Integrating Germline and Somatic Mutation Information for the Discovery of Biomarkers in Triple-Negative Breast Cancer



Abstract
1. Introduction
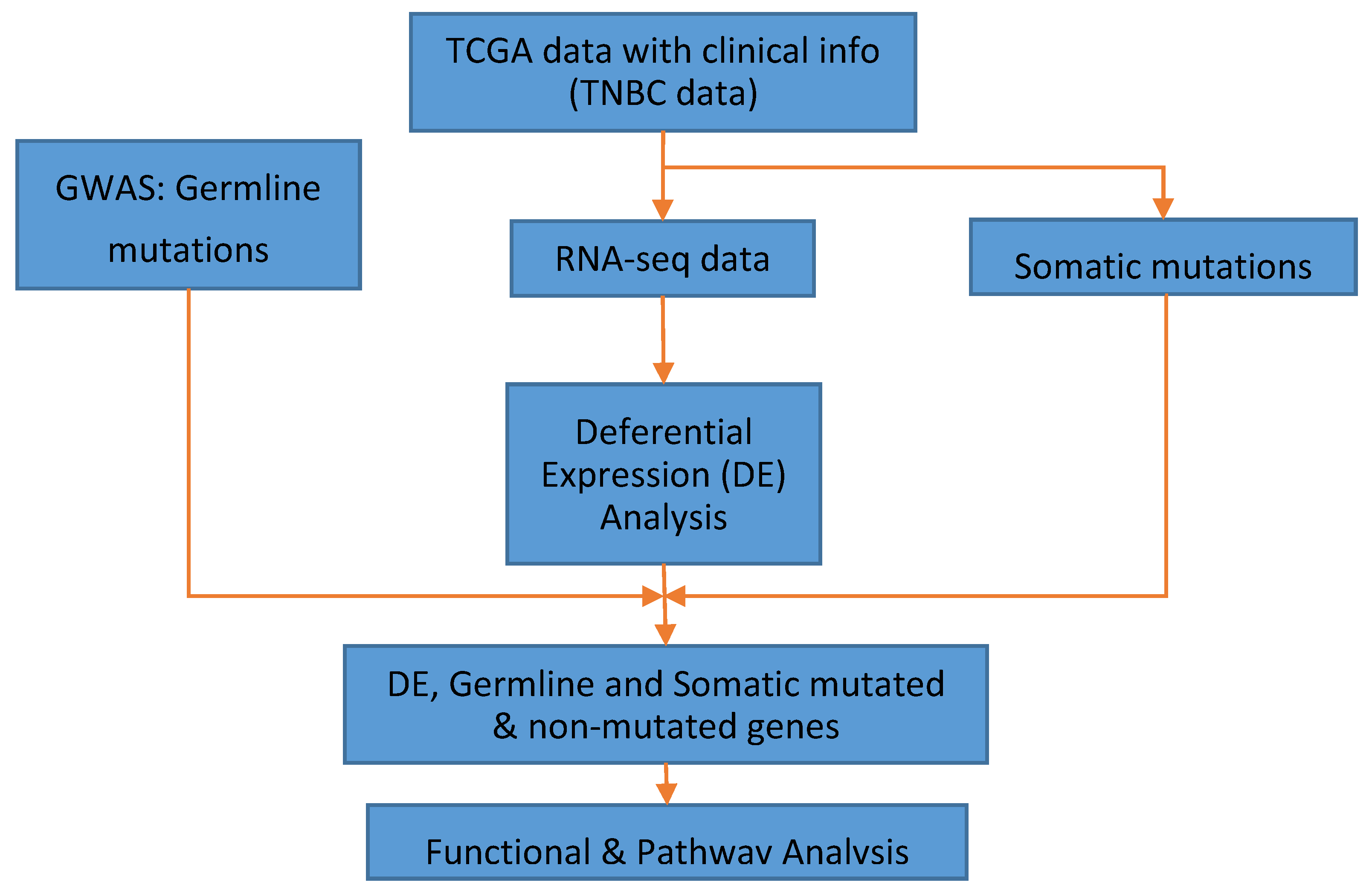
2. Materials and Methods
2.1. Germline Mutations and Associated Genes
2.2. Somatic Mutation Information and Gene Expression Data
2.3. Data Analysis
3. Results
3.1. Significant Differentially Expressed Mutated and Nonmutated Gene Signatures
3.2. Germline and Somatic Mutation Gene Signatures
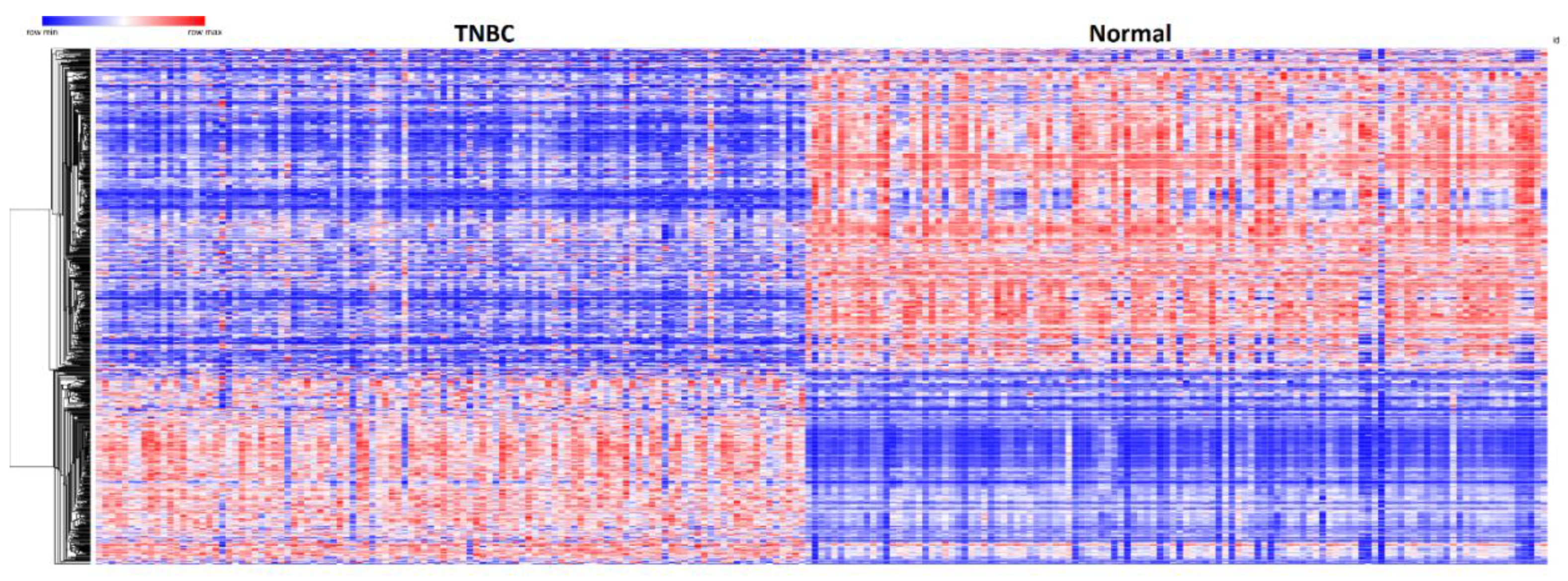
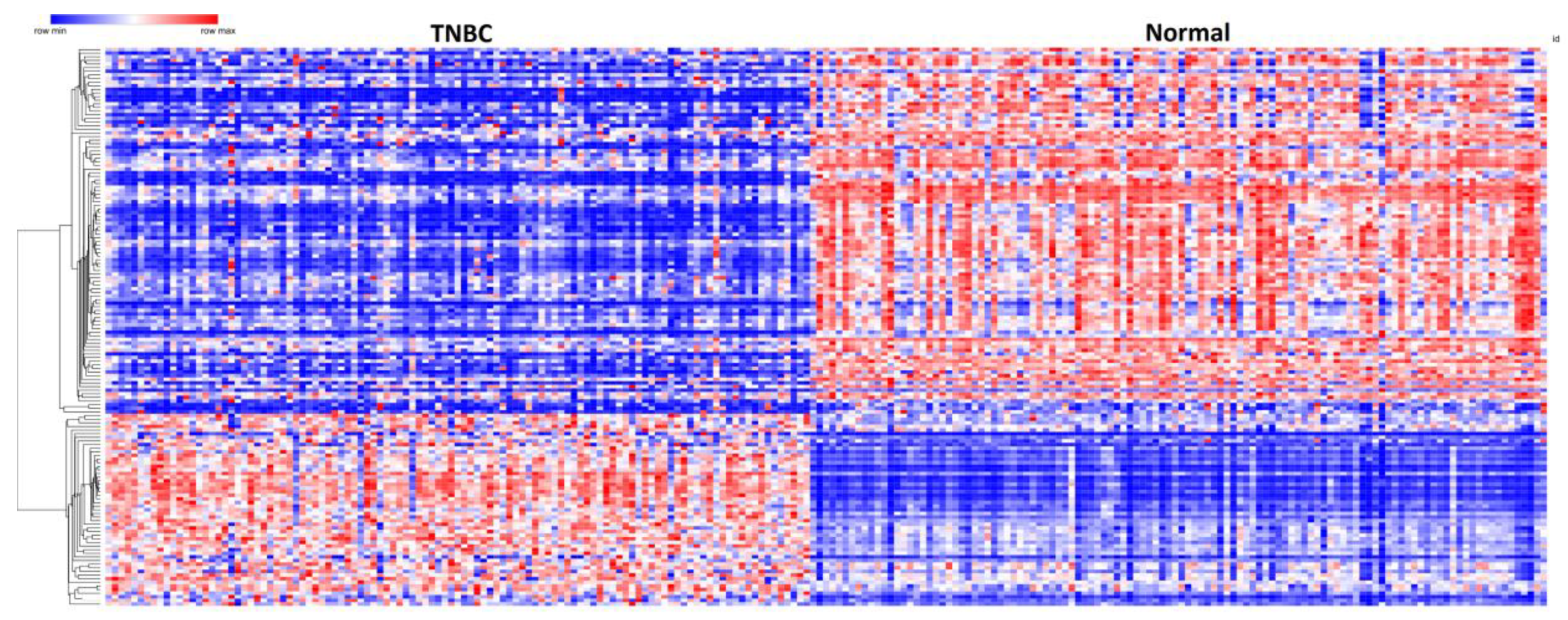
3.3. Patterns of Expression Profiles for Genes Containing Germline and Somatic Mutations
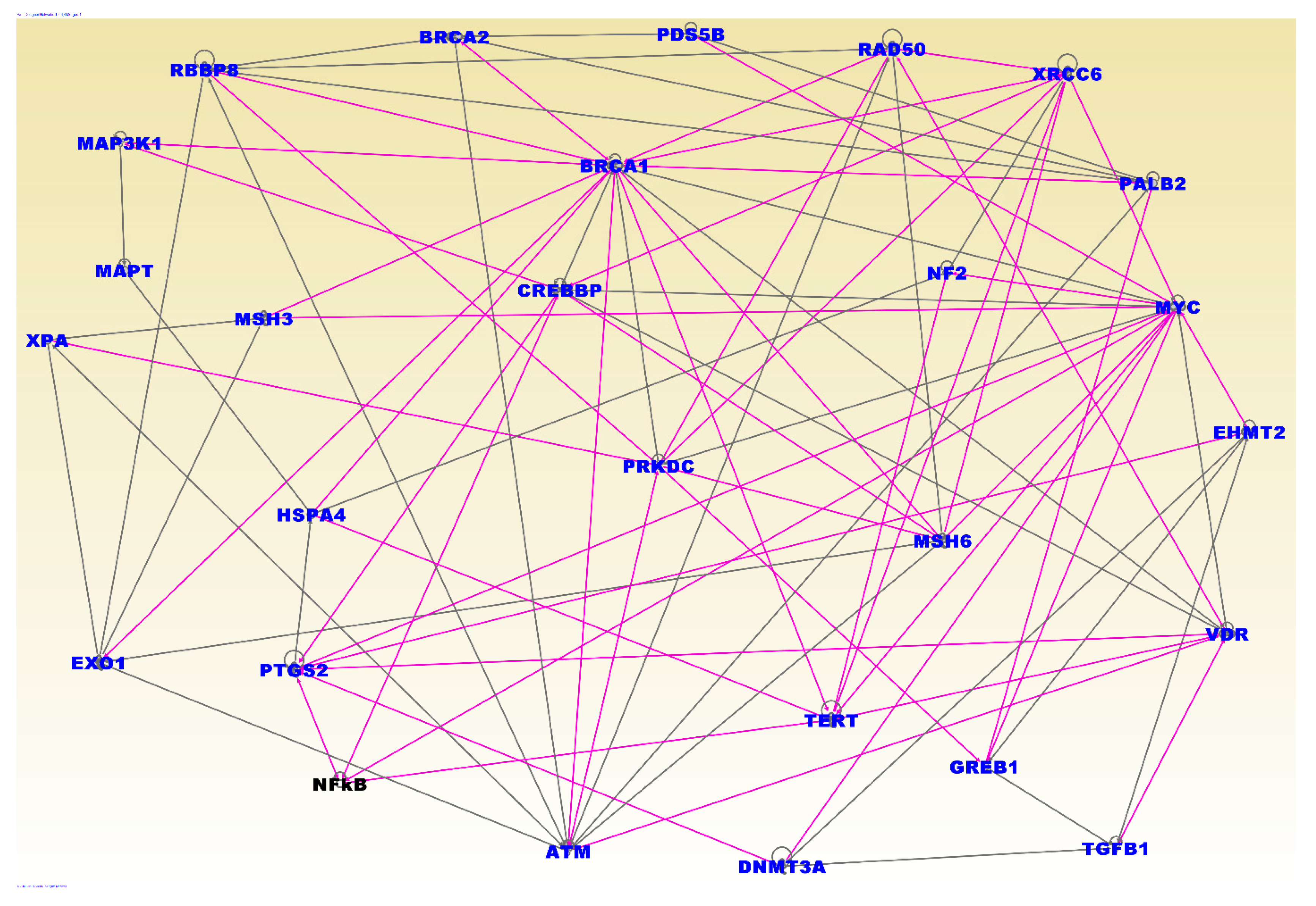
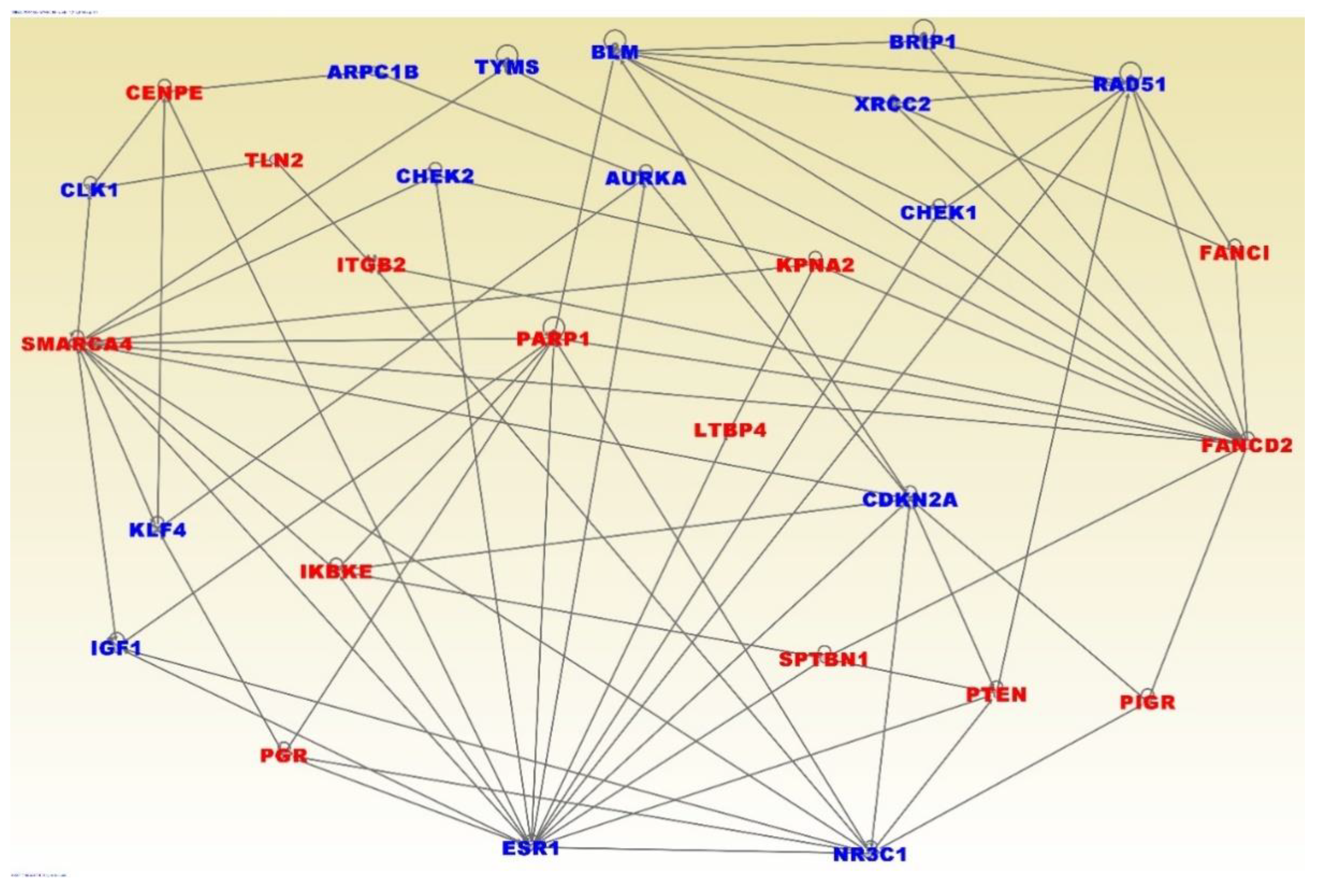
3.4. Molecular Networks and Biological Pathways
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Data Availability and Sharing
Conflicts of Interest
References
- Dietze, E.C.; Chavez, T.A.; Seewaldt, V.L. Obesity and Triple-Negative Breast Cancer. Am. J. Pathol. 