Asbestos, Smoking and Lung Cancer: An Update



{kind=link}
{kind=link}
Abstract
1. Introduction
Preliminary Remarks
- All commercial asbestos fiber types can be implicated in the causation of lung cancer, e.g. amphibole anthophyllite and including the noncommercial amphibole, tremolite. In this context, commercial amphiboles crocidolite and amosite appear to be about equipotent on a fiber-for-fiber basis for lung cancer induction, and chrysotile is also implicated, especially in the chrysotile textile industry [22,23]. (There remains an unexplained 30-fold to 50-fold differential in the risk of lung cancer among the Charleston textile workers who used commercial Canadian chrysotile almost exclusively, in comparison to a much smaller risk for the Quebec chrysotile miners and millers).
- Whilst histological criteria have been established, the differential diagnosis of malignant mesothelioma, primary lung carcinomas, and pleural metastases can cause problems, especially in the case of sarcomatoid tumors [24,25]. A comparison of the frequency of DNA copy number changes between mesothelioma and lung carcinoma using discriminant analysis suggests that they are genetically-different tumors [26]. In the case of malignant mesothelioma, asbestos is overwhelmingly the singular identifiable causal factor, with only rare cases related to other factors such as erionite or fluoro-edenite fiber inhalation, ionizing radiation (sometimes in association with asbestos exposure), and innate susceptibility factors such as germline mutations affecting the BAP1 gene. Tobacco is not implicated in the causation of mesothelioma.
- Lung carcinoma, on the other hand, is a multifactorial cancer for which tobacco (especially cigarette smoke) represents the most potent causal factor on a worldwide basis. There are other known causes such as ionizing radiation (including radon gas daughters); certain metals such as hexavalent chromium, nickel, cadmium, arsenic and beryllium; silica, diesel particulate, and heated cooking [27].
- There are no clinical, radiographic or pathologic features of the tumor that discriminate clearly between lung cancers for which asbestos exposure can be implicated versus those for which it cannot; that is, there are no differences in the anatomical distribution of lung cancers in asbestos-exposed individuals (such as the upper vs. lower lobe or a central vs. peripheral localization), and all major histological types of lung carcinoma occur in asbestos-exposed individuals in comparison to nonexposed subjects, with no significant differences in the immunophenotypes, and no clear or diagnostic differences in the molecular-genetic profiles (see later discussion).
- The relationship between asbestos and lung cancer in general is governed by a near-linear dose-response relationship, with no clearly delineated threshold, but the gradient of the dose-response line is less steep than the analogous dose-response line between asbestos and pleural malignant mesothelioma. From Gustavsson’s meticulous case-referent studies in Stockholm County [28,29], there appears to be some evidence that the dose-response gradient for lung cancer is steeper at low cumulative exposures than at higher exposure [29] (see also [17,30], and later discussion in this review). In their 2000 review of 17 cohort studies, Hodgson and Darnton commented that if a threshold does apply to lung cancer induction by amphibole asbestos, ‘it must be very low’, where as a threshold for chrysotile—a ‘zero or at least very low risk’—is ‘strongly arguable’ (but they commented that the asbestos-related dose-response effect for lung cancer in the Charleston textile cohort is ‘untypically high’ [31]). In contrast, some other studies have also found a higher dose-response effect for asbestos textile workers than for other chrysotile exposures [32,33].
- It is accepted that tobacco (especially cigarette) smoke and asbestos functionally interact in the causation of lung cancer; however, the type and strength of this interaction have occasionally been the issue if debate [34]. It is our opinion that the effect is synergistic, i.e., the combined effect is greater than the sum of the individual effects. By definition and in biological terms, the difference (the synergistic effect) cannot be apportioned back to each of these individual carcinogens; this issue is explored further in a later section of this review.
- It is our opinion on the balance of probabilities that asbestosis is not a necessary prerequisite for the attribution of lung cancer to asbestos in an asbestos-exposed smoker.
2. Materials and Methods
3. Discussion
3.1. Lung Cancer and Cumulative Asbestos Exposure, with or without Asbestosis-Source Epidemiological Data
