithivasan, P.; et al. Diagnostic Utility of Exome Sequencing for Kidney Disease. N. Engl. J. Med. 2019, 380, 142–151. [Google Scholar] [CrossRef]
- Daga, A.; Majmundar, A.J.; Braun, D.A.; Gee, H.Y.; Lawson, J.A.; Shril, S.; Jobst-Schwan, T.; Vivante, A.; Schapiro, D.; Tan, W.; et al. Whole exome sequencing frequently detects a monogenic cause in early onset nephrolithiasis and nephrocalcinosis. Kidney Int. 2018, 93, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Gee, H.Y.; Otto, E.A.; Hurd, T.W.; Ashraf, S.; Chaki, M.; Cluckey, A.; Vega-Warner, V.; Saisawat, P.; Diaz, K.A.; Fang, H.; et al. Whole-exome resequencing distinguishes cystic kidney diseases from phenocopies in renal ciliopathies. Kidney Int. 2014, 85, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Snoek, R.; Van Setten, J.; Keating, B.J.; Israni, A.; Jacobson, P.A.; Oetting, W.S.; Matas, A.J.; Mannon, R.B.; Zhang, Z.; Zhang, W.; et al. NPHP1 (Nephrocystin-1) gene deletions cause adult-onset ESRD. J. Am. Soc. Nephrol. 2018, 29, 1772–1779. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.A.; Schueler, M.; Halbritter, J.; Gee, H.Y.; Porath, J.D.; Lawson, J.A.; Airik, R.; Shril, S.; Allen, S.J.; Stein, D.; et al. Whole exome sequencing identifies causative mutations in the majority of consanguineous or familial cases with childhood-onset increased renal echogenicity. Kidney Int. 2016, 89, 468–475. [Google Scholar] [CrossRef]
- Armstrong, M.E.; Thomas, C.P. Diagnosis of monogenic chronic kidney diseases. Curr. Opin. Nephrol. Hypertens. 2019, 28, 183–194. [Google Scholar] [CrossRef]
- Mann, N.; Braun, D.A.; Amann, K.; Tan, W.; Shril, S.; Connaughton, D.M.; Nakayama, M.; Schneider, R.; Kitzler, T.M.; Van Der Ven, A.T.; et al. Whole-exome sequencing enables a precision medicine approach for kidney transplant recipients. J. Am. Soc. Nephrol. 2019, 30, 201–215. [Google Scholar] [CrossRef]
- Lata, S.; Marasa, M.; Li, Y.; Fasel, D.A.; Groopman, E.; Jobanputra, V.; Rasouly, H.; Mitrotti, A.; Westland, R.; Verbitsky, M.; et al. Whole-exome sequencing in adults with chronic kidney disease: A pilot study. Ann. Intern. Med. 2018, 168, 100–109. [Google Scholar] [CrossRef]
- Connaughton, D.M.; Kennedy, C.; Shril, S.; Mann, N.; Murray, S.L.; Williams, P.A.; Conlon, E.; Nakayama, M.; Van Der Ven, A.T.; Ityel, H.; et al. Monogenic causes of chronic kidney disease in adults. Kidney Int. 2019, 95, 914–928. [Google Scholar] [CrossRef]
- Gast, C.; Pengelly, R.J.; Lyon, M.; Bunyan, D.J.; Seaby, E.G.; Graham, N.; Venkat-Raman, G.; Ennis, S. Collagen (COL4A) mutations are the most frequent mutations underlying adult focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 2016, 31, 961–970. [Google Scholar] [CrossRef]
- Pierides, A.; Voskarides, K.; Athanasiou, Y.; Ioannou, K.; Damianou, L.; Arsali, M.; Zavros, M.; Pierides, M.; Vargemezis, V.; Patsias, C.; et al. Clinico-pathological correlations in 127 patients in 11 large pedigrees, segregating one of three heterozygous mutations in the COL4A3/ COL4A4 genes associated with familial haematuria and significant late progression to proteinuria and chronic kidney disease from focal segmental glomerulosclerosis. Nephrol. Dial. Transpl. 2009, 24, 2721–2729. [Google Scholar] [CrossRef]
- Fervenza, F.C. A patient with nephrotic-range proteinuria and focal global glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2013, 8, 1979–1987. [Google Scholar] [CrossRef] [PubMed]