2018, 188, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Dietze, E.C.; Sistrunk, C.; Miranda-Carboni, G.; O’Regan, R.; Seewaldt, V.L. Triple-negative breast cancer in African-American women: Disparities versus biology. Nat. Rev. Cancer 2015, 15, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M. Molecular Stratification of Triple-Negative Breast Cancers. Oncologist 2010, 15, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Eirew, P.; Mullaly, S.C.; Aparicio, S. The omics of triple-negative breast cancers. Clin. Chem. 2014, 60, 122–133. [Google Scholar] [CrossRef]
- Stevens, K.N.; Vachon, C.M.; Couch, F.J. Genetic susceptibility to triple-negative breast cancer. Cancer Res. 2013, 73, 2025–2030. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Pietenpol, J.A. Identification and use of biomarkers in treatment strategies for triple-negative breast cancer subtypes. J. Pathol. 2014, 232, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Jovanović, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef]
- Carter, H.; Marty, R.; Hofree, M.; Gross, A.M.; Jensen, J.; Fisch, K.M.; Wu, X.; DeBoever, C.; Van Nostrand, E.L.; Song, Y.; et al. Interaction Landscape of Inherited Polymorphisms with Somatic Events in Cancer. Cancer Discov. 2017, 7, 410–423. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Mukherjee, A.; Machiela, M.J.; Song, L.; Hua, X.; Shi, J.; Garcia-Closas, M.; Chanock, S.J.; Chatterjee, N. An investigation of the association of genetic susceptibility risk with somatic mutation burden in breast cancer. Br. J. Cancer 2016, 115, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Hicks, C.; Kumar, R.; Pannuti, A.; Backus, K.; Brown, A.; Monico, J.; Miele, L. An Integrative Genomics Approach for Associating GWAs Information with Triple-negative Breast cancer. Cancer Inform. 2013, 12, CIN-S10413. [Google Scholar] [CrossRef] [PubMed]
- Purrington, K.S.; Slager, S.; Eccles, D.; Yannoukakos, D.; Fasching, P.A.; Miron, P.; Carpenter, J.; Chang-Claude, J.; Martin, N.G.; Montgomery, G.W.; et al. Genome-wide association study identifies 25 known breast cancer susceptibility loci as risk factors for triple-negative breast cancer. Carcinogenesis 2014, 35, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012, 486, 395–399. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Hudson, T.J.; Anderson, W.; Aretz, A.; Barker, A.D.; Bell, C.; Bernabé, R.R.; Bhan, M.K.; Calvo, F.; Eerola, I.; Gerhard, D.S.; et al. International network of cancer genome projects. Nature 2010, 464, 993–998. [Google Scholar] [CrossRef] [PubMed]