3.2. A Peer Opinion Regarding the Requirement for Asbestosis for Attribution of Lung Cancer
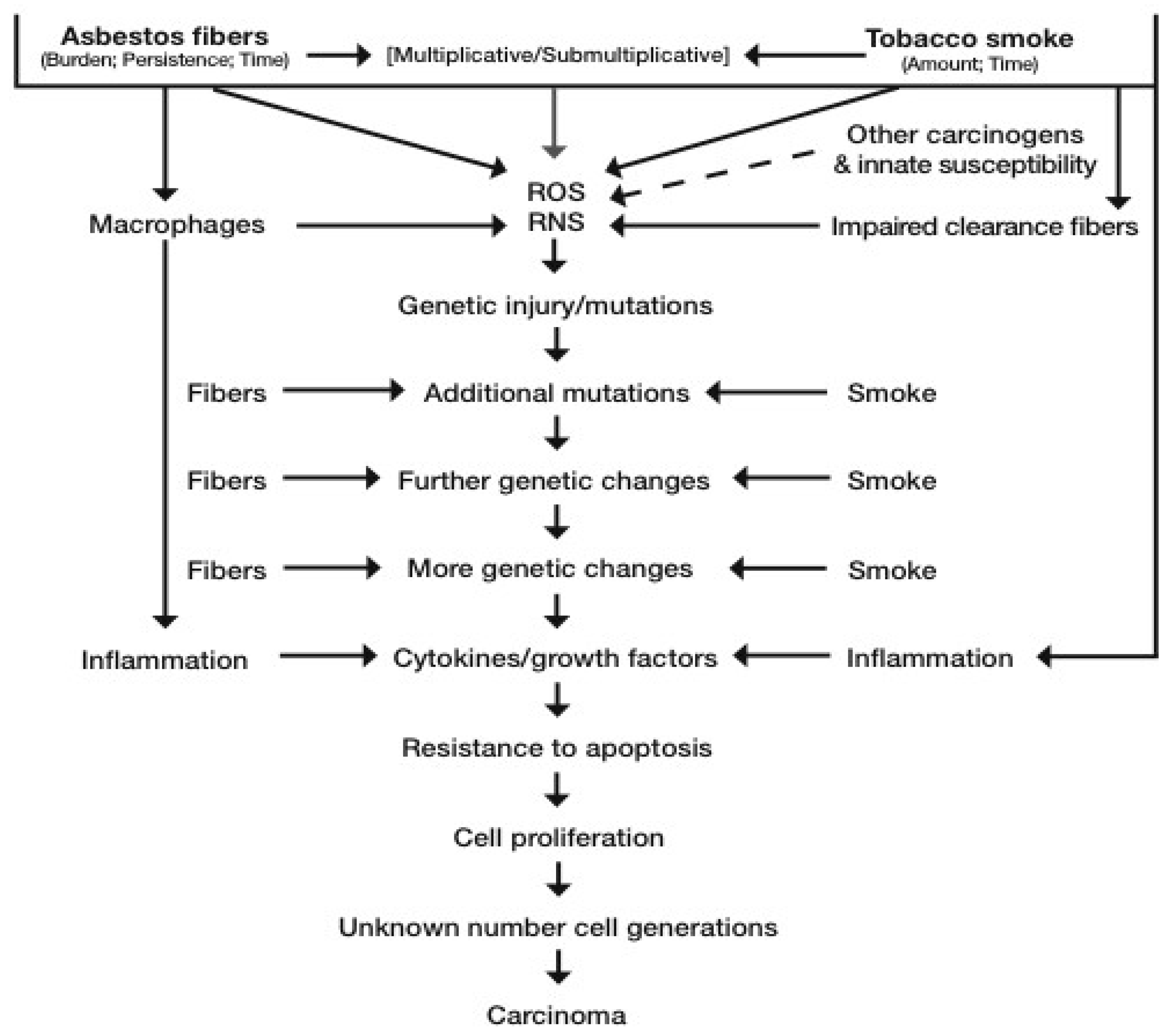
3.3. The Pathogenesis of, and Some Molecular Alterations in, Asbestos-Related Lung Cancer
3.4. The Synergy between Asbestos Fibers and Tobacco Smoke for Lung Cancer Causation Epidemiological Data
3.4.1. The Synergy between Asbestos Fibers and Tobacco Smoke for Lung Cancer Causation: Biological Data
- (1)
- asbestos contributes to improved uptake of chemical carcinogens in cigarette smoke and their metabolism to carcinogenic metabolites in lung epithelial cells
- (2)
- inhibition of clearance and retention of carcinogens
- (3)
- chronic inflammation that drives development and metastases of lung tumors
- (4)
- Direct synergistic effects on proliferation
3.4.2. The Synergy between Asbestos Fibers and Tobacco Smoke for Lung Cancer Causation–Animal Studies
3.4.3. The Synergy between Asbestos Fibers and Tobacco Smoke for Lung Cancer Causation—Studies in Humans
3.4.4. The Synergy between Asbestos Fibers and Tobacco Smoke for Lung Cancer Causation: Summary
3.5. Relevance of Estimates of Cumulative Asbestos Exposure to Causal Attribution and Lung Cancer Risk
- Dispute as to whether there is genuine interstitial fibrosis, even in high-resolution CT scans [132], or whether any changes are related to associated pleural fibrosis.
- Dispute as to whether interstitial fibrosis represents asbestosis or idiopathic pulmonary fibrosis (usual interstitial pneumonia [UIP]) or nonspecific interstitial pneumonia [NSIP] unrelated to asbestos, especially when pleural plaques are not demonstrable [133].
- Even on histologic examination, disagreement between pathologists as to whether there is genuine interstitial fibrosis in a distribution appropriate for asbestosis, i.e., a UIP pattern or the fibrotic variant of NSIP, or diffuse interstitial fibrosis which is not readily classifiable as either [133], or whether there are sufficient asbestos bodies for that diagnosis [134].
3.6. Problems with Numerical Assessments of Asbestos Exposure
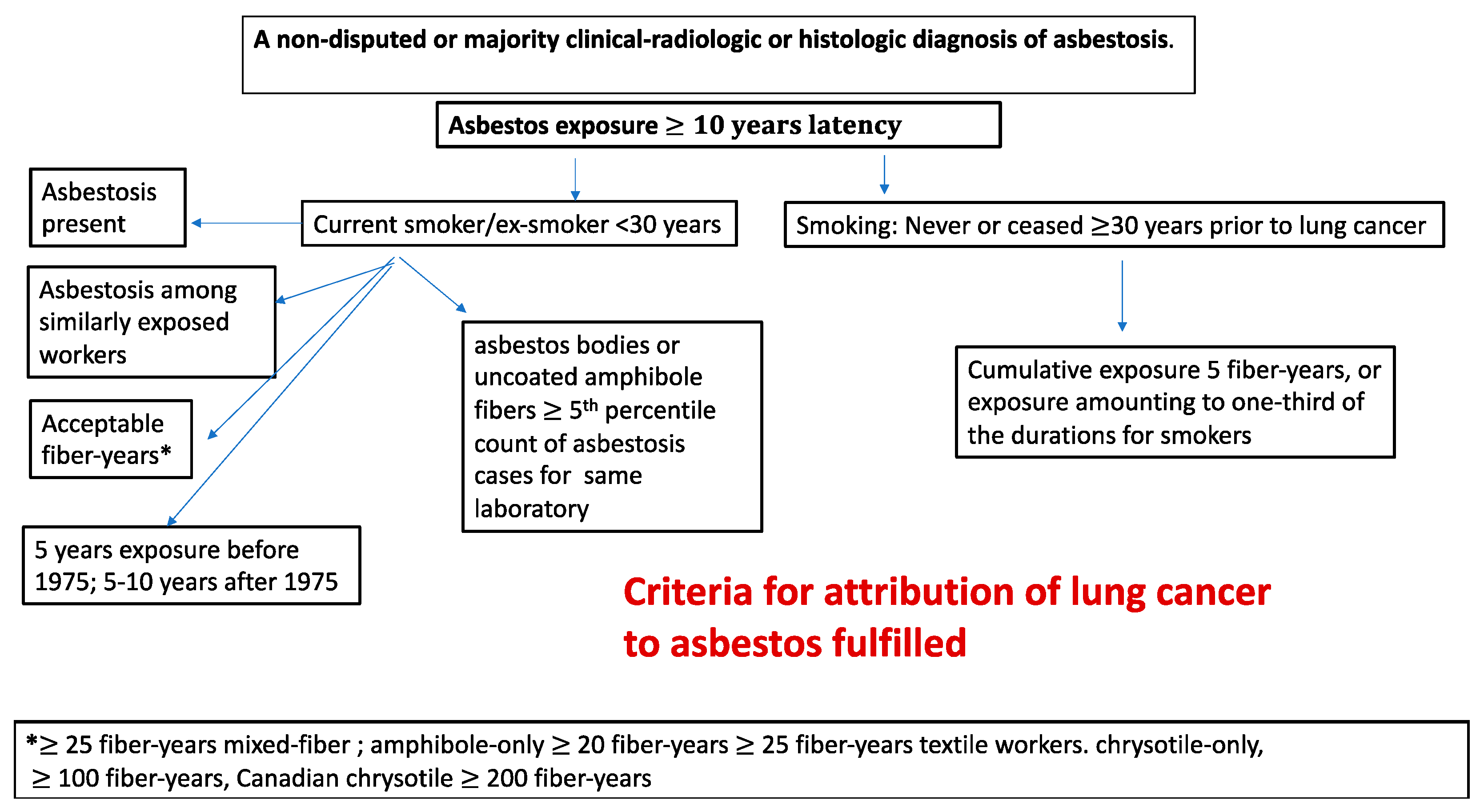
3.7. Proposed Criteria for Attribution of Lung Cancers to Exposure to Asbestos
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A
Definitions of Additive and Multiplicative Models
References
- Henderson, D.W.; Leigh, J. The history of asbestos utilization and recognition of asbestos-induced diseases. In Asbestos: Risk Assessment, Epidemiology and Health Effects, 2nd ed.; Dodson, R.F., Hammar, S.P., Eds.; CRC/Taylor&Francis: Boca Raton, FL, USA, 2011; pp. 1–22. [Google Scholar]
- Lynch, K.M.; Smith, W.A. Pulmonary asbestosis III: Carcinoma of lung in asbesto-silicosis. Am. J. Cancer 1935, 24, 56–64. [Google Scholar] [CrossRef]
- Gloyne, S.R. Two cases of squamous carcinoma of the lung occurring in asbestosis. Tubercle 1935, 17, 5–10. [Google Scholar] [CrossRef]
- Egbert, D.S.; Geiger, A.J. Pulmonary asbestosis and carcinoma. Report of a case with necropsy findings. Am. Rev. Tuberc. 1936, 34, 143–150. [Google Scholar]
- Gloyne, S.R. A case of oat cell carcinoma of the lung occurring in asbestosis. Tubercle 1936, 18, 100–101. [Google Scholar] [CrossRef]
- Proctor, R.N. The Nazi War on Cancer; Princeton University Press: Princeton, NJ, USA, 1999; pp. 73–119. [Google Scholar]
- Nordmann, M.; Sorge, A. Lungenkrebs durch Asbeststaub im Tierversuch. Z Krebsforsch 1941, 51, 170. [Google Scholar] [CrossRef]
- Enterline, P.E. Changing attitudes and opinions regarding asbestos and cancer 1934–1965. Am. J. Ind. Med. 1991, 20, 685–700. [Google Scholar] [CrossRef]
- Doll, R. Mortality from lung cancer in asbestos workers. Br. J. Ind. Med. 1955, 12, 81–86. [Google Scholar] [CrossRef]
- Gloyne, S.R. Pneumoconiosis. A histological survey of necropsy material in 1205 cases. Lancet 1951, 1, 810–814. [Google Scholar]
- Henderson, D.W.; Roggli, V.L.; Shilkin, K.B.; Hammar, S.P.; Leigh, J. Is asbestosis an obligate precursor for asbestos-induced lung cancer? In Sourcebook on Asbestos Diseases; Peters, G.A., Peters, B.J., Eds.; Michie Company: Charlottesville, VA, USA, 1995; Volume 11, pp. 97–168. [Google Scholar]
- Leigh, J.; Berry, G.; de Klerk, N.H.; Henderson, D.W. Asbestos-related lung cancer: apportionment of causation and damages to asbestos and tobacco smoke. In Sourcebook on Asbestos Diseases; Peters, G.A., Peters, B.J., Eds.; Michie: Charlottesville, VA, USA, 1996; Volume 13, pp. 141–166. [Google Scholar]
- Henderson, D.W.; de Klerk, N.H.; Hammar, S.P.; Hillerdal, G.; Huuskonen, M.; Leigh, J.; Pott, F.; Roggli, V.L.; Shilkin, K.B.; Tossavainen, A. Asbestos and lung cancer: is it attributable to asbestosis, or to asbestos fiber burden? In Pathology of Lung Tumors; Corrin, B., Ed.; Churchill Livingstone: New York, NY, USA, 1997; pp. 83–118. [Google Scholar]
