ARDEP, a Rapid Degenerate Primer Design Pipeline Based on k-mers for Amplicon Microbiome Studies


Abstract
1. Introduction
2. Materials and Methods
2.1. Analysis Platform of ARDEP
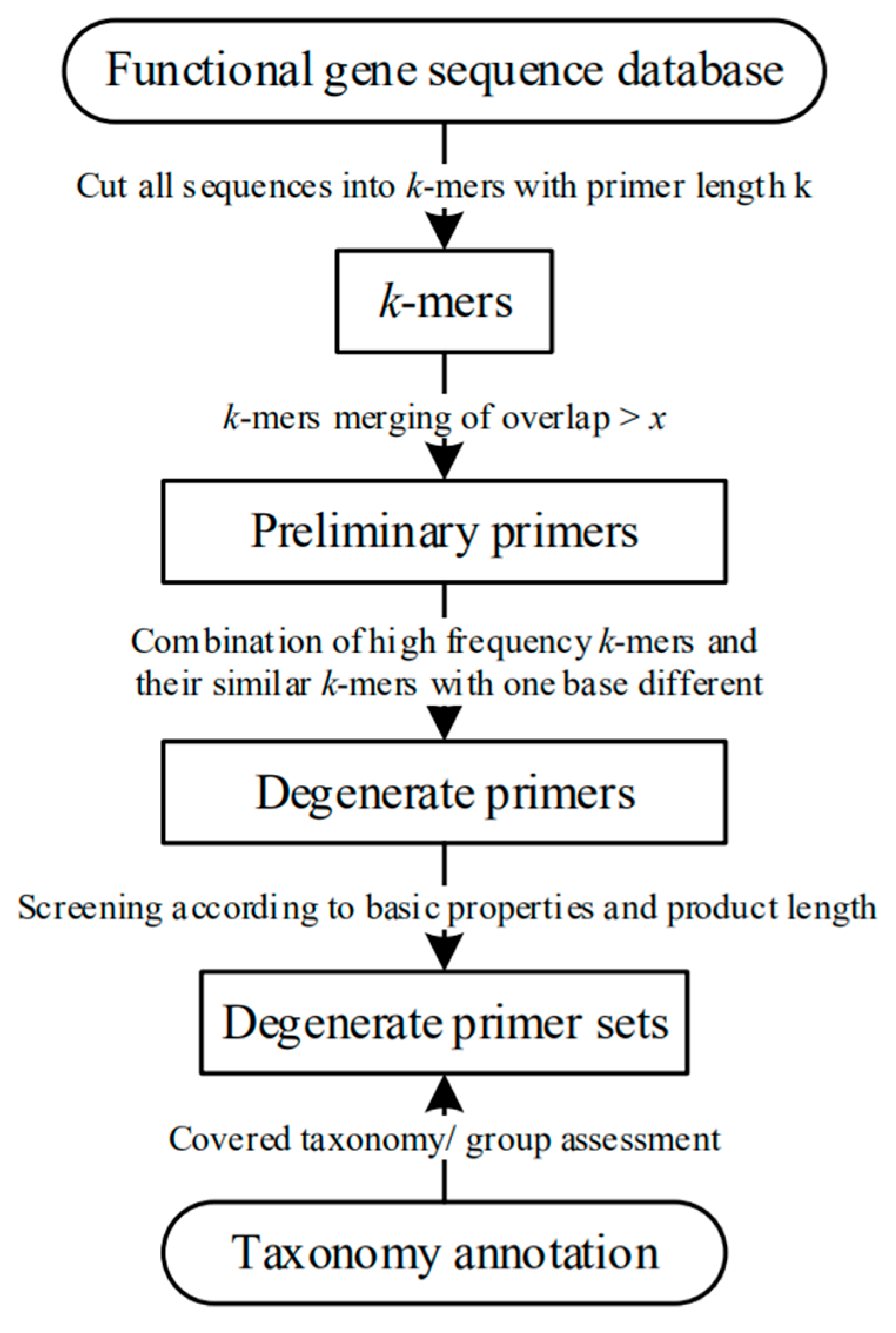
2.2. Algorithm of ARDEP
2.2.1. Division of Database Sequences by k-mer Program
2.2.2. Degenerate Primer Combination
2.2.3. Calculation of Basic Primer Properties
- The annealing temperature (Tm), secondary structure (ΔG of primer’s self-folding), and GC content (GC%) of primers are calculated as follows:
- GC% = (G + C)/primer length
- Annealing temperature (Tm): when the primer length is shorter than 20 bp: Tm = 4 (G + C) + 2 (A + T); when the primer length is longer than 20 bp, Tm = 62.3 °C + 0.41 °C (GC%) − 500/primer length.