- The NHGRI-EBI Catalog of Published Genome-Wide Association Studies. Available online: https://www.ebi.ac.uk/gwas/ (accessed on 14 March 2019).
- Ioannidis, J.P.; Boffetta, P.; Little, J.; O’Brien, T.R.; Uitterlinden, A.G.; Vineis, P.; Balding, D.J.; Chokkalingam, A.; Dolan, S.M.; Flanders, W.D.; et al. Assessment of cumulative evidence on genetic associations: Interim guidelines. Int. J. Epidemiol. 2008, 37, 120–132. [Google Scholar] [CrossRef]
- Khoury, M.J.; Bertram, L.; Boffetta, P.; Butterworth, A.S.; Chanock, S.J.; Dolan, S.M.; Fortier, I.; Garcia-Closas, M.; Gwinn, M.; Higgins, J.P.T.; et al. Genome-Wide Association Studies, Field Synopses, and the Development of the Knowledge Base on Genetic Variation and Human Diseases. Am. J. Epidemiol. 2009, 170, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Sagoo, G.S.; Little, J.; Higgins, J.P.T. Systematic Reviews of Genetic Association Studies. PLoS Med. 2009, 6, e1000028. [Google Scholar] [CrossRef]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; Group, T.P. Preferred Reporting Items for Systematic Reviews and Meta-Analyses: The PRISMA Statement. PLoS Med. 2009, 6, e1000097. [Google Scholar] [CrossRef]
- Liberati, A.; Altman, D.G.; Tetzlaff, J.; Mulrow, C.; Gøtzsche, P.C.; Ioannidis, J.P.A.; Clarke, M.; Devereaux, P.J.; Kleijnen, J.; Moher, D. The PRISMA Statement for Reporting Systematic Reviews and Meta-Analyses of Studies That Evaluate Health Care Interventions: Explanation and Elaboration. PLoS Med. 2009, 6, e1000100. [Google Scholar] [CrossRef]
- The Short Genetic Variations Database (dbSNP). Available online: https://www.ncbi.nlm.nih.gov/snp (accessed on 14 March 2019).
- Human Genome Nomenclature Committee (HGNC). Available online: https://www.genenames.org/ (accessed on 14 March 2019).
- NCI Genomics Data Commons. Available online: https://gdc.cancer.gov/ (accessed on 14 March 2019).
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate : A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Morpheus. Available online: https://software.broadinstitute.org/morpheus/ (accessed on 14 March 2019).