- Saida, K.; Kamijo, Y.; Matsuoka, D.; Noda, S.; Hidaka, Y.; Mori, T.; Shimojo, H.; Ehara, T.; Miura, K.; Takita, J.; et al. A case of adult Dent disease in Japan with advanced chronic kidney disease. CEN Case Rep. 2014, 3, 132–138. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Edwards, N.; Rice, S.J.; Raman, S.; Hynes, A.M.; Srivastava, S.; Moore, I.; Al-Hamed, M.; Xu, Y.; Santibanez-Koref, M.; Thwaites, D.T.; et al. A novel LMX1B mutation in a family with end-stage renal disease of ‘unknown cause’. Clin. Kidney J. 2015, 8, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Barua, M.; Stellacci, E.; Stella, L.; Weins, A.; Genovese, G.; Muto, V.; Caputo, V.; Toka, H.R.; Charoonratana, V.T.; Tartaglia, M.; et al. Mutations in PAX2 associate with adult-onset FSGS. J. Am. Soc. Nephrol. 2014, 25, 1942–1953. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Zhang, H.; Cao, S.; Xiao, H.; Yao, Y. Dent’s disease complicated by nephrotic syndrome: A case report. Intractable Rare Dis. Res. 2016, 5, 297–300. [Google Scholar] [CrossRef]
- Malone, A.F.; Phelan, P.J.; Hall, G.; Cetincelik, U.; Homstad, A.; Alonso, A.S.; Jiang, R.; Lindsey, T.B.; Wu, G.; Sparks, M.A.; et al. Rare hereditary COL4A3/COL4A4 variants maybe mistaken for familial focal segmental glomerulosclerosis. Kidney Int. 2014, 86, 1253–1259. [Google Scholar] [CrossRef]
- Vallés, P.; Peralta, M.; Carrizo, L.; Martin, L.; Principi, I.; Gonzalez, A.; Manucha, W. Follow-up of steroid-resistant nephrotic syndrome: Tubular proteinuria and enzymuria. Pediatr. Nephrol. 2000, 15, 252–258. [Google Scholar] [CrossRef]
- Wang, X.; Anglani, F.; Beara-Lasic, L.; Mehta, A.J.; Vaughan, L.E.; Hernandez, L.H.; Cogal, A.; Scheinman, S.J.; Ariceta, G.; Isom, R.; et al. Glomerular pathology in Dent disease and its association with kidney function. Clin. J. Am. Soc. Nephrol. 2016, 11, 2168–2176. [Google Scholar] [CrossRef]
- Du Moulin, M.; Koehn, A.; Golsari, A.; Dulz, S.; Atiskova, Y.; Patten, M.; Münch, J.; Avanesov, M.; Ullrich, K.; Muschol, N. The mutation p.D313Y is associated with organ manifestation in Fabry disease. Clin. Genet. 2017, 92, 528–533. [Google Scholar] [CrossRef]
- Köping, M.; Shehata-Dieler, W.; Cebulla, M.; Rak, K.; Oder, D.; Müntze, J.; Nordbeck, P.; Wanner, C.; Hagen, R.; Schraven, S. Cardiac and renal dysfunction is associated with progressive hearing loss in patients with Fabry disease. PLoS ONE 2017, 12, e0188103. [Google Scholar] [CrossRef]
- Torra, R.; Furlano, F.; Ars, E. How genomics reclassifies diseases: The case of Alport syndrome. Clin. Kidney J. 2020, 1–3. [Google Scholar] [CrossRef]
- Adam, J.; Connor, T.M.; Wood, K.; Lewis, D.; Naik, R.; Gale, D.P.; Sayer, J.A. Genetic testing can resolve diagnostic confusion in Alport syndrome. Clin. Kidney J. 2014, 7, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Wu, X.; Ren, H.; Wang, W.; Pan, X.; Hao, X.; Tong, J.; Ma, J.; Ye, Z.; Meng, G.; et al. COL4A3 mutations cause focal segmental glomerulosclerosis. J. Mol. Cell Biol. 2014, 6, 498–505, Erratum in 2015, 7, 184. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.; Udwan, K.; John, R.; Rana, A.; Haghighi, A.; Xu, L.; Hack, S.; Reich, H.N.; Hladunewich, M.A.; Cattran, D.C.; et al. Integration of Genetic Testing and Pathology for the Diagnosis of Adults with FSGS. Clin. J. Am. Soc. Nephrol. 2019, 14, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Riedhammer, K.M.; Braunisch, M.C.; Günthner, R.; Wagner, M.; Hemmer, C.; Strom, T.M.; Schmaderer, C.; Renders, L.; Tasic, V.; Gucev, Z.; et al. Exome sequencing and identification of phenocopies in patients with clinically presumed hereditary nephropathies. Am. J. Kidney Dis. 2020, 76, 460–470. [Google Scholar] [CrossRef]
