Salivary Microbiome and Cigarette Smoking: A First of Its Kind Investigation in Jordan


Abstract
1. Introduction
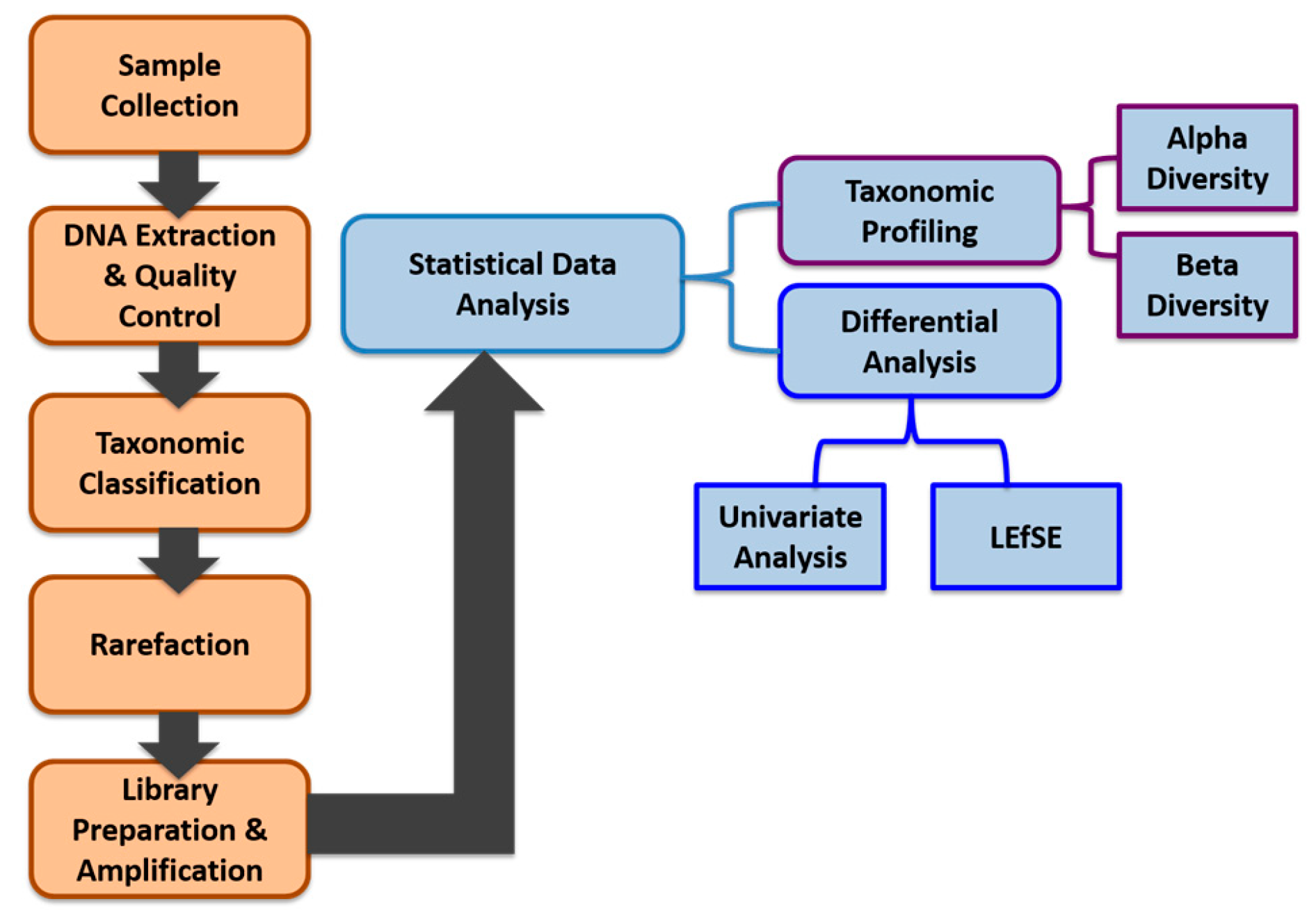
2. Materials and Methods
2.1. Study Subjects
2.2. Sample Collection, Processing, and Storage
2.3. DNA Extraction and Quality Controls
2.4. DNA Sequencing
2.5. Taxonomic Classification
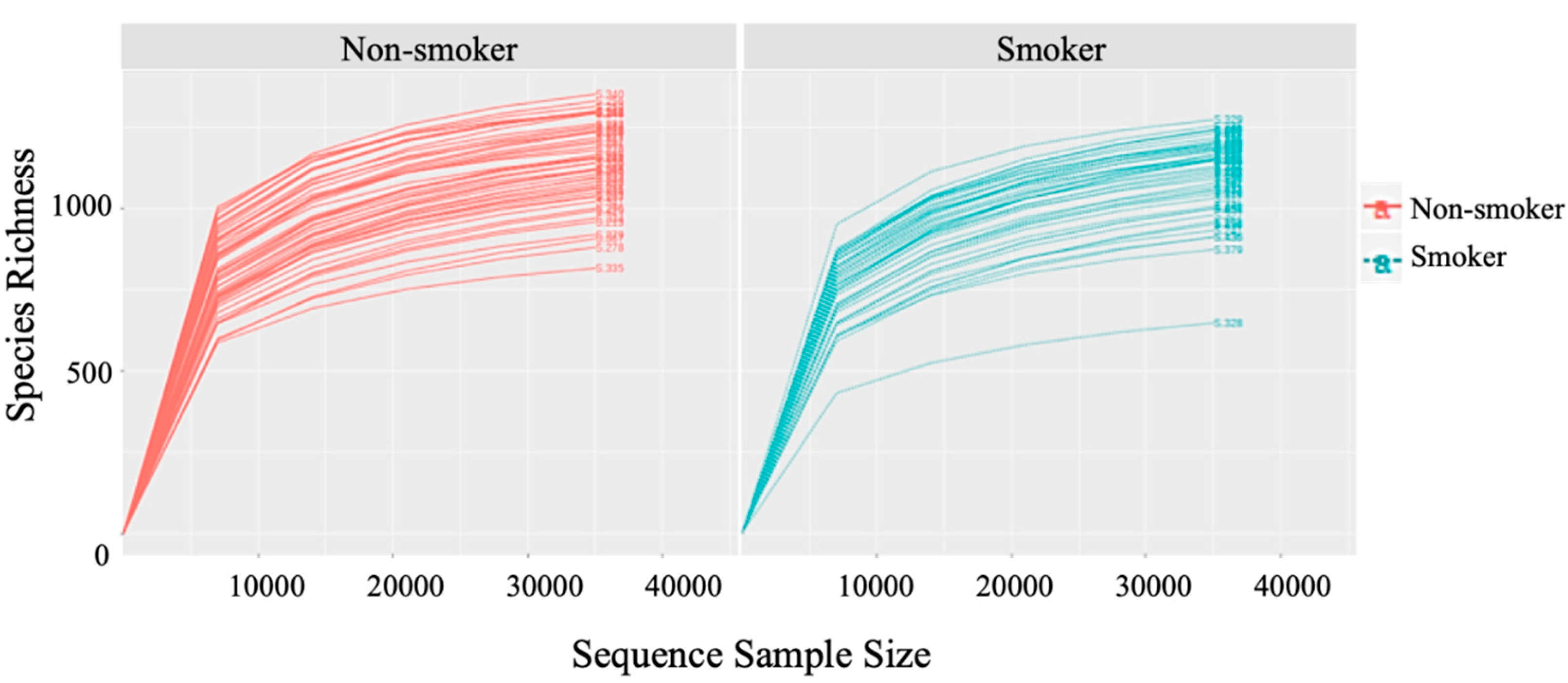
2.6. Rarefaction
2.7. Library Preparation and Sequence Amplification
2.8. Data Pre-Processing
2.9. Data Analysis
2.9.1. Alpha (α) Diversity and Beta (β) Diversity
2.9.2. Univariate Analysis
2.9.3. LDA Effect Size (LEfSe)
3. Results
3.1. Demographic Data of the Study Subjects
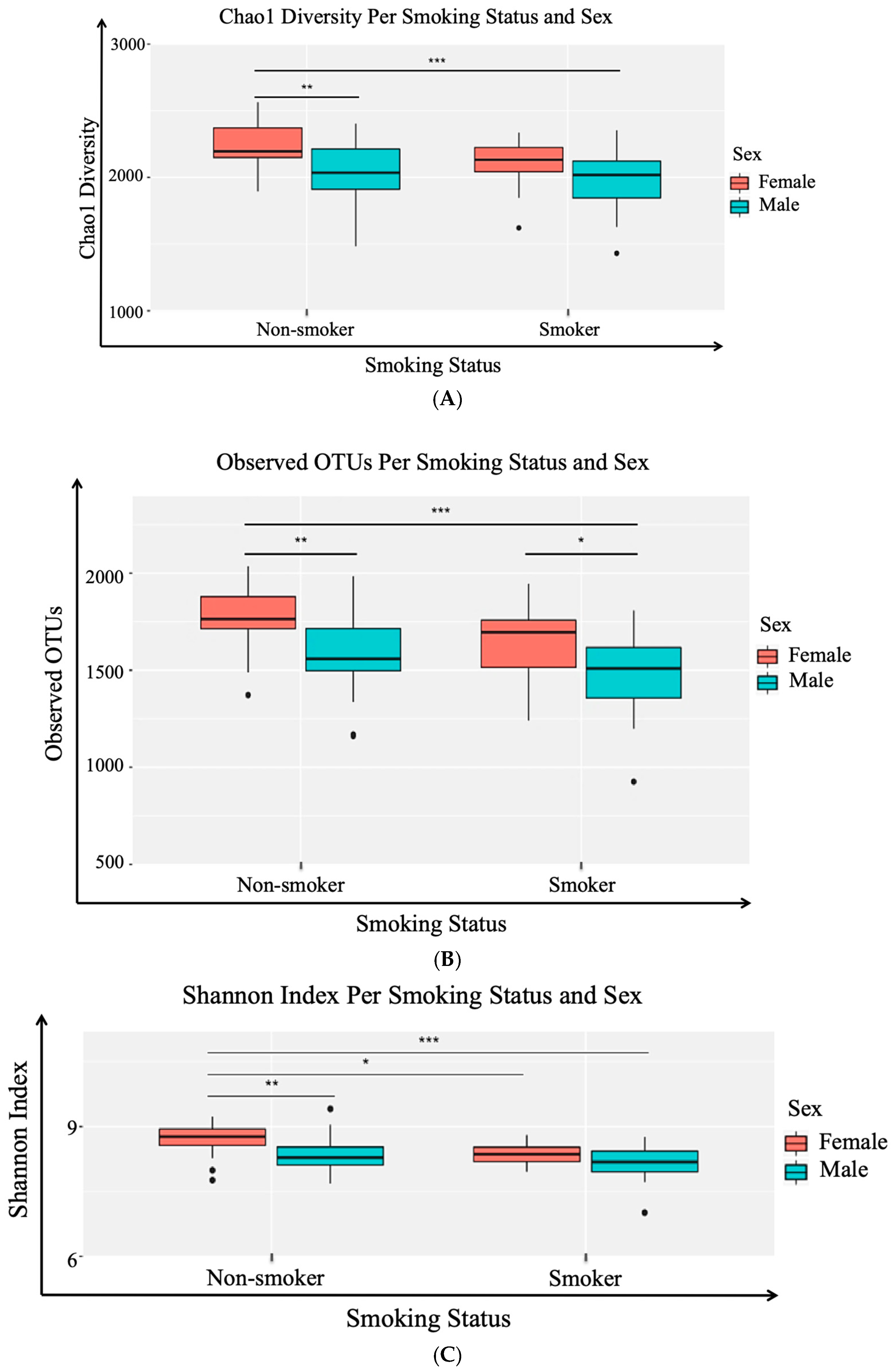
3.2. Alpha (α) Diversity Metrics
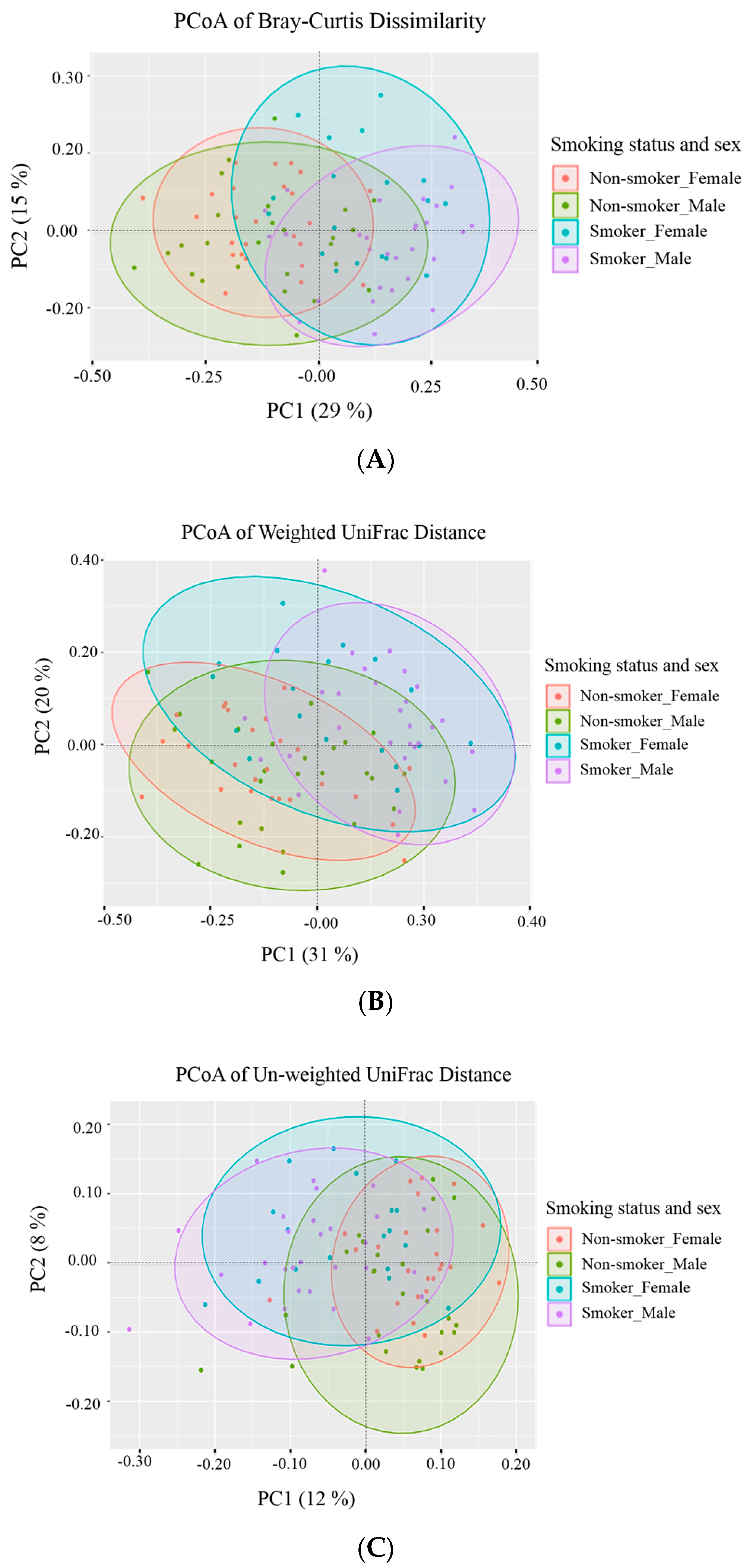
3.3. Beta (β) Diversity Metrics
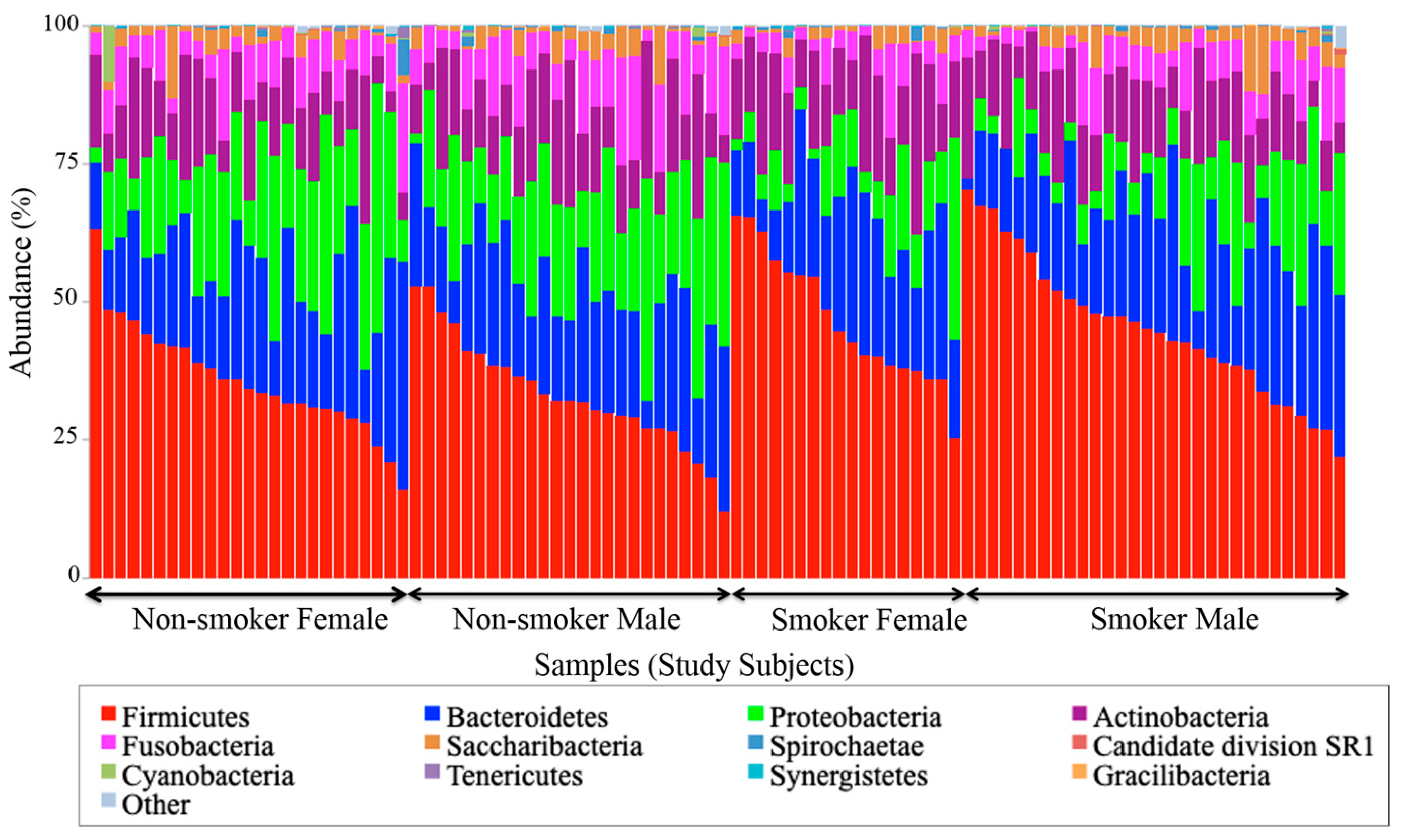
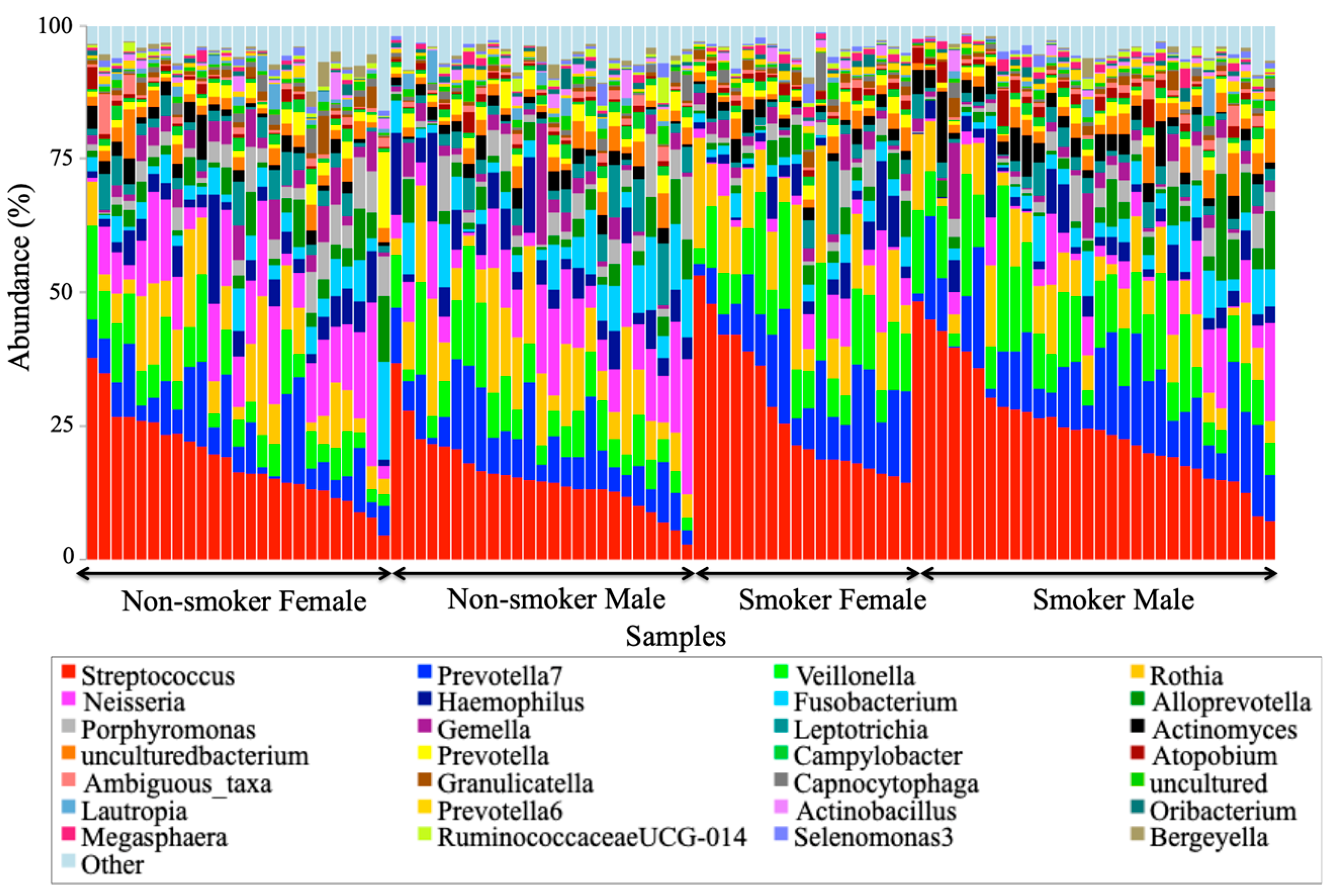
3.4. Visualization of the Taxonomic Profiling Results
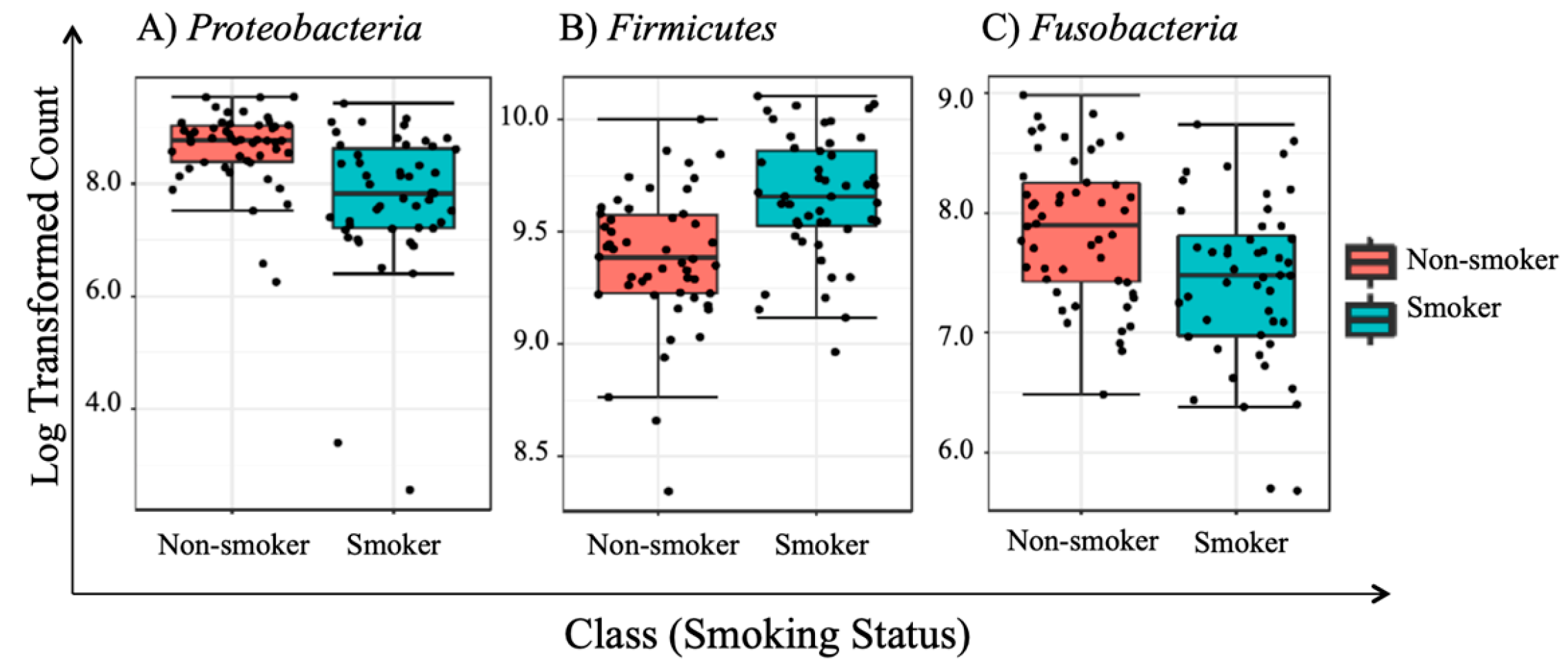
3.5. Univariate Analysis
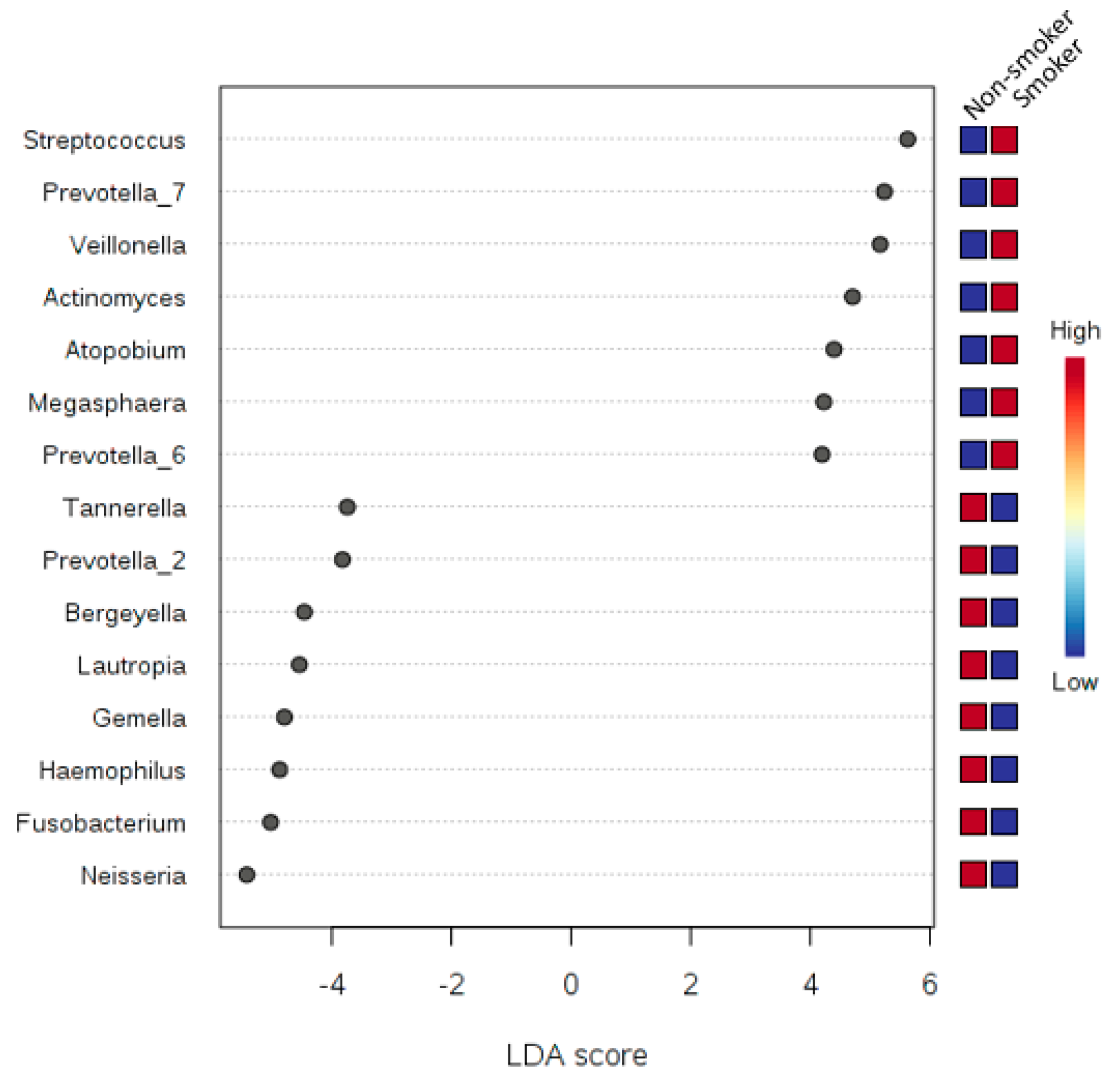
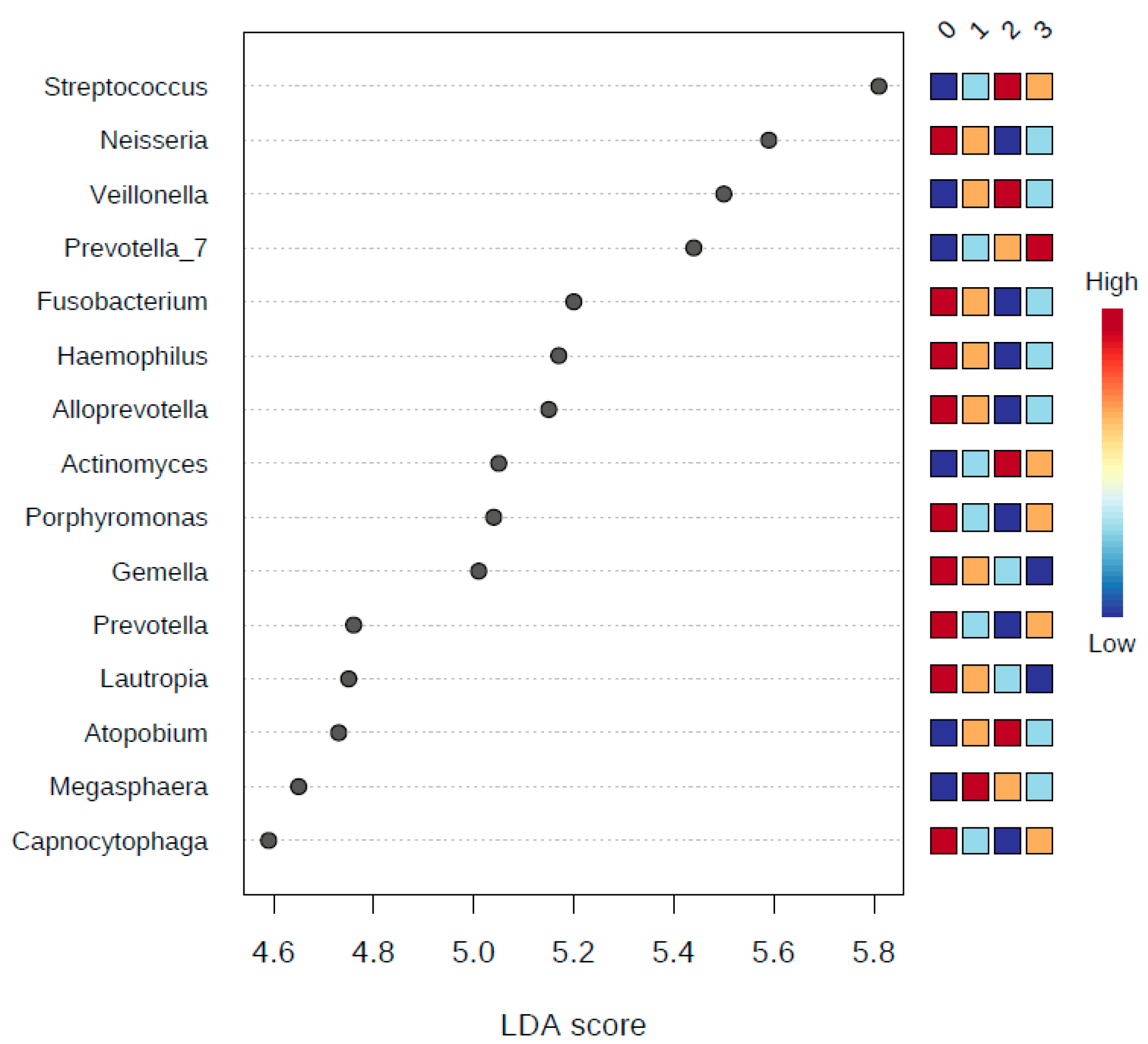
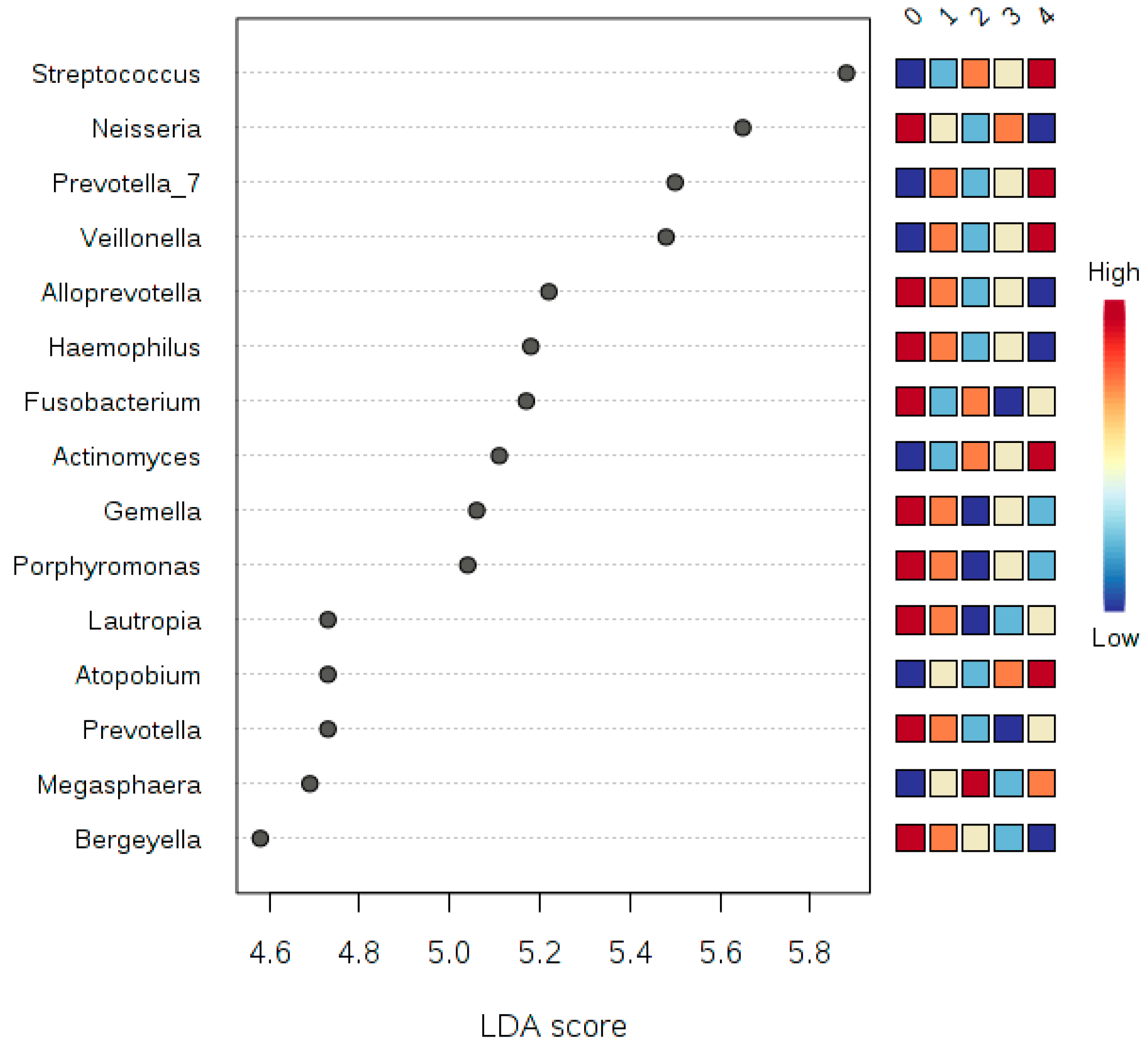
3.6. LDA Effect Size (LEfSe)
3.7. Multivariate Analysis by Linear Models (MaAsLin)
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Privacy and Availability Statement
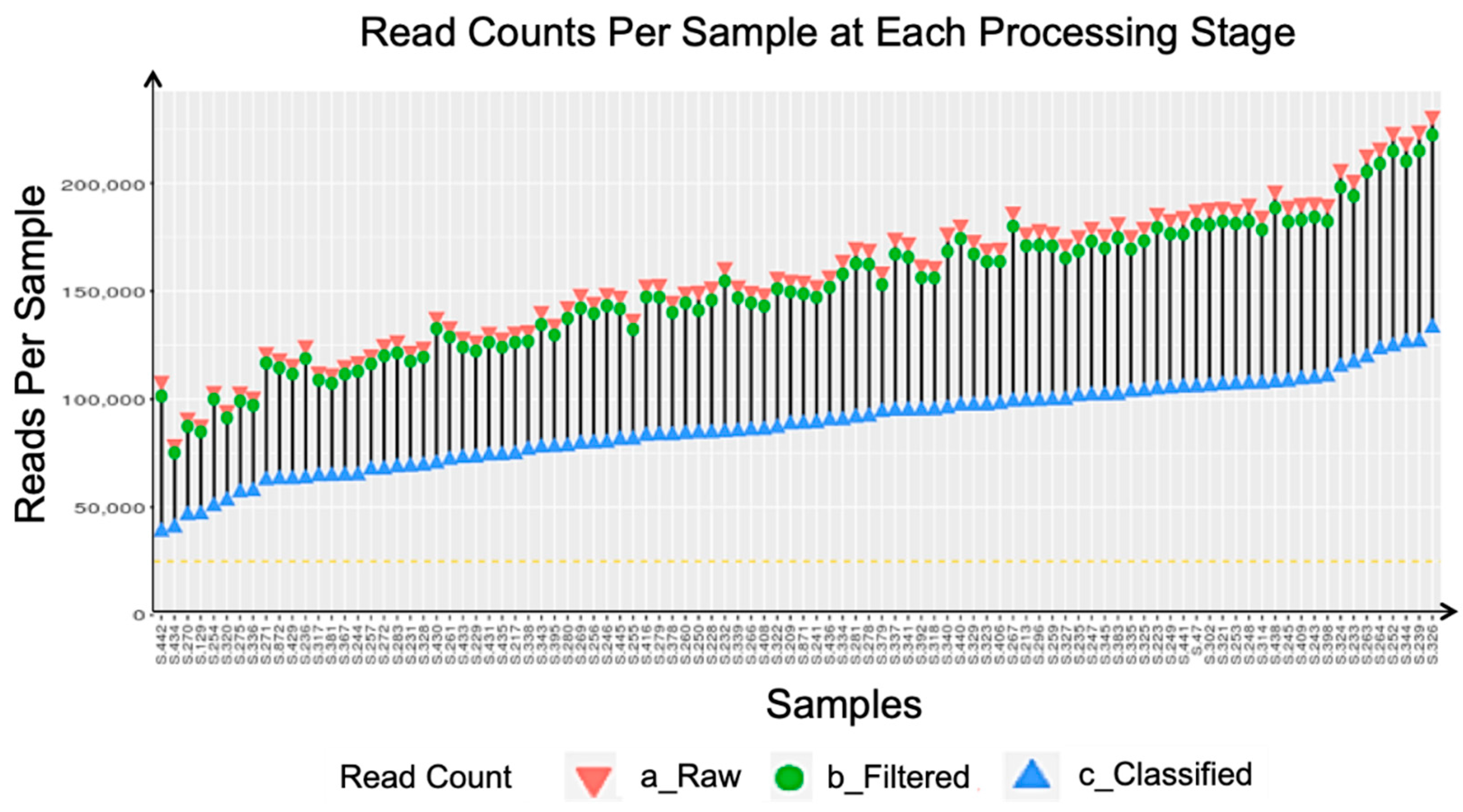
Appendix A. Sequencing Quality Metrics

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metric | Minimum Requirement | Results |
|---|---|---|
| % ≥ Q30 | ≥70% | 79.62% |
| Cluster Density | ≥800 k/mm2 | 914 ± 16 k/mm2 |
| Clusters Passing Filter (%) | ≥80% | 91.47 ± 0.75% |
| Sequencing Yield | ≥13.2 Gbp | 12.36 Gbp |
| PhiX Alignment (%) | 12%–18% | 19.01% |


References
- Saha, S.P.; Bhalla, D.K.; Whayne, T.F.; Gairola, C. Cigarette Smoke and Adverse Health Effects: An Overview of Research Trends and Future Needs. Int. J. Angiol. 2007, 16, 77–83. [Google Scholar] [CrossRef]