- Hillerdal, G.; Henderson, D.W. Asbestos, asbestosis, pleural plaques and lung cancer. Scand. J. Work Environ. Health 1997, 23, 93–103. [Google Scholar] [CrossRef]
- Henderson, D.W.; Leigh, J. Asbestos and lung cancer: A selective up-date to The Helsinki Criteria for individual attribution. In People and Work Research Reports 36; Finnish Institute for Occupational Health: Helsinki, Finland, 2000; Volume 36, pp. 3–18. [Google Scholar]
- Henderson, D.W.; Leigh, J. An Australian Perspective on The Helsinki Criteria for the attribution of lung cancer to asbestos. Pathol. Int. 2004, 54 (Suppl. 1), S442–S451. [Google Scholar]
- Henderson, D.W.; Rodelsperger, K.; Woitowitz, H.J.; Leigh, J. After Helsinki: A multidisciplinary review of the relationship between asbestos exposure and lung cancer, with emphasis on studies published during 1997–2004. Pathology 2004, 36, 517–550. [Google Scholar] [CrossRef] [PubMed]
- Leigh, J.; Henderson, D. Lung cancer related to asbestos exposure: Causation and compensation. J. Occup. Health Saf. 2006, 22, 449–462. [Google Scholar]
- Hammar, S.P.; Lemen, R.A.; Henderson, D.W.; Leigh, J. Asbestos and other cancers. In Asbestos: Risk Assessment, Epidemiology and Health Effects, 2nd ed.; Dodson, R.F., Hammar, S.P., Eds.; CRC Press/Taylor&Francis: Boca Raton, FL, USA, 2011; pp. 419–446. [Google Scholar]
- Klebe, S.; Henderson, D.W. The Molecular Pathogenesis of Asbestos-Related Disorders. In Asbestos: Risk, Assessment, Epidemiology, Health and Effects, 2nd ed.; Taylor & Francis: Boca Raton, FL, USA, 2011; pp. 109–130. [Google Scholar]
- Henderson, D.W. Commentary regarding the article by Fischer et al.: Fibre years, pulmonary asbestos burden and asbestosis. Int. J. Hyg. Environ. Health 2003, 206, 249–250. [Google Scholar] [CrossRef] [PubMed]
- Green, F.H.; Harley, R.; Vallyathan, V.; Althouse, R.; Fick, G.; Dement, J.; Mitha, R.; Pooley, F. Exposure and mineralogical correlates of pulmonary fibrosis in chrysotile asbestos workers. Occup. Environ. Med. 1997, 54, 549–559. [Google Scholar] [CrossRef]
- Dement, J.M.; Brown, D.P.; Okun, A. Follow-up study of chrysotile asbestos textile workers: Cohort mortality and case-control analyses. Am. J. Ind. Med. 1994, 26, 431–447. [Google Scholar] [CrossRef]
- Husain, A.N.; Colby, T.V.; Ordonez, N.G.; Allen, T.C.; Attanoos, R.L.; Beasley, M.B.; Butnor, K.J.; Chirieac, L.R.; Churg, A.M.; Dacic, S.; et al. Guidelines for Pathologic Diagnosis of Malignant Mesothelioma 2017 Update of the Consensus Statement From the International Mesothelioma Interest Group. Arch. Pathol. Lab. Med. 2018, 142, 89–108. [Google Scholar] [CrossRef]
- Marchevsky, A.M.; LeStang, N.; Hiroshima, K.; Pelosi, G.; Attanoos, R.; Churg, A.; Chirieac, L.; Dacic, S.; Husain, A.; Khoor, A.; et al. The differential diagnosis between pleural sarcomatoid mesothelioma and spindle cell/pleomorphic (sarcomatoid) carcinomas of the lung: Evidence-based guidelines from the International Mesothelioma Panel and the MESOPATH National Reference Center. Hum. Pathol. 2017, 67, 160–168. [Google Scholar] [CrossRef]
- Bjorkqvist, A.M.; Tammilehto, L.; Nordling, S.; Nurminen, M.; Anttila, S.; Mattson, K.; Knuutila, S. Comparison of DNA copy number changes in malignant mesothelioma, adenocarcinoma and large-cell anaplastic carcinoma of the lung. Br. J. Cancer 1998, 77, 260–269. [Google Scholar] [CrossRef]
- Driscoll, T.; Nelson, D.I.; Steenland, K.; Leigh, J.; Concha-Barrientos, M.; Fingerhut, M.; Pruss-Ustun, A. The global burden of disease due to occupational carcinogens. Am. J. Ind. Med. 2005, 48, 419–431. [Google Scholar] [CrossRef]
- Gustavsson, P.; Jakobsson, R.; Nyberg, F.; Pershagen, G.; Jarup, L.; Scheele, P. Occupational exposure and lung cancer risk: A population-based case-referent study in Sweden. Am. J. Epidemiol. 2000, 152, 32–40. [Google Scholar] [CrossRef]
- Gustavsson, P.; Nyberg, F.; Pershagen, G.; Scheele, P.; Jakobsson, R.; Plato, N. Low-dose exposure to asbestos and lung cancer: Dose-response relations and interaction with smoking in a population-based case-referent study in Stockholm, Sweden. Am. J. Epidemiol. 2002, 155, 1016–1022. [Google Scholar] [CrossRef]
- Henderson, D.W.; Leigh, J. Asbestos and carcinoma of the lung. In Asbestos: Risk Assessment, Epidemiology and Health Effects, 2nd ed.; Dodson, R.F., Hammar, S.P., Eds.; CRC Press/Taylor&Francis: Boca Raton, FL, USA, 2011; pp. 269–306. [Google Scholar]
- Hodgson, J.T.; Darnton, A. The quantitative risks of mesothelioma and lung cancer in relation to asbestos exposure. Ann. Occup. Hyg. 2000, 44, 565–601. [Google Scholar] [CrossRef]
- Pira, E.; Pelucchi, C.; Buffoni, L.; Palmas, A.; Turbiglio, M.; Negri, E.; Piolatto, P.G.; La Vecchia, C. Cancer mortality in a cohort of asbestos textile workers. Br. J. Cancer 2005, 92, 580–586. [Google Scholar] [CrossRef] [PubMed]