- Ingenuity Pathways Analysis (IPA) System. Redwood, CA: Ingenuity Systems. Available online: https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/ (accessed on 14 March 2019).
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Hahnen, E.; Lederer, B.; Hauke, J.; Loibl, S.; Kröber, S.; Schneeweiss, A.; Denkert, C.; Fasching, P.A.; Blohmer, J.U.; Jackisch, C.; et al. Germline Mutation Status, Pathological Complete Response, and Disease-Free Survival in Triple-Negative Breast Cancer: Secondary Analysis of the GeparSixto Randomized Clinical Trial. JAMA Oncol. 2017, 3, 1378–1385. [Google Scholar] [CrossRef]
- Petrucelli, N.; Daly, M.B.; Feldman, G.L. Hereditary breast and ovarian cancer due to mutations in BRCA1 and BRCA2. Genet. Med. 2010, 12, 245–259. [Google Scholar] [CrossRef] [PubMed]
- De Leeneer, K.; Coene, I.; Crombez, B.; Simkens, J.; Van den Broecke, R.; Bols, A.; Stragier, B.; Vanhoutte, I.; De Paepe, A.; Poppe, B.; et al. Prevalence of BRCA1/2 mutations in sporadic breast/ovarian cancer patients and identification of a novel de novo BRCA1 mutation in a patient diagnosed with late onset breast and ovarian cancer: Implications for genetic testing. Breast Cancer Res. Treat. 2012, 132, 87–95. [Google Scholar] [CrossRef]
- Engel, C.; Rhiem, K.; Hahnen, E.; Loibl, S.; Weber, K.E.; Seiler, S.; Zachariae, S.; Hauke, J.; Wappenschmidt, B.; Waha, A.; et al. Prevalence of pathogenic BRCA1/2 germline mutations among 802 women with unilateral triple-negative breast cancer without family cancer history. BMC Cancer 2018, 18, 265. [Google Scholar] [CrossRef]
- Hauke, J.; Horvath, J.; Groß, E.; Gehrig, A.; Honisch, E.; Hackmann, K.; Schmidt, G.; Arnold, N.; Faust, U.; Sutter, C.; et al. Gene panel testing of 5589 BRCA1/2-negative index patients with breast cancer in a routine diagnostic setting: Results of the German Consortium for Hereditary Breast and Ovarian Cancer. Cancer Med. 2018, 7, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Shimelis, H.; LaDuca, H.; Hu, C.; Hart, S.N.; Na, J.; Thomas, A.; Akinhanmi, M.; Moore, R.M.; Brauch, H.; Cox, A.; et al. Triple-Negative Breast Cancer Risk Genes Identified by Multigene Hereditary Cancer Panel Testing. J. Natl. Cancer Inst. 2018, 110, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Mavaddat, N.; Michailidou, K.; Dennis, J.; Lush, M.; Fachal, L.; Lee, A.; Tyrer, J.P.; Chen, T.-H.; Wang, Q.; Bolla, M.K.; et al. Polygenic Risk Scores for Prediction of Breast Cancer and Breast Cancer Subtypes. Am. J. Hum. Genet. 2019, 104, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Churbanov, A.; Vorechovský, I.; Hicks, C. A method of predicting changes in human gene splicing induced by genetic variants in context of cis-acting elements. BMC Bioinform. 2010, 11, 22. [Google Scholar] [CrossRef]
- Chen, H.; Li, C.; Peng, X.; Zhou, Z.; Weinstein, J.N.; Caesar-Johnson, S.J.; Demchok, J.A.; Felau, I.; Kasapi, M.; Ferguson, M.L.; et al. A Pan-Cancer Analysis of Enhancer Expression in Nearly 9000 Patient Samples. Cell 2018, 173, 386–399.e12. [Google Scholar] [CrossRef] [PubMed]
- Bonifaci, N.; Górski, B.; Masojć, B.; Wokołorczyk, D.; Jakubowska, A.; Dębniak, T.; Berenguer, A.; Serra Musach, J.; Brunet, J.; Dopazo, J.; et al. Exploring the link between germline and somatic genetic alterations in breast carcinogenesis. PLoS ONE 2010, 5, e14078. [Google Scholar] [CrossRef] [PubMed]