- Stämpfli, M.R.; Anderson, G.P. How Cigarette Smoke Skews Immune Responses to Promote Infection, Lung Disease and Cancer. Nat. Rev. Immunol. 2009, 9, 377–384. [Google Scholar] [CrossRef]
- Kumar, P.S.; Matthews, C.R.; Joshi, V.; de Jager, M.; Aspiras, M. Tobacco Smoking Affects Bacterial Acquisition and Colonization in Oral Biofilms. Infect. Immunol. 2011, 79, 4730–4738. [Google Scholar] [CrossRef]
- Warren, G.W.; Alberg, A.J.; Kraft, A.S.; Cummings, K.M. The 2014 Surgeon General’s Report: “The Health Consequences of Smoking-50 Years of Progress”: A Paradigm Shift in Cancer Care. Cancer 2014, 120, 1914–1916. [Google Scholar] [CrossRef]
- Yu, G.; Phillips, S.; Gail, M.H.; Goedert, J.J.; Humphrys, M.S.; Ravel, J.; Ren, Y.; Caporaso, N.E. The Effect of Cigarette Smoking on the Oral and Nasal Microbiota. Microbiome 2017, 5, 3. [Google Scholar] [CrossRef]
- Zarco, M.F.; Vess, T.J.; Ginsburg, G.S. The Oral Microbiome in Health and Disease and the Potential Impact on Personalized Dental Medicine. Oral Dis. 2012, 18, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Kato, I.; Vasquez, A.A.; Moyerbrailean, G.; Land, S.; Sun, J.; Lin, H.-S.; Ram, J.L. Oral Microbiome and History of Smoking and Colorectal Cancer. J. Epidemiol. Res. 2016, 2, 92. [Google Scholar] [CrossRef]
- Wu, J.; Peters, B.A.; Dominianni, C.; Zhang, Y.; Pei, Z.; Yang, L.; Ma, Y.; Purdue, M.P.; Jacobs, E.J.; Gapstur, S.M.; et al. Cigarette Smoking and the Oral Microbiome in a Large Study of American Adults. ISME J. 2016, 10, 2435–2446. [Google Scholar] [CrossRef]
- Littman, D.R.; Pamer, E.G. Role of the Commensal Microbiota in Normal and Pathogenic Host Immune Responses. Cell Host Microbe 2011, 10, 311–323. [Google Scholar] [CrossRef]
- Acharya, A.; Chan, Y.; Kheur, S.; Jin, L.J.; Watt, R.M.; Mattheos, N. Salivary Microbiome in Non-Oral Disease: A Summary of Evidence and Commentary. Arch. Oral Biol. 2017, 83, 169–173. [Google Scholar] [CrossRef]