- The stability parameter ΔG, which is the free energy needed for DNA double-strand formation, reflects the stability of the double-strand structure. If the complementary base of the primer is too long, a primer dimer or hairpin structure could be produced, which leads to high ΔG and the PCR reaction cannot proceed normally. This parameter is calculated by mfold software [14].
2.2.4. Coverage Verification and Covered Taxonomy/Group Calculation
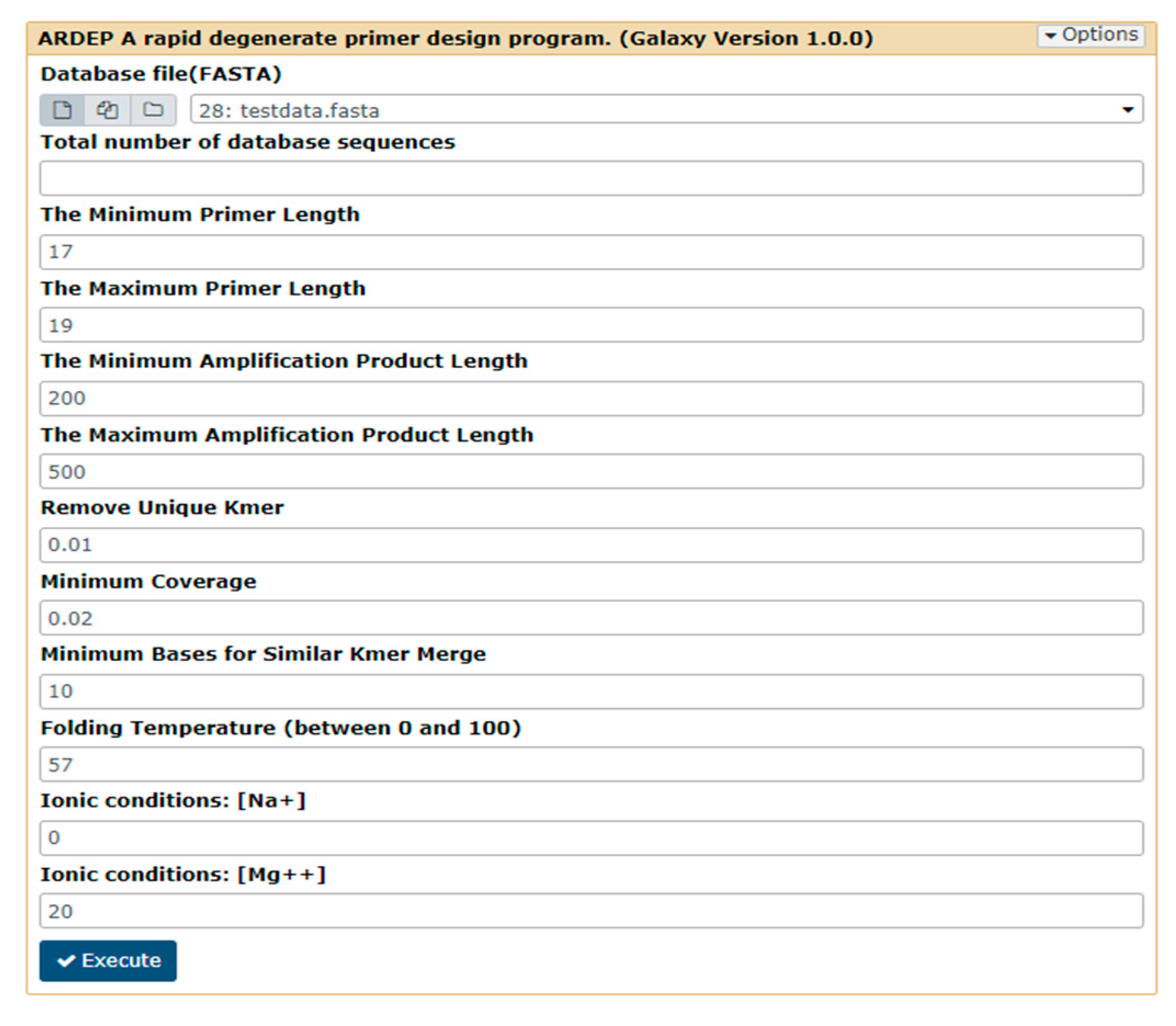
2.2.5. Parameters Required by ARDEP Platform
- Number of database sequences;
- Primer length (maximum and minimum, default 18–20 bp);
- Amplification product fragment length (maximum and minimum, default value is 200–500 bp);
- Minimum coverage users could accept of the output primer (the default value is 20%, i.e., 0.2);
- Overlap of high-frequency k-mers to be merged (the default value is 10);
- DNA folding temperature in PCR experiment (the default value is 57 °C);
- Ionic conditions of Mg++ (the default value is 20) and Na+ (the default value is 0) in PCR experiment (Figure 2).
2.3. Test Datasets
3. Results
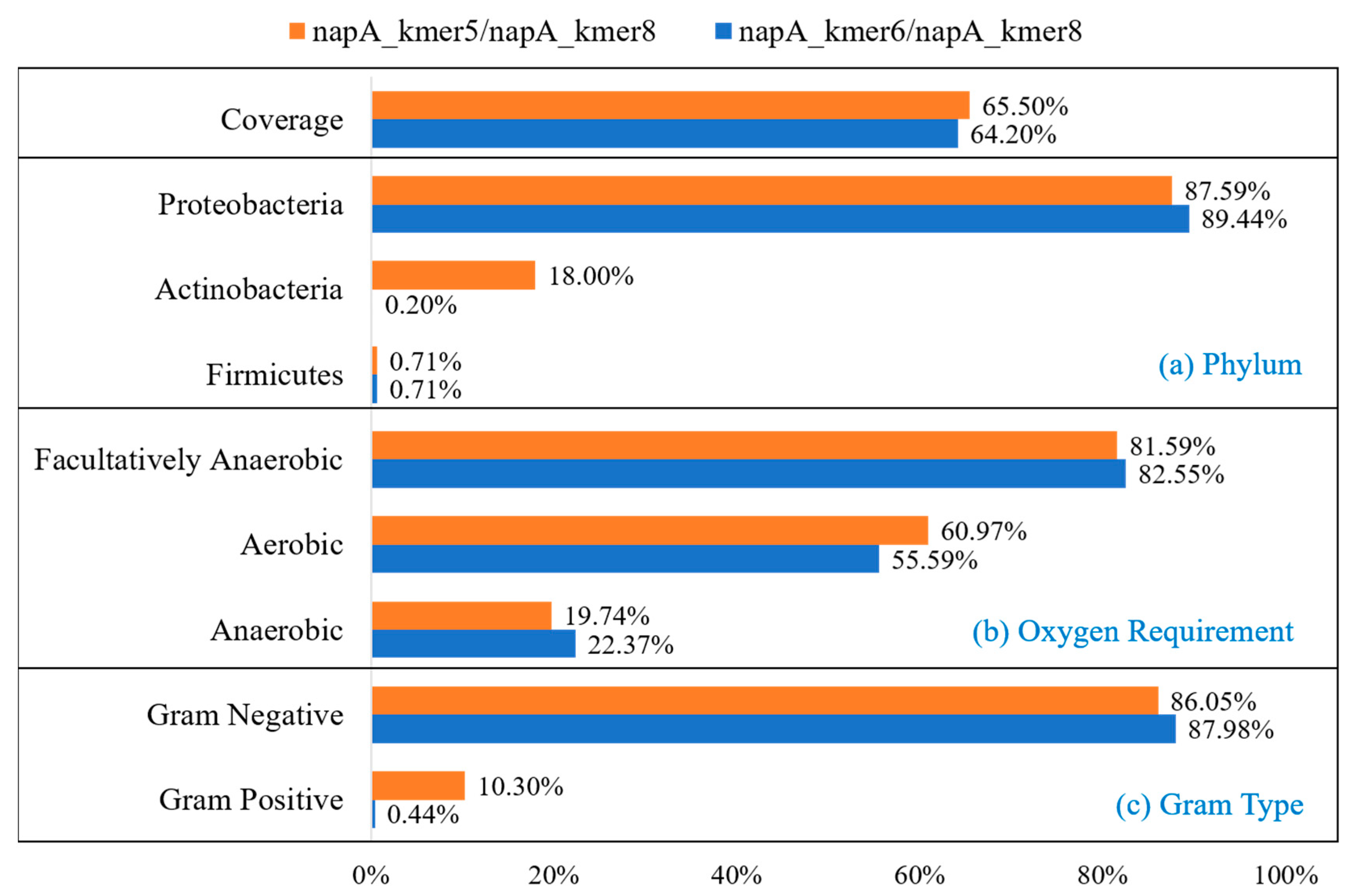
3.1. Primer Design of Functional Gene napA
3.2. Primer Design of Functional Gene amoA
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gilbert, J.A.; Meyer, F.; Jansson, J.; Gordon, J.; Pace, N.; Tiedje, J.; Ley, R.; Fierer, N.; Field, D.; Kyrpides, N. The earth microbiome project: Meeting report of the “1 st EMP meeting on sample selection and acquisition” at Argonne National Laboratory October 6th 2010. Stand. Genom. Sci. 2010, 3, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Hugenholtz, P. Exploring prokaryotic diversity in the genomic era. Genome Biol. 2002, 3, 1–8. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pettersson, E.; Lundeberg, J.; Ahmadian, A. Generations of sequencing technologies. Genomics 2009, 93, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Scharf, S.; Horn, G.; Erlich, H. Direct cloning and sequence analysis of enzymatically amplified genomic sequences. Science 1986, 233, 1076–1078. [Google Scholar] [CrossRef]
