Sulforaphane Cannot Protect Human Fibroblasts From Repeated, Short and Sublethal Treatments with Hydrogen Peroxide

 , ,
, ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Cell Lines and Treatments
2.2. Proliferation Assay
2.3. Cell Cycle Analysis
2.4. p53 Level of Expression by Flow Cytometry
2.5. Quantification of Intracellular ROS by H2DCFDA
2.6. Quantification of Numbers of Mitochondria
2.7. Quantification of Thiols in Proteins
2.8. Western Blot
2.9. Immunofluorescence
2.10. -H2AX Spots Counting and Nuclear Survivin
2.11. Statistical Analysis
3. Results
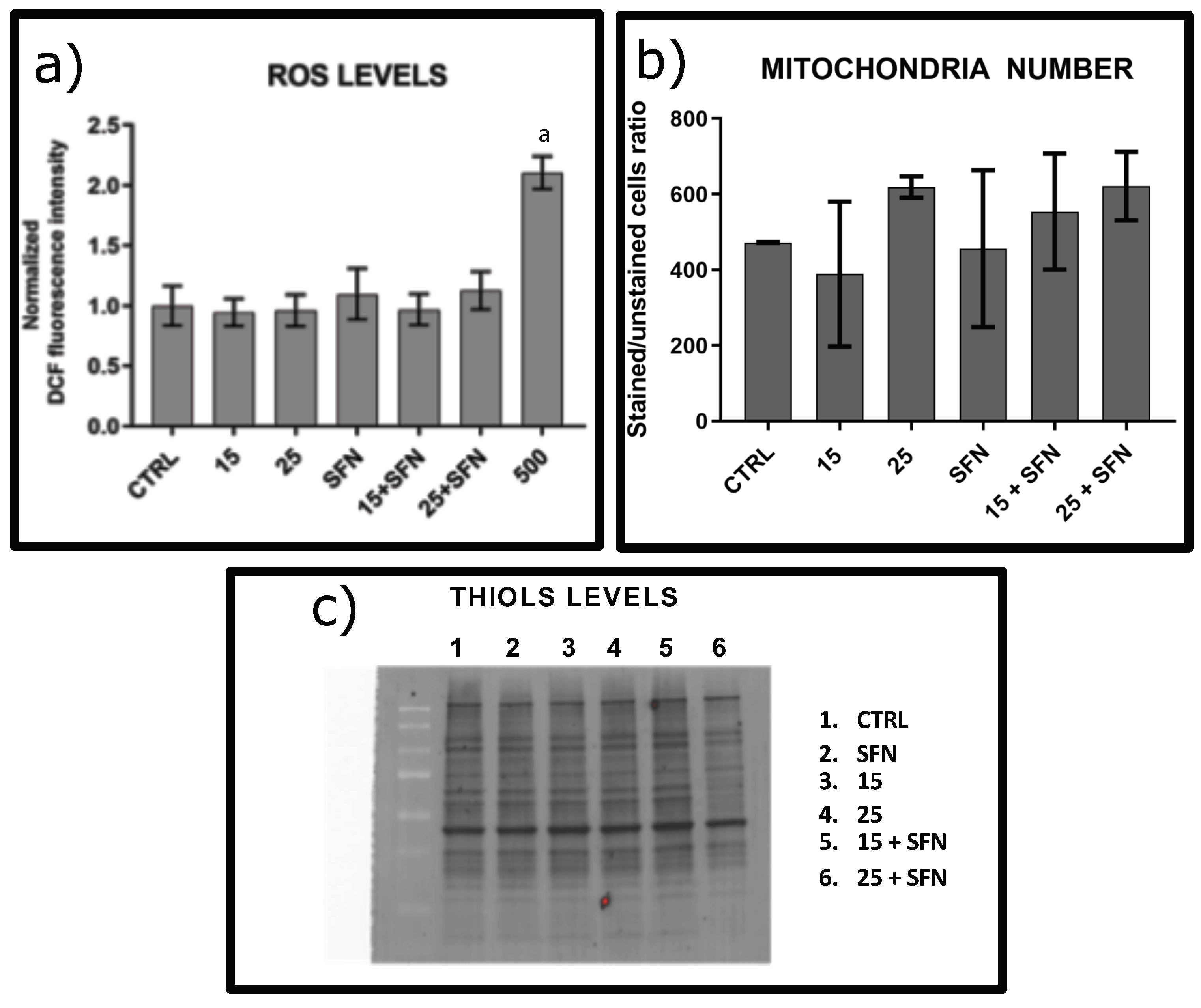
3.1. Effect of Short and Repeated Sublethal Treatment with Hydrogen Peroxide without or in Combination with SFN on the Oxidative Status of hSDF
3.2. SFN and Oxidative Stress Decrease Cell Vitality and Regulate Apoptosis
3.3. Effect of SFN Alone or with Hydrogen Peroxide on DNA Damage
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- LeBrasseur, N.K.; Tchkonia, T.; Kirkland, J.L. Cellular Senescence and the Biology of Aging, Disease, and Frailty. Nestlé Nutr. Inst. Workshop Ser. 2015, 83, 11–18. [Google Scholar] [PubMed]
- Caldini, R.; Chevanne, M.; Mocali, A.; Tombaccini, D.; Paoletti, F. Premature induction of aging in sublethally H2O2-treated young MRC5 fibroblasts correlates with increased glutathione peroxidase levels and resistance to DNA breakage. Mech. Ageing Dev. 1998, 105, 137–150. [Google Scholar] [CrossRef]
- Davalli, P.; Mitic, T.; Caporali, A.; Lauriola, A.; D’Arca, D. ROS, Cell Senescence, and Novel Molecular Mechanisms in Aging and Age-Related Diseases. Oxid. Med. Cell Longev. 2016, 2016, 3565127. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Schmeisser, S. Extending life span by increasing oxidative stress. Free Radic. Biol. Med. 2011, 51, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Cachat, J.; Deffert, C.; Hugues, S.; Krause, K.H. Phagocyte NADPH oxidase and specific immunity. Clin. Sci. 2015, 128, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Lawless, C.; Jurk, D.; Gillespie, C.S.; Shanley, D.; Saretzki, G.; von Zglinicki, T.; Passos, J.F. A stochastic step model of replicative senescence explains ROS production rate in aging cell populations. PLoS ONE 2012, 7, e32117. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.J. Positive oxidative stress in aging and aging-related disease tolerance. Redox Biol. 2014, 2, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Yin, P.H.; Lu, C.Y.; Chi, C.W.; Wei, Y.H. Increase of mitochondria and mitochondrial DNA in response to oxidative stress in human cells. Biochem. J. 2000, 348, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Spagnuolo, C.; Russo, G.L.; Skalicka-Woźniak, K.; Daglia, M.; Sobarzo-Sánchez, E.; Nabavi, S.F.; Nabavi, S.M. Nrf2 targeting by sulforaphane: A potential therapy for cancer treatment. Crit. Rev. Food Sci. Nutr. 2018, 58, 1391–1405. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Hung, C.M.; Yang, Y.R.; Lee, M.J.; Hsu, Y.C. Sulforaphane induced cell cycle arrest in the G2/M phase via the blockade of cyclin B1/CDC2 in human ovarian cancer cells. J. Ovarian Res. 2013, 6. [Google Scholar] [CrossRef] [PubMed]