- Jayasinghe, K.; Dm, Z.S.; Kerr, P.G.; Gaff, C.; Martyn, M.; Whitlam, J.; Creighton, B.; Bn, E.D.; Hunter, M.; Jarmolowicz, A.; et al. Clinical impact of genomic testing in patients with suspected monogenic kidney disease. Genet. Med. 2020. [Google Scholar] [CrossRef]
- Bullich, G.; Domingo-Gallego, A.; Vargas, I.; Ruiz, P.; Lorente-Grandoso, L.; Furlano, M.; Fraga, G.; Madrid, Á.; Ariceta, G.; Borregán, M.; et al. A kidney-disease gene panel allows a comprehensive genetic diagnosis of cystic and glomerular inherited kidney diseases. Kidney Int. 2018, 94, 363–371. [Google Scholar] [CrossRef]
- De Haan, A.; Eijgelsheim, M.; Vogt, L.; Knoers, N.V.A.M.; de Borst, M.H. Diagnostic yield of next-generation sequencing in patients with chronic kidney disease of unknown etiology. Front. Genet. 2019, 10, 1264. [Google Scholar] [CrossRef]
- Choi, M.; Scholl, U.I.; Ji, W.; Liu, T.; Tikhonova, I.R.; Zumbo, P.; Nayir, A.; Bakkaloğlu, A.; Özen, S.; Sanjad, S.; et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc. Natl. Acad. Sci. USA 2009, 106, 19096–19101. [Google Scholar] [CrossRef]
- Ahram, D.F.; Aggarwal, V.S.; Sanna-Cherch, S. Phenocopies, phenotypic expansion and coincidental diagnosis: Time to abandon targeted gene panels? Am. J. Kidney Dis. 2020, 76, 451–453. [Google Scholar] [CrossRef]
- Ars, E.; Torra, R. Rare diseases, rare presentations: Recognizing atypical inherited kidney disease phenotype in the age of genomics. Clin. Kidney J. 2017, 10, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Barwell, J.G.; O’Sullivan, R.B.G.; Mansbridge, L.K.; Lowry, J.M.; Dorkins, H.R. Challenges in implementing genomic medicine: The 100,000 Genomes Project. J. Transl. Genet. Genom. 2018, 2, 13. [Google Scholar] [CrossRef]
- Hunter, D.J.; Longo, D.L. The precision of evidence needed to practice “Precision Medicine”. N. Engl. J. Med. 2019, 380, 2472–2474. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Disease (Phenotype) | Causative Gene/s | Phenocopy | Mechanism of Phenocopy Determination | Ref. |
|---|---|---|---|---|
| Pendred’s syndrome | SLC26A4 | Endemic cretinism | Environmental | [34] |
| Monogenic skeletal disorders (e.g., Holt–Oram syndrome, radius aplasia-thrombocytopenia syndrome) | e.g., TBX5, RBM8A | Thalidomide embryopathy | Environmental | [34,38] |
| North Carolina macular dystrophy (NCMD) grade 3 | locus chr5p15-p13 | Congenital toxoplasmosis | Environmental | [41] |
| North Carolina macular dystrophy (NCMD) grade 1 | DHS6S1 | Foveal hypoplasia | Unknown | [41] |
| North Carolina macular dystrophy (NCMD) grade 2 | PROM1 | Torpedo maculopathy | Unknown | [41] |
| Hypertrophic cardiomyopathy | Sarcomeric genes | Glycogen storage diseases (e.g., Danon disease) | Monogenic | [42,43,44] |
| Lisosomal storage diseases (e.g., Fabry disease) | Monogenic | |||
| Mitocondrial cytopathies (e.g., MELAS) | Monogenic | |||
| AL amyloidosis | Unknown | |||
| Autoimmune lymphoproliferative syndrome (ALPS) | FAS, FASLG, CASP10 | ALPS phenocopies | Somatic mutations in FAS (non-Mendelian genetic mechanism) | [47,48,49] |