- Pace, N.R. Analyzing natural microbial populations by rRNA sequences. ASM News 1985, 51, 4–12. [Google Scholar]
- Li, S.; Deng, Y.; Wang, Z.; Zhang, Z.; Kong, X.; Zhou, W.; Yi, Y.; Qu, Y. Exploring the accuracy of amplicon-based internal transcribed spacer markers for a fungal community. Mol. Ecol. Resour. 2020, 20, 170–184. [Google Scholar] [CrossRef]
- Linhart, C.; Shamir, R. The degenerate primer design problem. Bioinformatics 2002, 18, S172–S181. [Google Scholar] [CrossRef]
- Walters, W.A.; Caporaso, J.G.; Lauber, C.L.; Berg-Lyons, D.; Fierer, N.; Knight, R. PrimerProspector: De novo design and taxonomic analysis of barcoded polymerase chain reaction primers. Bioinformatics 2011, 27, 1159–1161. [Google Scholar] [CrossRef]
- Hugerth, L.W.; Wefer, H.A.; Lundin, S.; Jakobsson, H.E.; Lindberg, M.; Rodin, S.; Engstrand, L.; Andersson, A.F. DegePrime, a program for degenerate primer design for broad-taxonomic-range PCR in microbial ecology studies. Appl. Environ. Microbiol. 2014, 80, 5116–5123. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Benoit, G.; Peterlongo, P.; Mariadassou, M.; Drezen, E.; Schbath, S.; Lavenier, D.; Lemaitre, C. Multiple comparative metagenomics using multiset k-mer counting. PeerJ Comput. Sci. 2016, 2, e94. [Google Scholar] [CrossRef]
- Ward, T.; Larson, J.; Meulemans, J.; Hillmann, B.; Lynch, J.; Sidiropoulos, D.; Spear, J.; Caporaso, G.; Blekhman, R.; Knight, R. BugBase predicts organism level microbiome phenotypes. BioRxiv 2017, 133462. [Google Scholar]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Yin, H.; Van Nostrand, J.D.; Voordeckers, J.W.; Tu, Q.; Deng, Y.; Yuan, M.; Zhou, A.; Zhang, P.; Xiao, N. Functional Gene Array-Based Ultrasensitive and Quantitative Detection of Microbial Populations in Complex Communities. MSystems 2019, 4. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef]