- World Trade Organization (WTO). European Communities-Measures Concerning Asbestos and Asbestos-containing Products; WTO: Geneva, Switzerland, 2000. [Google Scholar]
- El Zoghbi, M.; Salameh, P.; Stücker, I.; Brochard, P.; Delva, F.; Lacourt, A. Absence of multiplicative interactions between occupational lung carcinogens and tobacco smoking: A systematic review involving asbestos, crystalline silica and diesel engine exhaust emissions. BMC Public Health 2017, 17, 156. [Google Scholar] [CrossRef] [PubMed]
- Hessel, P.A.; Gamble, J.F.; McDonald, J.C. Asbestos, asbestosis, and lung cancer: A critical assessment of the epidemiological evidence. Thorax 2005, 60, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, L.S.; Baelum, J.; Rasmussen, J.; Dahl, S.; Olsen, K.E.; Albin, M.; Hansen, N.C.; Sherson, D. Occupational asbestos exposure and lung cancer—A systematic review of the literature. Arch. Environ. Occup. Health 2014, 69, 191–206. [Google Scholar] [CrossRef]
- Markowitz, S.B.; Levin, S.M.; Miller, A.; Morabia, A. Asbestos, asbestosis, smoking, and lung cancer. New findings from the North American insulator cohort. Am. J. Respir. Crit. Care Med. 2013, 188, 90–96. [Google Scholar] [CrossRef]
- Ross, R. Asbestosis, not asbestos exposure, is the primary risk factor for lung cancer. Am. J. Respir. Crit. Care Med. 2014, 189, 114–115. [Google Scholar] [CrossRef]
- Geyer, S.J. Asbestos, asbestosis, smoking, and lung cancer: Study bias and confounding issues that complicate the interpretation of the results. Am. J. Respir. Crit. Care Med. 2014, 189, 115–116. [Google Scholar] [CrossRef]
- Markowitz, S.; Morabia, A.; Miller, A. Reply: To PMID 23590275. Am. J. Respir. Crit. Care Med. 2014, 189, 116–117. [Google Scholar] [CrossRef]
- Balmes, J.R. Asbestos and lung cancer: What we know. Am. J. Respir. Crit. Care Med. 2013, 188, 8–9. [Google Scholar] [CrossRef] [PubMed]
- Frost, G.; Darnton, A.; Harding, A.H. The effect of smoking on the risk of lung cancer mortality for asbestos workers in Great Britain (1971-2005). Ann. Occup. Hyg. 2011, 55, 239–247. [Google Scholar] [CrossRef][Green Version]
- Olsson, A.C.; Vermeulen, R.; Schuz, J.; Kromhout, H.; Pesch, B.; Peters, S.; Behrens, T.; Portengen, L.; Mirabelli, D.; Gustavsson, P.; et al. Exposure-Response Analyses of Asbestos and Lung Cancer Subtypes in a Pooled Analysis of Case-Control Studies. Epidemiology 2017, 28, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Karjalainen, A.; Anttila, S.; Vanhala, E.; Vainio, H. Asbestos exposure and the risk of lung cancer in a general urban population. Scand. J. Work Environ. Health 1994, 20, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Churg, A. Neoplastic asbestos induced disease. In Pathology of Occupational Lung Disease, 2nd ed.; Churg, A., Green, F.H., Eds.; Williams & Wilkins: Baltimore, MD, USA, 1998; pp. 339–391. [Google Scholar]
- Tossaavainen, A. Asbestos, asbestosis, and cancer: The Helsinki criteria for diagnosis and attribution. Scand. J. Work Environ. Health 1997, 23, 311–316. [Google Scholar]
- Weiss, W. Asbestos-related pleural plaques and lung cancer. Chest 1993, 103, 1854–1859. [Google Scholar] [CrossRef]
- Nurminen, M.; Tossavainen, A. Is there an association between pleural plaques and lung cancer without asbestosis? Scand. J. Work Environ. Health 1994, 20, 62–64. [Google Scholar] [CrossRef]
- Nurminen, M.; Tossavainen, A. Asbestos-related pleural plaques and lung cancer. Chest 1994, 106, 648–649. [Google Scholar] [CrossRef]
- Weiss, W. Asbestosis: A marker for the increased risk of lung cancer among workers exposed to asbestos. Chest 1999, 115, 536–549. [Google Scholar] [CrossRef]
- Cagle, P.T. Criteria for attributing lung cancer to asbestos exposure. Am. J. Clin. Pathol. 2002, 117, 9–15. [Google Scholar] [CrossRef]
- Gibbs, A.; Attanoos, R.L.; Churg, A.; Weill, H. The Helsinki criteria for attribution of lung cancer to asbestos exposure: How robust are the criteria? Arch. Pathol. Lab. Med. 2007, 131, 181–183. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.M.; Weill, H. Asbestosis as a precursor of asbestos related lung cancer: Results of a prospective mortality study. Br. J. Ind. Med. 1991, 48, 229–233. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Banks, D.E.; Shi, R.; McLarty, J.; Cowl, C.T.; Smith, D.; Tarlo, S.M.; Daroowalla, F.; Balmes, J.; Baumann, M. American College of Chest Physicians consensus statement on the respiratory health effects of asbestos. Results of a Delphi study. Chest 2009, 135, 1619–1627. [Google Scholar] [CrossRef] [PubMed]
- Arsenic, Metals, Fibres and Dust International Agency for Research on Cancer (IARC) Monographs; International Agency for Research on Cancer: Lyon, France, 2012.