- Grünewald, T.G.P.; Delattre, O. Cooperation between somatic mutations and germline susceptibility variants in tumorigenesis—A dangerous liaison. Mol. Cell. Oncol. 2016, 3, e1086853. [Google Scholar] [CrossRef] [PubMed]
- Waszak, S.M.; Tiao, G.; Zhu, B.; Rausch, T.; Muyas, F.; Rodriguez-Martin, B.; Rabionet, R.; Yakneen, S.; Escaramis, G.; Li, Y.; et al. Germline Determinants of the Somatic Mutation Landscape in 2642 Cancer Genomes; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 2017. [Google Scholar]
- Wallden, B.; Storhoff, J.; Nielsen, T.; Dowidar, N.; Schaper, C.; Ferree, S.; Liu, S.; Leung, S.; Geiss, G.; Snider, J.; et al. Development and verification of the PAM50-based Prosigna breast cancer gene signature assay. BMC Med. Genom. 2015, 8, 54. [Google Scholar] [CrossRef]
- Liu, M.C.; Pitcher, B.N.; Mardis, E.R.; Davies, S.R.; Friedman, P.N.; Snider, J.E.; Vickery, T.L.; Reed, J.P.; DeSchryver, K.; Singh, B.; et al. PAM50 gene signatures and breast cancer prognosis with adjuvant anthracycline- and taxane-based chemotherapy: Correlative analysis of C9741 (Alliance). NPJ Breast Cancer 2016, 2, 15023. [Google Scholar] [CrossRef] [PubMed]
- Machiela, M.J.; Ho, B.M.; Fisher, V.A.; Hua, X.; Chanock, S.J. Limited evidence that cancer susceptibility regions are preferential targets for somatic mutation. Genome Biol. 2015, 16, 193. [Google Scholar] [CrossRef]
- Njiaju, U.O.; Olopade, O.I. Genetic Determinants of Breast Cancer Risk: A Review of Current Literature and Issues Pertaining to Clinical Application. Breast J. 2012, 18, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Landry, L.G.; Ali, N.; Williams, D.R.; Rehm, H.L.; Bonham, V.L. Lack Of Diversity In Genomic Databases Is A Barrier To Translating Precision Medicine Research Into Practice. Health Aff. 2018, 37, 780–785. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Chromosome Position | Expression p-Value | Number of Mutation Events |
|---|---|---|---|
| TTN * | 2q31.2 | 0.012712726 | 27 |
| MUC16 | 19p13.2 | 9.79 × 106 | 11 |
| OBSCN | 1q42.13 | 0.001208653 | 10 |
| SPTA1 | 1q23.1 | 4.58 × 10−5 | 9 |
| SYNE1 * | 6q25.2 | 1.11 × 10−44 | 9 |
| DNAH17 * | 17q25.3 | 4.74 × 10−5 | 8 |
| DST | 6p12.1 | 1.88 × 10−50 | 8 |
| MUC5B | 11p15.5 | 8.87 × 10−21 | 8 |
| PIK3CA * | 3q26.32 | 2.59 × 10−9 | 8 |
| AHCTF1 * | 1q44 | 2.54 × 10−9 | 7 |
| ASPM | 1q31.3 | 3.27 × 10−68 | 7 |
| CREBBP | 16p13.3 | 3.83 × 10−6 | 7 |
| CSMD2 | 1p35.1 | 1.38 × 10−12 | 7 |
| FLG | 1q21.3 | 0.018204904 | 7 |
| KMT2D * | 12q13.12 | 9.52 × 10−7 | 7 |
| PLEC | 8q24.3 | 0.000153621 | 7 |
| SMG1 | 16p12.3 | 0.040114062 | 7 |
| USP34 | 2p15 | 0.000850126 | 7 |
| AHNAK | 11q12.3 | 2.29 × 10−61 | 6 |
| ARID1B | 6q25.3 | 7.25 × 10−5 | 6 |
| CACNA1B | 9q34.3 | 9.79 × 10−8 | 6 |
| CENPE | 4q24 | 2.29 × 10−60 | 6 |
| COL18A1 | 21q22.3 | 0.007574248 | 6 |
| F5 | 1q24.2 | 6.47 × 10−8 | 6 |
| IGSF10 * | 3q25.1 | 2.63 × 10−38 | 6 |
| KIF26B | 1q44 | 9.80 × 10−26 | 6 |
| LAMA3 | 18q11.2 | 3.55 × 10−41 | 6 |
| LRP1 * | 12q13.3 | 5.49 × 10−33 | 6 |
| LYST | 1q42.3 | 1.39 × 10−21 | 6 |
| MFI2 | 3p29 | 5.09 × 10−13 | 6 |
| SAGE1 | Xq26.3 | 0.045910304 | 6 |
| SPTBN1 | 2p16.2 | 2.23 × 10−34 | 6 |