- Alzyoud, S.; Kheirallah, K.; Weglicki, L.; Ward, K.; Al-Khawaldeh, A.; Shotar, A. Tobacco Smoking Status and Perception of Health among a Sample of Jordanian Students. Int. J. Environ. Res. Public Health 2014, 11, 7022–7035. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, K.; Chen, T.; Paster, B. A Practical Guide to the Oral Microbiome and Its Relation to Health and Disease. Oral Dis. 2017, 23, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Nasidze, I.; Li, J.; Quinque, D.; Tang, K.; Stoneking, M. Global Diversity in the Human Salivary Microbiome. Genome Res. 2009, 19, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-A. A Literature Analysis of the Risk Factors for Oral Cancer. In Oral Cancer; InTech: Vienna, Austria, 2012; pp. 68–78. [Google Scholar] [CrossRef]
- Söder, B.; Jin, L.J.; Wickholm, S. Granulocyte Elastase, Matrix Metalloproteinase-8 and Prostaglandin E2 in Gingival Crevicular Fluid in Matched Clinical Sites in Smokers and Non-Smokers with Persistent Periodontitis. J. Clin. Periodontol. 2002, 29, 384–391. [Google Scholar] [CrossRef]
- Barbour, S.E.; Nakashima, K.; Zhang, J.B.; Tangada, S.; Hahn, C.L.; Schenkein, H.A.; Tew, J.G. Tobacco and Smoking: Environmental Factors That Modify the Host Response (Immune System) and Have an Impact on Periodontal Health. Crit. Rev. Oral Biol. Med. 1997, 8, 437–460. [Google Scholar] [CrossRef]
- Heikkinen, A.M. Oral Health, Smoking and Adolescence, Helsingin Yliopisto. Ph.D. Thesis, Helsinki University, Helsinki, Finland, 2011. [Google Scholar]
- Dewhirst, F.E.; Chen, T.; Izard, J.; Paster, B.J.; Tanner, A.C.R.; Yu, W.H.; Lakshmanan, A.; Wade, W.G. The Human Oral Microbiome. J. Bacteriol. 2010, 192, 5002–5017. [Google Scholar] [CrossRef]
- Danser, M.M.; Gomez, S.M.; Van der Weijden, G.A. Tongue Coating and Tongue Brushing: A Literature Review. Int. J. Dent. Hyg. 2003, 1, 151–158. [Google Scholar] [CrossRef]