- Lan, H.; Yuan, H.; Lin, C. Sulforaphane induces p53-deficient SW480 cell apoptosis via the ROS-MAPK signaling pathway. Mol. Med. Rep. 2017, 16, 7796–7804. [Google Scholar] [CrossRef] [PubMed]
- Coccè, V.; Vitale, A.; Colombo, S.; Bonomi, A.; Sisto, F.; Ciusani, E.; Alessandri, G.; Parati, E.; Brambilla, P.; Brambilla, M.; La Porta, C.A.; Pessina, A. Human skin-derived fibroblasts used as a ’Trojan horse’ for drug delivery. Clin. Exp. Dermatol. 2016, 41, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Orellana, E.A.; Kasinski, A.L. Sulforhodamine B (SRB) Assay in Cell Culture to Investigate Cell Proliferation. Bio-protocol 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Ormerod, M.; Tribukait, B.; Giaretti, W. Consensus report of the task force on standardisation of DNA flow cytometry in clinical pathology. Anal. Cell. Pathol. 1998, 17, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Hill, B.G.; Reily, C.; Oh, J.Y.; Johnson, M.S.; Landar, A. Methods for the determination and quantification of the reactive thiol proteome. Free Radic. Biol. Med. 2009, 47, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Rivero-Gutiérrez, B.; Anzola, A.; Martínez-Augustin, O.; de Medina, F.S. Stain-free detection as loading control alternative to Ponceau and housekeeping protein immunodetection in Western blotting. Anal. Biochem. 2014, 467, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Giampietro, C.; Lionetti, M.C.; Costantini, G.; Mutti, F.; Zapperi, S.; La Porta, C.A. Cholesterol impairment contributes to neuroserpin aggregation. Sci. Rep. 2017, 7, 43669. [Google Scholar] [CrossRef] [PubMed]
- Thimmulappa, R.K.; Mai, K.H.; Srisuma, S.; Kensler, T.W.; Yamamoto, M.; Biswal, S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002, 62, 5196–5203. [Google Scholar] [PubMed]
- Wagner, A.E.; Ernst, I.; Iori, R.; Desel, C.; Rimbach, G. Sulforaphane but not ascorbigen, indole-3-carbinole and ascorbic acid activates the transcription factor Nrf2 and induces phase-2 and antioxidant enzymes in human keratinocytes in culture. Exp. Dermatol. 2010, 19, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Deneke, S.M. Thiol-based antioxidants. Curr. Top. Cell Regul. 2000, 36, 151–180. [Google Scholar] [PubMed]
- Włodek, L. Beneficial and harmful effects of thiols. Pol. J. Pharmacol. 2002, 54, 215–223. [Google Scholar] [PubMed]
- Rossi, R.; Giustarini, D.; Milzani, A.; Dalle-Donne, I. Cysteinylation and homocysteinylation of plasma protein thiols during aging of healthy human beings. J. Cell. Mol. Med. 2009, 13, 3131–3140. [Google Scholar] [CrossRef] [PubMed]
- Giustarini, D.; Dalle-Donne, I.; Lorenzini, S.; Selvi, E.; Colombo, G.; Milzani, A.; Fanti, P.; Rossi, R. Protein thiolation index (PTI) as a biomarker of oxidative stress. Free Radic. Biol. Med. 2012, 53, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Colombo, G.; Reggiani, F.; Podestà, M.A.; Garavaglia, M.L.; Portinaro, N.M.; Milzani, A.; Badalamenti, S.; Dalle-Donne, I. Plasma protein thiolation index (PTI) as a biomarker of thiol-specific oxidative stress in haemodialyzed patients. Free Radic. Biol. Med. 2015, 89, 443–4451. [Google Scholar] [CrossRef] [PubMed]