| Cryopyrinopathies | NLRP3 | Cryopyrinopathies phenocopies | Somatic mutations in NLRP3 (non-Mendelian genetic mechanism) | [47] |
| Hereditary angioedema | SERPING1 | Acquired angioedema | Autoantibodies anti-C1-inhibitor (complex mechanism) | [50] |
| Chronic mucocutaneous candidiasis | Genes encoding IL-17, IL-22 | Recurrent fungal infections | Autoantibodies anti-IL17 or IL-22 (complex mechanism) | [51] |
| Familial Parkinson’s disease | SNCA, LRRK2, PRKN, PINK1 and others | Familial Parkinson’s disease negative for mutations in known genes | Unknown | [53,54] |
| Familial breast cancer | BRCA1, BRCA2 | Familial breast cancer negative for mutations in BRCA genes | Unknown (probably genetic) | [56,57] |
| Huntington disease (HD) | HTT | HD phenocopies | Monogenic (mutations in genes different from HTT) | [58,59] |
| Brugada syndrome | SCN5A | Brugada phenocopies (e.g., metabolic abnormalities, ischemia, mechanic compression, myocardial and pericardial diseases) | Unknown (probably complex) | [60,61,62] |
| Gene | Phenotype | Ref. | |
|---|---|---|---|
| CLCN5 | CHLORIDE CHANNEL 5 | Dent disease | [3,4,76,77,91] |
| COL4A3 | COLLAGEN, TYPE IV, ALPHA-3 | Alport syndrome | [3,4,72,81,88,89,90] |
| COL4A4 | COLLAGEN, TYPE IV, ALPHA-4 | Alport syndrome | [72,74,81,89] |
| COL4A5 | COLLAGEN, TYPE IV, ALPHA-5 | Alport syndrome | [3,72,89,90] |
| GLA | GALACTOSIDASE, ALPHA | Fabry disease | [3,4,90] |
| AGXT | ALANINE-GLYOXYLATE AMINOTRANSFERASE | Hyperoxaluria, primary, type 1 | [3] |
| CTNS | CYSTINOSIN | Cystinosis | [3,4] |
| FN1 | FIBRONECTIN 1 | Glomerulopathy with fibronectin deposits 2 | [3] |
| WDR19 | WD REPEAT-CONTAINING PROTEIN 19 | Nephronophthisis 13 | [3] |
| LAMB2 | LAMININ, BETA-2 | Pierson syndrome | [4] |
| FAT1 | FAT ATYPICAL CADHERIN 1 | FAT1-related glomerulo-tubular nephropathy | [4] |
| FAT4 | FAT ATYPICAL CADHERIN 4 | Van Maldergem syndrome 2 | [4] |
| PAX2 | PAIRED BOX GENE 2 | Papillo-renal syndrome/FSGS | [4] |
| LMX1B | LIM HOMEOBOX TRANSCRIPTION FACTOR 1, BETA | Nail–patella syndrome | [4] |
| KANK1 | KN MOTIF- AND ANKYRIN REPEAT DOMAIN-CONTAINING PROTEIN 1 | Cerebral palsy | [4] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Becherucci, F.; Landini, S.; Cirillo, L.; Mazzinghi, B.; Romagnani, P. Look Alike, Sound Alike: Phenocopies in Steroid-Resistant Nephrotic Syndrome. Int. J. Environ. Res. Public Health 2020, 17, 8363. https://doi.org/10.3390/ijerph17228363
Becherucci F, Landini S, Cirillo L, Mazzinghi B, Romagnani P. Look Alike, Sound Alike: Phenocopies in Steroid-Resistant Nephrotic Syndrome. International Journal of Environmental Research and Public Health. 2020; 17(22):8363. https://doi.org/10.3390/ijerph17228363
Chicago/Turabian StyleBecherucci, Francesca, Samuela Landini, Luigi Cirillo, Benedetta Mazzinghi, and Paola Romagnani. 2020. "Look Alike, Sound Alike: Phenocopies in Steroid-Resistant Nephrotic Syndrome" International Journal of Environmental Research and Public Health 17, no. 22: 8363. https://doi.org/10.3390/ijerph17228363
APA StyleBecherucci, F., Landini, S., Cirillo, L., Mazzinghi, B., & Romagnani, P. (2020). Look Alike, Sound Alike: Phenocopies in Steroid-Resistant Nephrotic Syndrome. International Journal of Environmental Research and Public Health, 17(22), 8363. https://doi.org/10.3390/ijerph17228363