- Al-Ghalith, G.A.; Montassier, E.; Ward, H.N.; Knights, D. NINJA-OPS: Fast Accurate Marker Gene Alignment Using Concatenated Ribosomes. PLoS Comput. Biol. 2016, 12, e1004658. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J. MicrobiomeAnalyst: A Web-Based Tool for Comprehensive Statistical, Visual and Meta-Analysis of Microbiome Data. Nucleic Acids Res. 2017, 45, W180–W188. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Magoc, T.; Salzberg, S.L. FLASH: Fast Length Adjustment of Short Reads to Improve Genome Assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME Allows Analysis of High-Throughput Community Sequencing Data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Bray, J.R.; Curtis, J.T. An Ordination of the Upland Forest Communities of Southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic Biomarker Discovery and Explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Morgan, X.C.; Tickle, T.L.; Sokol, H.; Gevers, D.; Devaney, K.L.; Ward, D.V.; Reyes, J.A.; Shah, S.A.; LeLeiko, N.; Snapper, S.B.; et al. Dysfunction of the Intestinal Microbiome in Inflammatory Bowel Disease and Treatment. Genome Biol. 2012, 13, R79. [Google Scholar] [CrossRef]
- Morris, A.; Beck, J.M.; Schloss, P.D.; Campbell, T.B.; Crothers, K.; Curtis, J.L.; Flores, S.C.; Fontenot, A.P.; Ghedin, E.; Huang, L.; et al. Comparison of the Respiratory Microbiome in Healthy Nonsmokers and Smokers. Am. J. Respir. Crit. Care Med. 2013, 187, 1067–1075. [Google Scholar] [CrossRef]
- Tanaka, S.; Yoshida, M.; Murakami, Y.; Ogiwara, T.; Shoji, M.; Kobayashi, S.; Watanabe, S.; Machino, M.; Fujisawa, S. The Relationship of Prevotella Intermedia, Prevotella Nigrescens and Prevotella Melaninogenica in the Supragingival Plaque of Children, Caries and Oral Malodor. J. Clin. Pediatr. Dent. 2008, 32, 195–200. [Google Scholar] [CrossRef]
- Porter, S.R.; Scully, C. Oral Malodour (Halitosis). Br. Med. J. 2006, 333, 632–635. [Google Scholar] [CrossRef]