- Sayers, E.W.; Beck, J.; Brister, J.R.; Bolton, E.E.; Canese, K.; Comeau, D.C.; Funk, K.; Ketter, A.; Kim, S.; Kimchi, A. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2020, 48, D9. [Google Scholar] [CrossRef]
- Fish, J.A.; Chai, B.; Wan, Q.; Sun, Y.; Titus Brown, C.; Tiedje, J.M.; Cole, J.R. FunGene: The functional gene pipeline and repository. Front. Microbiol. 2013, 4, 291. [Google Scholar] [CrossRef]
- Katoh, K.; Asimenos, G.; Toh, H. Multiple alignment of DNA sequences with MAFFT. In Bioinformatics for DNA Sequence Analysis; Springer: Boston, MA, USA, 2009; pp. 39–64. [Google Scholar]
- Mancabelli, L.; Milani, C.; Lugli, G.A.; Fontana, F.; Turroni, F.; van Sinderen, D.; Ventura, M. The Impact of Primer Design on Amplicon-Based Metagenomic Profiling Accuracy: Detailed Insights into Bifidobacterial Community Structure. Microorganisms 2020, 8, 131. [Google Scholar] [CrossRef]
- Wei, Z.; Feng, K.; Li, S.; Zhang, Y.; Chen, H.; Yin, H.; Xu, M.; Deng, Y. Exploring abundance, diversity and variation of a widespread antibiotic resistance gene in wastewater treatment plants. Environ. Int. 2018, 117, 186–195. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Tu, Q.; Lin, L.; Cheng, L.; Deng, Y.; He, Z. NCycDB: A curated integrative database for fast and accurate metagenomic profiling of nitrogen cycling genes. Bioinformatics 2018, 35, 1040–1048. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequence | Product Length | Coverage | Specificity | Primer Properties | ||||
|---|---|---|---|---|---|---|---|---|---|
| narG | nt | ΔG | GC% | Tm | |||||
| napA_kmer5 | TTYTAYGACTGGTAYKSYGA | 421 | 80.01% | 65.50% | 0.82% | <0.01% | (−2.07)–(1.70) | 30.00–55.00% | 52.00–62.00 |