- Purvis, J.E.; Karhohs, K.W.; Mock, C.; Batchelor, E.; Loewer, A.; Lahav, G. p53 dynamics control cell fate. Science 2012, 336, 1440–1444. [Google Scholar] [CrossRef] [PubMed]
- Hovest, M.G.; Brüggenolte, N.; Hosseini, K.S.; Krieg, T.; Herrmann, G. Senescence of human fibroblasts after psoralen photoactivation is mediated by ATR kinase and persistent DNA damage foci at telomeres. Mol. Biol. Cell. 2006, 17, 1758–1767. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, D.; Roedl, D.; Gordon, L.B.; Djabali, K. Sulforaphane enhances progerin clearance in Hutchinson-Gilford progeria fibroblasts. Aging Cell 2015, 14, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Burnett, J.; Lim, G.; Li, Y.; Shah, R.; Lim, R.; Paholak, H.; McDermott, S.; Sun, L.; Tsume, Y.; Bai, S.; et al. Sulforaphane enhances the anticancer activity of taxanes against triple negative breast cancer by killing cancer stem cells. Cancer Lett. 2017, 394, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Zanichelli, F.; Capasso, S.; Cipollaro, M.; Pagnotta, E.; Cartenì, M.; Casale, F.; Iori, R.; Galderisi, U. Dose-dependent effects of R-sulforaphane isothiocyanate on the biology of human mesenchymal stem cells, at dietary amounts, it promotes cell proliferation and reduces senescence and apoptosis, while at anti-cancer drug doses, it has a cytotoxic effect. Age 2012, 34, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Sauer, H.; Wartenberg, M.; Hescheler, J. Reactive oxygen species as intracellular messengers during cell growth and differentiation. Cell. Physiol. Biochem. 2001, 11, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Schmeisser, K. Mitohormesis: Promoting health and lifespan by increased levels of reactive oxygen species (ROS). Dose-Response 2014, 12, 288–341. [Google Scholar] [CrossRef] [PubMed]
- Shimi, T.; Goldman, R.D. Nuclear lamins and oxidative stress in cell proliferation and longevity. In Cancer Biology and the Nuclear Envelope; Springer: New York, NY, USA, 2014; pp. 415–430. [Google Scholar]
- Helmut Sies, C.B.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef] [PubMed]
- Pickering, A.M.; Vojtovich, L.; Tower, J.; Davies, K.J. Oxidative stress adaptation with acute, chronic, and repeated stress. Free Radic. Biol. Med. 2013, 55, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.; Na, W.; Kabir, M.; Yi, E.; Kwon, S.; Yeom, J.; Ahn, J.W.; Choi, H.H.; Lee, Y.; Seo, K.; et al. WIP1, a Homeostatic Regulator of the DNA Damage Response, Is Targeted by HIPK2 for Phosphorylation and Degradation. Mol. Cell 2013, 51, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.P.; Liu, F.; Wang, W. Two-phase dynamics of p53 in the DNA damage response. Proc. Natl. Acad. Sci. USA 2011, 108, 8990–8995. [Google Scholar] [CrossRef] [PubMed]