- Vartoukian, S.R.; Palmer, R.M.; Wade, W.G. Diversity and Morphology of Members of the Phylum “Synergistetes” in Periodontal Health and Disease. Appl. Environ. Microbiol. 2009, 75, 3777–3786. [Google Scholar] [CrossRef]
- Davis, J.P.; Youssef, N.H.; Elshahed, M.S. Assessment of the Diversity, Abundance, and Ecological Distribution of Members of Candidate Division SR1 Reveals a High Level of Phylogenetic Diversity but Limited Morphotypic Diversity †. Appl. Environ. Microbiol. 2009, 75, 4139–4148. [Google Scholar] [CrossRef] [PubMed]
- Said, H.S.; Suda, W.; Nakagome, S.; Chinen, H.; Oshima, K.; Kim, S.; Kimura, R.; Iraha, A.; Ishida, H.; Fujita, J.; et al. Dysbiosis of Salivary Microbiota in Inflammatory Bowel Disease and Its Association with Oral Immunological Biomarkers. DNA Res. 2014, 21, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, E.; Söder, B.; Andersson, L.C.; Meurman, J.H.; Söder, P.Ö. History of Dental Infections Associates with Cancer in Periodontally Healthy Subjects: 24-Year Follow-up Study from Sweden. J. Cancer 2014, 5, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Zhang, Q.; Hua, H.; Chen, F. Changes in the Salivary Microbiota of Oral Leukoplakia and Oral Cancer. Oral Oncol. 2016, 56, e6–e8. [Google Scholar] [CrossRef]
- Leake, S.L.; Pagni, M.; Falquet, L.; Taroni, F.; Greub, G. The Salivary Microbiome for Differentiating Individuals: Proof of Principle. Microbes Infect. 2016, 18, 399–405. [Google Scholar] [CrossRef]









| Gender (n a) | Age in Years (mean ± SD) | Ethnicity | Education | Tobacco Smokers versus Nonsmokers (n) | Smoking Duration (n) | Number of Cigarettes (n) | Teeth Brushing |
|---|---|---|---|---|---|---|---|
| Male (57) | 23.9 ± 6.20 | White (95) | School (20) | Smokers (49) | <1 Year (2) | 1–10 (15) | Brush (89) |
| Female (43) | 27.1 ± 7.57 | Black (4) | Bachelor (76) | Non-smokers (51) | 1–5 Years (15) | 10–20 (20) | Do not Brush (9) |
| Caucasian (1) | Master (4) | Male non-smokers (28) | 6–10 Years (20) | 21–30 (6) | |||
| Female non-smokers (23) | >10 Years (9) | >30 (5) | |||||