| napA_kmer8 | ACSTGGGADAYNCADCADAC | 67.91% | (−3.15)–(2.10) | 40.00–65.00% | 56.00–66.00 | ||||
| napA_kmer6 | TGGGGYGAVCADACNGAYGT | 466 | 77.77% | 64.20% | 0.80% | <0.01% | (−1.65)–(1.24) | 45.00–70.00% | 58.00–68.00 |
| napA_kmer8 | ACSTGGGADAYNCADCADAC | 67.91% | (−3.15)–(2.10) | 40.00–65.00% | 56.00–66.00 | ||||
| Primer | Coverage | Product Length | Specificity | ||
|---|---|---|---|---|---|
| Forward | Reverse | Mode | pmoA | nt | |
| AOA_kmer4 | AOA_kmer14 | 66.07% | 427 | <0.01% | <0.01% |
| AOA_kmer2 | AOA_kmer9 | 62.90% | 202 | <0.01% | 0.06% |
| AOA_kmer2 | AOA_kmer5 | 56.08% | 433 | <0.01% | 0.06% |
| AOA_kmer4 | AOA_kmer5 | 55.65% | 385 | <0.01% | 0.06% |
| AOA_kmer8 | AOA_kmer14 | 55.18% | 472 | <0.01% | <0.01% |
| AOA_kmer2 | AOA_kmer15 | 52.71% | 407 | <0.01% | <0.01% |
| AOA_kmer4 | AOA_kmer6 | 52.14% | 211 | <0.01% | 0.05% |
| AOA_kmer2 | AOA_kmer11 | 52.09% | 229 | <0.01% | <0.01% |
| AOA_kmer4 | AOA_kmer15 | 51.35% | 359 | <0.01% | <0.01% |
| Primer | Coverage | Product Length | Specificity | ||
|---|---|---|---|---|---|
| Forward | Reverse | Mode | pmoA | nt | |
| AOB_kmer3 | AOB_kmer6 | 55.50% | 204 | 0.06% | 0.05% |
| AOB_kmer1 | AOB_kmer22 | 53.66% | 439 | <0.01% | <0.01% |
| AOB_kmer6 | AOB_kmer26 | 51.83% | 218 | <0.01% | <0.01% |
| AOB_kmer3 | AOB_kmer17 | 51.63% | 204 | 0.06% | <0.01% |
| AOB_kmer1 | AOB_kmer5 | 51.30% | 448 | <0.01% | 0.04% |
| AOB_kmer6 | AOB_kmer22 | 51.12% | 294 | <0.01% | <0.01% |
| AOB_kmer4 | AOB_kmer6 | 50.87% | 228 | <0.01% | 0.04% |
| AOB_kmer1 | AOB_kmer27 | 50.79% | 451 | <0.01% | <0.01% |
| AOB_kmer17 | AOB_kmer26 | 50.10% | 218 | <0.01% | <0.01% |
| Primer | Sequence | Coverage | Specificity | DeltaG | GC% | Tm | Product Length (Mode) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| pmoA | nt | ||||||||||