- Kennedy, A.R.; Little, J.B. The transport and localization of benzo(a)pyrene-hematite and hematite-210Po in the hamster lung following intratracheal instillation. Cancer Res. 1974, 34, 1344–1352. [Google Scholar] [PubMed]
- Eastman, A.; Mossman, B.T.; Bresnick, E. Formation and removal of benzo(a)pyrene adducts of DNA in hamster tracheal epithelial cells. Cancer Res. 1981, 41, 2605–2610. [Google Scholar]
- Mossman, B.T.; Eastman, A.; Landesman, J.M.; Bresnick, E. Effects of crocidolite and chrysotile asbestos on cellular uptake and metabolism of benzo(a)pyrene in hamster tracheal epithelial cells. Environ. Health Perspect. 1983, 51, 331–335. [Google Scholar] [CrossRef]
- Eastman, A.; Mossman, B.T.; Bresnick, E. Influence of asbestos on the uptake of benzo(a)pyrene and DNA alkylation in hamster tracheal epithelial cells. Cancer Res. 1983, 43, 1251–1255. [Google Scholar]
- Churg, A. The uptake of mineral particles by pulmonary epithelial cells. Am. J. Respir. Crit. Care Med. 1996, 154, 1124–1140. [Google Scholar] [CrossRef]
- Churg, A.; Sun, J.; Zay, K. Cigarette smoke increases amosite asbestos fiber binding to the surface of tracheal epithelial cells. Am. J. Physiol. 1998, 275, L502–L508. [Google Scholar] [CrossRef]
- McFadden, D.; Wright, J.; Wiggs, B.; Churg, A. Cigarette smoke increases the penetration of asbestos fibers into airway walls. Am. J. Pathol. 1986, 123, 95–99. [Google Scholar]
- McFadden, D.; Wright, J.L.; Wiggs, B.; Churg, A. Smoking inhibits asbestos clearance. Am. Rev. Respir. Dis. 1986, 133, 372–374. [Google Scholar] [CrossRef] [PubMed]
- Gulino, G.R.; Polimeni, M.; Prato, M.; Gazzano, E.; Kopecka, J.; Colombatto, S.; Ghigo, D.; Aldieri, E. Effects of Chrysotile Exposure in Human Bronchial Epithelial Cells: Insights into the Pathogenic Mechanisms of Asbestos-Related Diseases. Environ. Health Perspect 2016, 124, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Nelson, H.H.; Christiani, D.C.; Wiencke, J.K.; Mark, E.J.; Wain, J.C.; Kelsey, K.T. k-ras mutation and occupational asbestos exposure in lung adenocarcinoma: Asbestos-related cancer without asbestosis. Cancer Res. 1999, 59, 4570–4573. [Google Scholar] [PubMed]
- Kettunen, E.; Aavikko, M.; Nymark, P.; Ruosaari, S.; Wikman, H.; Vanhala, E.; Salmenkivi, K.; Pirinen, R.; Karjalainen, A.; Kuosma, E.; et al. DNA copy number loss and allelic imbalance at 2p16 in lung cancer associated with asbestos exposure. Br. J. Cancer 2009, 100, 1336–1342. [Google Scholar] [CrossRef]
- Wolff, H.; Vehmas, T.; Oksa, P.; Rantanen, J.; Vainio, H. Asbestos, asbestosis, and cancer, the Helsinki criteria for diagnosis and attribution 2014: Recommendations. Scand. J. Work Environ. Health 2015, 41, 5–15. [Google Scholar] [CrossRef]
- Selikoff, I.J.; Hammond, E.C.; Churg, J. Asbestos exposure, smoking, and neoplasia. JAMA 1968, 204, 106–112. [Google Scholar] [CrossRef]
- Ngamwong, Y.; Tangamornsuksan, W.; Lohitnavy, O.; Chaiyakunapruk, N.; Scholfield, C.N.; Reisfeld, B.; Lohitnavy, M. Additive Synergism between Asbestos and Smoking in Lung Cancer Risk: A Systematic Review and Meta-Analysis. PLoS ONE 2015, 10, e0135798. [Google Scholar] [CrossRef]
- Hammond, E.C.; Selikoff, I.J.; Seidman, H. Asbestos exposure, cigarette smoking and death rates. Ann. New York Acad. Sci. 1979, 330, 473–490. [Google Scholar] [CrossRef]
- Nurminen, M.; Karjalainen, A. Epidemiologic estimate of the proportion of fatalities related to occupational factors in Finland. Scand. J. Work Environ. Health 2001, 27, 161–213. [Google Scholar] [CrossRef]
- Richie, G.; Tony, B.; John, E.; Robert, K.; Bernard, W.S. Occupational Cancer: A Giude to Prevention, Assesment and Investigation; The royal Australasian college of physicians: Sydney, NSW, Australia, 2003; p. 36. [Google Scholar]
- Vainio, H.; Boffetta, P. Mechanisms of the combined effect of asbestos and smoking in the etiology of lung cancer. Scand. J. Work Environ. Health 1994, 20, 235–242. [Google Scholar] [CrossRef]
- Liddell, F.D. Joint action of smoking and asbestos exposure on lung cancer. Occup. Environ. Med. 2002, 59, 494–495. [Google Scholar] [CrossRef]
- Liddell, F.D.; Armstrong, B.G. The combination of effects on lung cancer of cigarette smoking and exposure in quebec chrysotile miners and millers. Ann. Occup. Hyg. 2002, 46, 5–13. [Google Scholar] [CrossRef]
- Lee, P.N. Relation between exposure to asbestos and smoking jointly and the risk of lung cancer. Occup. Environ. Med. 2001, 58, 145–153. [Google Scholar] [CrossRef]
- Jockel, K.H.; Ahrens, W.; Jahn, I.; Pohlabeln, H.; Bolm-Audorff, U. Occupational risk factors for lung cancer: A case-control study in West Germany. Int. J. Epidemiol. 1998, 27, 549–560. [Google Scholar] [CrossRef]