| Male smokers (29) | |||||||
| Female smokers (20) | |||||||
| p-value | 0.0245 * (age) | 0.6693 (smokers versus non-smokers) |
| Group | Bray-Curtis Dissimilarity | Weighted UniFrac | Unweighted UniFrac | |||
|---|---|---|---|---|---|---|
| p-Value | p-Adjusted | p-Value | p-Adjusted | p-Value | p-Adjusted | |
| Male non-smokers versus male smokers | 0.001 | 0.006 * | 0.001 | 0.006 * | 0.001 | 0.006 * |
| Male non-smokers versus female non-smokers | 0.074 | 0.444 | 0.171 | 1.000 | 0.003 | 0.018 * |
| Male non-smokers versus female smokers | 0.001 | 0.006 * | 0.001 | 0.006 * | 0.001 | 0.006 * |
| Male smokers versus female non-smokers | 0.001 | 0.006 * | 0.001 | 0.006 * | 0.001 | 0.006 * |
| Male smokers versus female smokers | 0.005 | 0.030 * | 0.013 | 0.078 | 0.020 | 0.120 |
| Female non-smokers versus female smokers | 0.001 | 0.006 * | 0.001 | 0.006 * | 0.001 | 0.006 * |
| # | Phylum | p-Values | FDR |
|---|---|---|---|
| 1 | Proteobacteria | 2.80 × 10−6 * | 2.52 × 10−5 * |
| 2 | Firmicutes | 7.16 × 10−6 * | 3.22 × 10−5 * |
| 3 | Fusobacteria | 0.0017 * | 0.0053 * |
| 4 | Spirochaetae | 0.2924 | 0.6579 |
| 5 | Synergistetes | 0.4506 | 0.8112 |
| 6 | Actinobacteria | 0.5616 | 0.8424 |
| 7 | Candidate_division_SR1 | 0.7550 | 0.8939 |
| 8 | Saccharibacteria | 0.7946 | 0.8939 |
| 9 | Bacteroidetes | 0.9579 | 0.9579 |
| # | Genus | p-Values | FDR |
|---|---|---|---|
| 1 | Neisseria | 4.58 × 10−6 * | 0.0003 * |
| 2 | Streptococcus | 3.27 × 10−5 * | 0.0011 * |
| 3 | Prevotella | 0.0013 * | 0.0299 * |
| 4 | Veillonella | 0.0016 * | 0.0299 * |
| 5 | Bergeyella | 0.0043 | 0.0522 |
| 6 | Eikenella | 0.0049 | 0.0522 |
| 7 | Johnsonella | 0.0050 | 0.0522 |
| 8 | Fusobacterium | 0.0062 | 0.0566 |
| 9 | Megasphaera | 0.0076 | 0.0591 |
| Smoker versus Non-Smoker | ||||||||
| Phylum | p-Value * | FDR | Abundance | LDA Score | ||||
| Non-Smoker | Smoker | |||||||
| Proteobacteria | 6.06 × 10−6 | 5.45 × 10−5 | 1,828,000 | 1,003,200 | −5.62 | |||