| STAB1 | 3p21.1 | 2.20 × 10−6 | 6 |
| ZNF512B | 20q13.33 | 2.73 × 10−8 | 6 |
| Genes | Chromosome Position | Genetic Variant | GWAS p-Value | Expression p-Value | Mutation Event |
|---|---|---|---|---|---|
| CREBBP | 16p13.3 | rs12920416 | 8.00 × 10−7 | 3.83 × 10−6 | 7 |
| ARID1B * | 6q25.3 | rs140842923 | 3.00 × 10−6 | 7.25 × 10−5 | 6 |
| BRCA1 * | 17q21.31 | rs6558174 | 3.00 × 10−6 | 3.95 × 10−7 | 5 |
| ERBB4 * | 2q34 | rs13393577 | 9.00 × 10−14 | 9.03 × 10−41 | 5 |
| FHOD3 | 18q12.2 | rs9956546 | 2.90 × 10−6 | 1.62 × 10−19 | 5 |
| TNRC6B | 22q13.1 | rs12483853 | 1.00 × 10−18 | 9.59 × 10−6 | 5 |
| ARHGAP24 | 4q21.23-q21.3 | rs71599425 | 6.00 × 10−6 | 4.05 × 10−44 | 4 |
| ARHGAP5 * | 14q12 | rs140783387 | 3.00 × 10−7 | 6.22 × 10−15 | 4 |
| CNTNAP2 | 7q35-q36.1 | rs72826962 | 5.00 × 10−9 | 2.35 × 10−5 | 4 |
| DMD | Xp21.2-p21.1 | rs145455135 | 9.00 × 10−6 | 2.42 × 10−40 | 4 |
| EFR3B * | 2p23.3 | rs1971136 | 5.009 | 0.000451 | 4 |
| MSH3 | 5q14.1 | rs1863333 | 0.0056 | 2.45 × 10−30 | 4 |
| MYO10 | 5p15.1 | rs2562343 | 0.0092 | 1.56 × 10−25 | 4 |
| MYT1 | 20q13.33 | rs6062356 | 3.00 × 10−6 | 0.00559 | 4 |
| RELN | 7q22.1 | rs17157903 | 0.0006 | 8.36E-21 | 4 |
| SPAG17 | 1p12 | rs1962373 | 1.00 × 10−6 | 7.67 × 10−5 | 4 |
| TRIM46 | 1q22 | rs4971059 | 5.00 × 10−11 | 1.14 × 10−17 | 4 |
| ZFPM2 | 8q23 | rs12546444 | 8.00 × 10−11 | 3.46 × 10−20 | 4 |
| ADCY9 | 16p13.3 | rs56278937 | 1.00 × 10−6 | 1.31 × 10−25 | 3 |
| AKAP9 * | 7q21.2 | rs10644111 | 3.00 × 10−11 | 2.70 × 10−6 | 3 |
| ASH1L * | 1q22 | rs10796944 | 7.00 × 10−10 | 5.39 × 10−6 | 3 |
| ASXL2 | 2p23.3 | rs144079028 | 9.00 × 10−6 | 0.000116 | 3 |
| ATM * | 11q22.3 | rs1801516 | 0.0002 | 1.35 × 10−8 | 3 |
| ATXN1 | 6p22.3 | rs3819405 | 2.00 × 10−8 | 3.92 × 10−6 | 3 |
| BAHCC1 * | 17q25.3 | rs8074440 | 3.00 × 10−6 | 0.02797 | 3 |
| CASZ1 | 1p36.22 | rs199867187 | 1.00 × 10−6 | 0.00068 | 3 |
| CHST9 * | 18q11.2 | rs1436904 | 3.00 × 10−8 | 1.52 × 10−11 | 3 |
| CNTNAP1 | 17q21.2 | rs72826962 | 5.00 × 10−9 | 8.69 × 10−9 | 3 |
| DNAH11 | 7p15.3 | rs7971 | 2.00 × 10−8 | 3.32 × 10−8 | 3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, J.; Mamidi, T.K.K.; Zhang, L.; Hicks, C. Integrating Germline and Somatic Mutation Information for the Discovery of Biomarkers in Triple-Negative Breast Cancer. Int. J. Environ. Res. Public Health 2019, 16, 1055. https://doi.org/10.3390/ijerph16061055
Wu J, Mamidi TKK, Zhang L, Hicks C. Integrating Germline and Somatic Mutation Information for the Discovery of Biomarkers in Triple-Negative Breast Cancer. International Journal of Environmental Research and Public Health. 2019; 16(6):1055. https://doi.org/10.3390/ijerph16061055
Chicago/Turabian StyleWu, Jiande, Tarun Karthik Kumar Mamidi, Lu Zhang, and Chindo Hicks. 2019. "Integrating Germline and Somatic Mutation Information for the Discovery of Biomarkers in Triple-Negative Breast Cancer" International Journal of Environmental Research and Public Health 16, no. 6: 1055. https://doi.org/10.3390/ijerph16061055
APA StyleWu, J., Mamidi, T. K. K., Zhang, L., & Hicks, C. (2019). Integrating Germline and Somatic Mutation Information for the Discovery of Biomarkers in Triple-Negative Breast Cancer. International Journal of Environmental Research and Public Health, 16(6), 1055. https://doi.org/10.3390/ijerph16061055