- Keeling, B.; Hobson, J.; Churg, A. Effects of cigarette smoke on epithelial uptake of non-asbestos mineral particles in tracheal organ culture. Am. J. Respir. Cell Mol. Biol. 1993, 9, 335–340. [Google Scholar] [CrossRef]
- Michael, A.; John, A.B.; Ross, C.B.; Patricia, A.B. Tobacco Smoking and Involuntary Smoking International Agency for Research on Cancer (IARC) Monographs; International Agency for Research on Cancer: Lyon, France, 2004. [Google Scholar]
- Conway, E.M.; Pikor, L.A.; Kung, S.H.; Hamilton, M.J.; Lam, S.; Lam, W.L.; Bennewith, K.L. Macrophages, Inflammation, and Lung Cancer. Am. J. Respir. Crit. Care Med. 2016, 193, 116–130. [Google Scholar] [CrossRef]
- Jaurand, M.C. Mechanisms of fiber-induced genotoxicity. Environ. Health Perspect. 1997, 105 (Suppl. 5), 1073–1084. [Google Scholar] [CrossRef]
- Kamp, D.W.; Weitzman, S.A. The molecular basis of asbestos induced lung injury. Thorax 1999, 54, 638–652. [Google Scholar] [CrossRef]
- Hei, T.K.; Wu, L.J.; Piao, C.Q. Malignant transformation of immortalized human bronchial epithelial cells by asbestos fibers. Environ. Health Perspect. 1997, 105 (Suppl. 5), 1085–1088. [Google Scholar] [CrossRef]
- Mossman, B.T.; Lippmann, M.; Hesterberg, T.W.; Kelsey, K.T.; Barchowsky, A.; Bonner, J.C. Pulmonary endpoints (lung carcinomas and asbestosis) following inhalation exposure to asbestos. J. Toxicol. Environ. Health B Crit. Rev. 2011, 14, 76–121. [Google Scholar] [CrossRef]
- Harrison, P.T.; Hoskins, J.A.; Brown, R.C.; Pigott, G.H.; Hext, P.M.; Mugglestone, M.A. Hyperplastic and Neoplastic Changes in the Lungs of Rats Treated Concurrently with Chrysotile Asbestos and N-Nitrosoheptamethyleneimine. Inhal. Toxicol. 2000, 12 (Suppl. 3), 167–172. [Google Scholar] [CrossRef]
- Loli, P.; Topinka, J.; Georgiadis, P.; Dusinska, M.; Hurbankova, M.; Kovacikova, Z.; Volkovova, K.; Wolff, T.; Oesterle, D.; Kyrtopoulos, S.A. Benzo[a]pyrene-enhanced mutagenesis by asbestos in the lung of lambda-lacI transgenic rats. Mutat. Res. 2004, 553, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Nelson, H.H.; Kelsey, K.T. The molecular epidemiology of asbestos and tobacco in lung cancer. Oncogene 2002, 21, 7284–7288. [Google Scholar] [CrossRef] [PubMed]
- John, C.B.; Peter, R.B.; Robert, G.C.; Arthur, F.; Robert, F.H.; Karl, T.K. Asbestos: Slelected Cancers; US National Academy of Sciences Committee: Washington, DC, USA, 2006. [Google Scholar]
- Guidotti, T.L. Apportionment in asbestos-related disease for purposes of compensation. Ind. Health 2002, 40, 295–311. [Google Scholar] [CrossRef] [PubMed]
- Churg, A.; Stevens, B. Enhanced retention of asbestos fibers in the airways of human smokers. Am. J. Respir. Crit. Care Med. 1995, 151, 1409–1413. [Google Scholar] [CrossRef] [PubMed]
- Valavanidis, A.; Balomenou, H.; Macropoulou, I.; Zarodimos, I. A study of the synergistic interaction of asbestos fibers with cigarette tar extracts for the generation of hydroxyl radicals in aqueous buffer solution. Free Radic. Biol. Med. 1996, 20, 853–858. [Google Scholar] [CrossRef]
- Spitz, D.R.; Hauer-Jensen, M. Ionizing radiation-induced responses: Where free radical chemistry meets redox biology and medicine. Antioxid. Redox Signal. 2014, 20, 1407–1409. [Google Scholar] [CrossRef]
- Hammar, S.P.; Henderson, D.W.; Klebe, S.; Dodson, R.F. Neoplasms of the pleural. In Dail and Hammar’s Pulmonary Pathology, 3rd ed.; Tomashefski, J.F., Ed.; Springer: New York, NY, USA, 2008; pp. 558–734. [Google Scholar]
- Kimizuka, G.; Azuma, M.; Ishibashi, M.; Shinozaki, K.; Hayashi, Y. Co-carcinogenic effect of chrysotile and amosite asbestos with benzo(a)pyrene in the lung of hamsters. Acta Pathol. Jpn. 1993, 43, 149–153. [Google Scholar] [CrossRef]
- Irani, Y.D.; Pulford, E.; Mortimer, L.; Irani, S.; Butler, L.; Klebe, S.; Williams, K.A. Sex differences in corneal neovascularization in response to superficial corneal cautery in the rat. PLoS ONE 2019, 14, e0221566. [Google Scholar] [CrossRef]
- NHMRC. Best Practice Methodology in the Use of Animals for Scientific Purposes; Australian Research Council, Ed.: Canberra, Austria, 2017. [Google Scholar]
- Topinka, J.; Loli, P.; Dusinska, M.; Hurbankova, M.; Kovacikova, Z.; Volkovova, K.; Kazimirova, A.; Barancokova, M.; Tatrai, E.; Wolff, T.; et al. Mutagenesis by man-made mineral fibres in the lung of rats. Mutat. Res. 2006, 595, 174–183. [Google Scholar] [CrossRef]
- Morris, G.F.; Danchuk, S.; Wang, Y.; Xu, B.; Rando, R.J.; Brody, A.R.; Shan, B.; Sullivan, D.E. Cigarette smoke represses the innate immune response to asbestos. Physiol. Rep. 2015, 3, e12652. [Google Scholar] [CrossRef] [PubMed]
- Kettunen, E.; Hernandez-Vargas, H.; Cros, M.P.; Durand, G.; Le Calvez-Kelm, F.; Stuopelyte, K.; Jarmalaite, S.; Salmenkivi, K.; Anttila, S.; Wolff, H.; et al. Asbestos-associated genome-wide DNA methylation changes in lung cancer. Int. J. Cancer 2017, 141, 2014–2029. [Google Scholar] [CrossRef] [PubMed]