| Firmicutes | 9.99 × 10−5 | 4.50 × 10−4 | 3,539,800 | 4,517,700 | 5.69 | |||
| Fusobacteria | 2.54 × 10−3 | 7.62 × 10−3 | 882,530 | 583,700 | −5.17 | |||
| Candidate_division_SR1 | 7.27 × 10−2 | 1.64 × 10−1 | 21,233 | 18,901 | −3.07 | |||
| Synergistetes | 1.27 × 10−1 | 2.28 × 10−1 | 1706.6 | 1317.4 | −2.29 | |||
| Actinobacteria | 2.73 × 10−1 | 4.10 × 10−1 | 1,334,300 | 1,422,600 | 4.65 | |||
| Spirochaetae | 3.87 × 10−1 | 4.97 × 10−1 | 13,132 | 6343.5 | −3.53 | |||
| Saccharibacteria | 5.71 × 10−1 | 6.42 × 10−1 | 265,320 | 288,340 | 4.06 | |||
| Bacteroidetes | 6.44 × 10−1 | 6.44 × 10−1 | 2,114,000 | 2,157,900 | 4.34 | |||
| Binned Number of Years of Smoking | ||||||||
| Phylum | p-value * | FDR | Abundance | LDA Score | ||||
| Class 0 | Class 1 | Class 2 | Class 3 | |||||
| Proteobacteria | 1.11 × 10−9 | 9.99 × 10−9 | 1,968,900 | 1,072,400 | 619,710 | 830,020 | 5.83 | |
| Firmicutes | 2.26 × 10−7 | 1.02 × 10−6 | 3,368,600 | 4,624,900 | 5,071,300 | 4,371,100 | 5.93 | |
| Fusobacteria | 4.06 × 10−4 | 1.15 × 10−3 | 910,010 | 652,060 | 500,500 | 487,050 | 5.33 | |
| Candidate_division_SR1 | 5.10 × 10−4 | 1.15 × 10−3 | 32,448 | 2398.8 | 8833.8 | 5397.3 | 4.18 | |
| Actinobacteria | 4.17 × 10−2 | 7.51 × 10−2 | 1,244,500 | 1,287,900 | 1,664,300 | 1,606,300 | 5.32 | |
| Synergistetes | 2.12 × 10−1 | 3.17 × 10−1 | 1579.1 | 538.51 | 799.61 | 3512.6 | 3.17 | |
| Spirochaetae | 2.55 × 10−1 | 3.27 × 10−1 | 13,425 | 4161.2 | 5597.2 | 8196 | 3.67 | |
| Bacteroidetes | 4.70 × 10−1 | 5.28 × 10−1 | 2,203,300 | 2,055,600 | 1,867,200 | 2,337,300 | 5.37 | |
| Saccharibacteria | 8.80 × 10−1 | 8.80 × 10−1 | 257,200 | 300,020 | 261,740 | 351,060 | 4.67 | |
| Binned Number of Cigarettes | ||||||||
| Phylum | p-value * | FDR | Abundance | LDA Score | ||||
| Class 0 | Class 1 | Class 2 | Class 3 | Class 4 | ||||
| Proteobacteria | 5.63 × 10−9 | 5.06 × 10−8 | 1,968,900 | 1,096,000 | 799,060 | 865,290 | 545,000 | 5.85 |