| amoA_kmer7 | ATYAAYGCAGGRGACTAYAT | 27.62% | 22.90% | <0.01% | <0.01% | 0.06% | <0.01% | (−1.65)–(−0.74) | 30.00–50.00% | 52.00–60.00 | 427 |
| amoA_kmer15 | AADTTCTAYAAYAGYCCHG | 36.45% | <0.01% | <0.01% | (−0.03)–(1.29) | 26.32–52.63% | 48.00–58.00 | ||||
| amoA_kmer6 | GTVTGGTGGTAYYTTGGYAA | 22.40% | 20.95% | 0.17% | 0.03% | 0.05% | <0.01% | (−0.49)–(1.86) | 35.00–55.00% | 54.00–62.00 | 202 |
| amoA_kmer20 | TYTAYCCNGGYAACTGGMC | 36.82% | 35.83% | <0.01% | (−4.91)–(0.43) | 42.11–68.42% | 54.00–64.00 | ||||
| amoA_kmer2 | GGTTTCTACTGGTGGTCVCA | 23.93% | 20.28% | <0.01% | <0.01% | 0.05% | <0.01% | (−0.15)–(0.40) | 50.00–55.00% | 60.00–62.00 | 448 |
| amoA_kmer17 | GAAGAAGGCTTTSCMGAGG | 22.84% | <0.01% | <0.01% | (−0.66)–(−0.47) | 52.63–57.89% | 58.00–60.00 | ||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.; Feng, K.; Wei, Z.; Wang, Z.; Deng, Y. ARDEP, a Rapid Degenerate Primer Design Pipeline Based on k-mers for Amplicon Microbiome Studies. Int. J. Environ. Res. Public Health 2020, 17, 5958. https://doi.org/10.3390/ijerph17165958
Wu Y, Feng K, Wei Z, Wang Z, Deng Y. ARDEP, a Rapid Degenerate Primer Design Pipeline Based on k-mers for Amplicon Microbiome Studies. International Journal of Environmental Research and Public Health. 2020; 17(16):5958. https://doi.org/10.3390/ijerph17165958
Chicago/Turabian StyleWu, Yueni, Kai Feng, Ziyan Wei, Zhujun Wang, and Ye Deng. 2020. "ARDEP, a Rapid Degenerate Primer Design Pipeline Based on k-mers for Amplicon Microbiome Studies" International Journal of Environmental Research and Public Health 17, no. 16: 5958. https://doi.org/10.3390/ijerph17165958
APA StyleWu, Y., Feng, K., Wei, Z., Wang, Z., & Deng, Y. (2020). ARDEP, a Rapid Degenerate Primer Design Pipeline Based on k-mers for Amplicon Microbiome Studies. International Journal of Environmental Research and Public Health, 17(16), 5958. https://doi.org/10.3390/ijerph17165958