- Torrente, L.; Sanchez, C.; Moreno, R.; Chowdhry, S.; Cabello, P.; Isono, K.; Koseki, H.; Honda, T.; Hayes, J.; Dinkova-Kostova, A.; et al. Crosstalk between NRF2 and HIPK2 shapes cytoprotective responses. Oncogene 2017, 36, 6204–6212. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Egner, P.A.; Agyeman, A.S.; Visvanathan, K.; Groopman, J.D.; Chen, J.G.; Chen, T.Y.; Fahey, J.W.; Talalay, P. Keap1–nrf2 signaling: A target for cancer prevention by sulforaphane. In Natural Products in Cancer Prevention and Therapy; Springer: Berlin/Heidelberg, Germany, 2012; pp. 163–177. [Google Scholar]
- Takekawa, M.; Adachi, M.; Nakahata, A.; Nakayama, I.; Itoh, F.; Tsukuda, H.; Taya, Y.; Imai, K. p53-inducible Wip1 phosphatase mediates a negative feedback regulation of p38 MAPK-p53 signaling in response to UV radiation. EMBO J. 2000, 19, 6517–6526. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, E.; Mock, C.S.; Bhan, I.; Loewer, A.; Lahav, G. Recurrent initiation: a mechanism for triggering p53 pulses in response to DNA damage. Mol. Cell 2008, 30, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.H.; Lim, Y.; Kim, S.J.; Yoo, K.D.; Yoo, H.S.; Hong, J.T.; Lee, M.Y.; Yun, Y.P. Sulforaphane inhibits PDGF-induced proliferation of rat aortic vascular smooth muscle cell by up-regulation of p53 leading to G1/S cell cycle arrest. Vascular Pharmacol. 2013, 59, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Negrette-Guzmán, M.; Huerta-Yepez, S.; Medina-Campos, O.; Zatarain-Barrón, Z.; Hernández-Pando, R.; Torres, I.; Tapia, E.; Pedraza-Chaverri, J. Sulforaphane attenuates gentamicin-induced nephrotoxicity: Role of mitochondrial protection. Evid.-Based Complement. Altern. Med. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lionetti, M.C.; Mutti, F.; Soldati, E.; Fumagalli, M.R.; Coccé, V.; Colombo, G.; Astori, E.; Miani, A.; Milzani, A.; Dalle-Donne, I.; et al. Sulforaphane Cannot Protect Human Fibroblasts From Repeated, Short and Sublethal Treatments with Hydrogen Peroxide. Int. J. Environ. Res. Public Health 2019, 16, 657. https://doi.org/10.3390/ijerph16040657
Lionetti MC, Mutti F, Soldati E, Fumagalli MR, Coccé V, Colombo G, Astori E, Miani A, Milzani A, Dalle-Donne I, et al. Sulforaphane Cannot Protect Human Fibroblasts From Repeated, Short and Sublethal Treatments with Hydrogen Peroxide. International Journal of Environmental Research and Public Health. 2019; 16(4):657. https://doi.org/10.3390/ijerph16040657
Chicago/Turabian StyleLionetti, Maria Chiara, Federico Mutti, Erica Soldati, Maria Rita Fumagalli, Valentina Coccé, Graziano Colombo, Emanuela Astori, Alessandro Miani, Aldo Milzani, Isabella Dalle-Donne, and et al. 2019. "Sulforaphane Cannot Protect Human Fibroblasts From Repeated, Short and Sublethal Treatments with Hydrogen Peroxide" International Journal of Environmental Research and Public Health 16, no. 4: 657. https://doi.org/10.3390/ijerph16040657
APA StyleLionetti, M. C., Mutti, F., Soldati, E., Fumagalli, M. R., Coccé, V., Colombo, G., Astori, E., Miani, A., Milzani, A., Dalle-Donne, I., Ciusani, E., Costantini, G., & La Porta, C. A. M. (2019). Sulforaphane Cannot Protect Human Fibroblasts From Repeated, Short and Sublethal Treatments with Hydrogen Peroxide. International Journal of Environmental Research and Public Health, 16(4), 657. https://doi.org/10.3390/ijerph16040657