| Firmicutes | 4.48 × 10−7 | 2.01 × 10−6 | 3,368,600 | 4,546,800 | 5,000,800 | 4,423,500 | 4,993,400 | 5.91 |
| Fusobacteria | 1.53 × 10−3 | 4.60 × 10−3 | 910,010 | 538,570 | 647,930 | 492,040 | 505,900 | 5.32 |
| Candidate_division_SR1 | 4.04 × 10−3 | 9.10 × 10−3 | 32,448 | 5620.1 | 5607.6 | 7110.8 | 4922.3 | 4.14 |
| Actinobacteria | 6.14 × 10−2 | 1.10 × 10−1 | 1,244,500 | 1,279,700 | 1,438,900 | 1,882,600 | 1,458,900 | 5.50 |
| Spirochaetae | 8.35 × 10−2 | 1.25 × 10−1 | 13,425 | 4146.5 | 4018.8 | 4911.9 | 10,250 | 3.67 |
| Bacteroidetes | 3.95 × 10−1 | 5.08 × 10−1 | 2,203,300 | 2,298,200 | 1,761,300 | 1,958,300 | 2,234,600 | 5.43 |
| Synergistetes | 9.04 × 10−1 | 9.97 × 10−1 | 1579.1 | 856.72 | 778.84 | 885.28 | 3302.3 | 3.10 |
| Saccharibacteria | 9.97 × 10−1 | 9.97 × 10−1 | 257,200 | 230,080 | 341,600 | 365,390 | 243,750 | 4.83 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Zyoud, W.; Hajjo, R.; Abu-Siniyeh, A.; Hajjaj, S. Salivary Microbiome and Cigarette Smoking: A First of Its Kind Investigation in Jordan. Int. J. Environ. Res. Public Health 2020, 17, 256. https://doi.org/10.3390/ijerph17010256
Al-Zyoud W, Hajjo R, Abu-Siniyeh A, Hajjaj S. Salivary Microbiome and Cigarette Smoking: A First of Its Kind Investigation in Jordan. International Journal of Environmental Research and Public Health. 2020; 17(1):256. https://doi.org/10.3390/ijerph17010256
Chicago/Turabian StyleAl-Zyoud, Walid, Rima Hajjo, Ahmed Abu-Siniyeh, and Sarah Hajjaj. 2020. "Salivary Microbiome and Cigarette Smoking: A First of Its Kind Investigation in Jordan" International Journal of Environmental Research and Public Health 17, no. 1: 256. https://doi.org/10.3390/ijerph17010256
APA StyleAl-Zyoud, W., Hajjo, R., Abu-Siniyeh, A., & Hajjaj, S. (2020). Salivary Microbiome and Cigarette Smoking: A First of Its Kind Investigation in Jordan. International Journal of Environmental Research and Public Health, 17(1), 256. https://doi.org/10.3390/ijerph17010256