- Nelson, H.H.; Wiencke, J.K.; Gunn, L.; Wain, J.C.; Christiani, D.C.; Kelsey, K.T. Chromosome 3p14 alterations in lung cancer: Evidence that FHIT exon deletion is a target of tobacco carcinogens and asbestos. Cancer Res. 1998, 58, 1804–1807. [Google Scholar] [PubMed]
- Doll, N.J.; Peto, J. Asbestos: Effects of health of exposure to asbestos. A report to the health an safety commission. Occup. Environ. Med. 1996, 53, 160–167. [Google Scholar]
- Burdorf, A.; Swuste, P. An expert system for the evaluation of historical asbestos exposure as diagnostic criterion in asbestos-related diseases. Ann. Occup. Hyg. 1999, 43, 57–66. [Google Scholar] [CrossRef]
- Dement, J.M.; Harris Jr, R.L.; Symons, M.J.; Shy, C.M. Exposures and mortality among chrysotile asbestos workers. Part II: Mortality. Am. J. Ind. Med. 1983, 4, 421–433. [Google Scholar] [CrossRef]
- De Vuyst, P. Asbestos, Asbestosis and Lung Cancer: Guidelines for the Attribution of Lung Cancer to Asbestos; Finnish Institute of Occupational Health: Helsinki, Finland, 1997; pp. 92–96. [Google Scholar]
- BK-Report. Faserjahre. Bearbeitungshinweises Zur Berufskrankheit Nr 4104; Hessische Verwaltung für Bodenmanagement und Geoinformation: Wiesbaden, Germany, 1994. [Google Scholar]
- Lash, T.L.; Crouch, E.A.; Green, L.C. A meta-analysis of the relation between cumulative exposure to asbestos and relative risk of lung cancer. Occup. Environ. Med. 1997, 54, 254–263. [Google Scholar] [CrossRef]
- Stayner, L.; Smith, R.; Bailer, J.; Gilbert, S.; Steenland, K.; Dement, J.; Brown, D.; Lemen, R. Exposure-response analysis of risk of respiratory disease associated with occupational exposure to chrysotile asbestos. Occup. Environ. Med. 1997, 54, 646–652. [Google Scholar] [CrossRef]
- McDonald, A.D.; Fry, J.S.; Wooley, A.J.; Mcdonald, J.C. Dust exposure and mortality in an American chrysotile asbestos textile plant. Br. J. Ind. Med. 1983, 40, 361–367. [Google Scholar]
- Robins, J.M.; Greenland, S. Estimability and estimation of excess and etiologic fractions. Stat. Med. 1989, 8, 845–859. [Google Scholar] [CrossRef]
- Greenland, S. Relation of probability of causation to relative risk and doubling dose: A methodologic error that has become a social problem. Am. J. Public Health 1999, 89, 1166–1169. [Google Scholar] [CrossRef]
- Bryant, A.H.; Reinert, A. Epidemiology in the legal arena and the search for truth. Am. J. Epidemiol. 2001, 154, S27–S35. [Google Scholar] [CrossRef]
- Soskolne, C.J.; Lilienfeld, D.E.; Black, B.; Ingvarsen, S. Advances in Epidemiology in Legal Proceedings in the United States. In Modern Environmental Toxicology; Mehlman, M.A., Upton, A., Eds.; National Academical Press: Washington, DC, USA, 1994; Volumes Vl–XXII, pp. 101–115. [Google Scholar]
- Armstrong, B.; Theriault, G. Compensating lung cancer patients occupationally exposed to coal tar pitch volatiles. Occup. Environ. Med. 1996, 53, 160–167. [Google Scholar] [CrossRef]
- Begin, R. Asbestos exposure and pleuropulmonary cancer. Rev. Mal. Respir. 1998, 15, 723–730. [Google Scholar]
- Attfield, M.D.; Borm, P.J.; Checkoway, H.; Donaldson, K.; Dosemeci, M.; Feron, V.J. Silica, Some Silocates, Coal Dust and Para-Aramid Fibrils; International Agency for Research on Cancer: Lyon, France, 1997. [Google Scholar]
- David M., D.; Karam, E.B.; Eric, G.; Per, G.; Uwe, H.; Charles, W.J.; Christopher, J.P. Diesel and Gasoline Engine Exhausts and Some Nitroarenes; International Agency for Research on Cancer: Lyon, France, 2013. [Google Scholar]
- Bradford, C.M. A discussion on the association of disease with environment with special reference to Great Britain. R. Inst. Public Health Hyg. J. 1965, 28, 203–218. [Google Scholar]
- Rothman, K.J.; Greenland, S. Modern Epidemiology; Lippincott-Raven: Philadelphia, PA, USA, 1998. [Google Scholar]
- Seidman, H.; Selikoff, I.J.; Gelb, S.K. Mortality experience of amosite asbestos factory workers: Dose-response relationships 5 to 40 years after onset of short-term work exposure. Am. J. Ind. Med. 1986, 10, 479–514. [Google Scholar] [CrossRef]
- Rodelsperger, K.; Woitowitz, H.J. Airborne fibre concentrations and lung burden compared to the tumour response in rats and humans exposed to asbestos. Ann. Occup. Hyg. 1995, 39, 715–725. [Google Scholar] [CrossRef]
- Van der Bij, S.; Koffijberg, H.; Lenters, V.; Portengen, L.; Moons, K.G.; Heederik, D.; Vermeulen, R.C. Lung cancer risk at low cumulative asbestos exposure: Meta-regression of the exposure-response relationship. Cancer Causes Control 2013, 24, 1–12. [Google Scholar] [CrossRef]
- Pohlabeln, H.; Wild, P.; Schill, W.; Ahrens, W.; Jahn, I.; Bolm-Audorff, U.; Jockel, K.H. Asbestos fibreyears and lung cancer: A two phase case-control study with expert exposure assessment. Occup. Environ. Med. 2002, 59, 410–414. [Google Scholar] [CrossRef]
- Berman, D.W.; Crump, K.S. A meta-analysis of asbestos-related cancer risk that addresses fiber size and mineral type. Crit. Rev. Toxicol. 2008, 38 (Suppl. 1), 49–73. [Google Scholar] [CrossRef]
- Lenters, V.; Vermeulen, R.; Dogger, S.; Stayner, L.; Portengen, L.; Burdorf, A.; Heederik, D. A meta-analysis of asbestos and lung cancer: Is better quality exposure assessment associated with steeper slopes of the exposure-response relationships? Environ. Health Perspect. 2011, 119, 1547–1555. [Google Scholar] [CrossRef] [PubMed]
- Yano, E.; Wang, X.; Wang, M.; Qiu, H.; Wang, Z. Lung cancer mortality from exposure to chrysotile asbestos and smoking: A case-control study within a cohort in China. Occup. Environ. Med. 2010, 67, 867–871. [Google Scholar] [CrossRef] [PubMed]
- Henderson, D.W.; Jones, M.L.; De Klerk, N.; Leigh, J.; Musk, A.W.; Shilkin, K.B.; Williams, V.M. The diagnosis and attribution of asbestos-related diseases in an Australian context: Report of the Adelaide Workshop on Asbestos-Related Diseases. October 6–7, 2000. Int. J. Occup. Environ. Health 2004, 10, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Offermans, N.S.; Vermeulen, R.; Burdorf, A.; Goldbohm, R.A.; Kauppinen, T.; Kromhout, H.; van den Brandt, P.A. Occupational asbestos exposure and risk of pleural mesothelioma, lung cancer, and laryngeal cancer in the prospective Netherlands cohort study. J. Occup. Environ. Med. 2014, 56, 6–19. [Google Scholar] [CrossRef]
- Ioannidis, J.; Schmid, C.H.; Lau, J. Meta-analysis approaches for epidemiologic research on asbestos. In Sourcebook on Asbestos Diseases; Peters, G.A., Peters, B.J., Eds.; Lexis: Charlottesville, VA, USA, 1998; pp. 93–116. [Google Scholar]
- Leigh, J.; Henderson, D. The epidemiology of malignant mesothelioima. J. Occup. Health Safety Aust. NZ. 2006, 22, 441–447. [Google Scholar]
- Berry, G.; Liddell, F.D. The interaction of asbestos and smoking in lung cancer: A modified measure of effect. Ann. Occup. Hyg. 2004, 48, 459–462. [Google Scholar] [CrossRef]
- De Vuyst, P.; Karjalainen, A.; Dumortier, P.; Pairon, J.C.; Monso, E.; Brochard, P.; Teschler, H.; Tossavainen, A.; Gibbs, A. Guidelines for mineral fibre analyses in biological samples: Report of the ERS Working Group. European Respiratory Society. Eur. Respir. J. 1998, 11, 1416–1426. [Google Scholar] [CrossRef]
- Adams, H.; Crane, M.D. Radiological features of the asbestos-associated diseases. In Asbestos and Its Diseases; Oxford University Press: Oxford, UK, 2008; pp. 269–298. [Google Scholar]
- Roggli, V.L.; Gibbs, A.R.; Attanoos, R.; Churg, A.; Popper, H.; Cagle, P.; Corrin, B.; Franks, T.J.; Galateau-Salle, F.; Galvin, J.; et al. Pathology of asbestosis—An update of the diagnostic criteria: Report of the asbestosis committee of the college of american pathologists and pulmonary pathology society. Arch. Pathol. Lab. Med. 2010, 134, 462–480. [Google Scholar] [CrossRef]
- Hammar, S.P. Asbestosis. In Asbestos: Risk Assesment, Epidemiology and Health Effects; Taylor & Francis: Boca Raton, FL, USA, 2011; pp. 447–479. [Google Scholar]
- Baur, X.; Woitowitz, H.J.; Budnik, L.T.; Egilman, D.; Oliver, C.; Frank, A.; Soskolne, C.L.; Landrigan, P.J.; Lemen, R.A. Asbestos, asbestosis, and cancer: The Helsinki criteria for diagnosis and attribution. Critical need for revision of the 2014 update. Am. J. Ind. Med. 2017, 60, 411–421. [Google Scholar] [CrossRef]
- Wei, S.; Wang, L.E.; McHugh, M.K.; Han, Y.; Xiong, M.; Amos, C.I.; Spitz, M.R.; Wei, Q.W. Genome-wide gene-environment interaction analysis for asbestos exposure in lung cancer susceptibility. Carcinogenesis 2012, 33, 1531–1537. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klebe, S.; Leigh, J.; Henderson, D.W.; Nurminen, M. Asbestos, Smoking and Lung Cancer: An Update. Int. J. Environ. Res. Public Health 2020, 17, 258. https://doi.org/10.3390/ijerph17010258
Klebe S, Leigh J, Henderson DW, Nurminen M. Asbestos, Smoking and Lung Cancer: An Update. International Journal of Environmental Research and Public Health. 2020; 17(1):258. https://doi.org/10.3390/ijerph17010258
Chicago/Turabian StyleKlebe, Sonja, James Leigh, Douglas W. Henderson, and Markku Nurminen. 2020. "Asbestos, Smoking and Lung Cancer: An Update" International Journal of Environmental Research and Public Health 17, no. 1: 258. https://doi.org/10.3390/ijerph17010258
APA StyleKlebe, S., Leigh, J., Henderson, D. W., & Nurminen, M. (2020). Asbestos, Smoking and Lung Cancer: An Update. International Journal of Environmental Research and Public Health, 17(1), 258. https://doi.org/10.3390/ijerph17010258