Influence of Iron on Cytotoxicity and Gene Expression Profiles Induced by Arsenic in HepG2 Cells

Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment
2.2. Cytotoxicity Test
2.3. Mitochondrial Membrane Potential Assay
2.4. Oxidative Stress Analysis
2.5. Microarray Analysis
2.6. Real-Time Quantitative PCR (qPCR)
2.7. Statistical Analysis
3. Results and Discussion
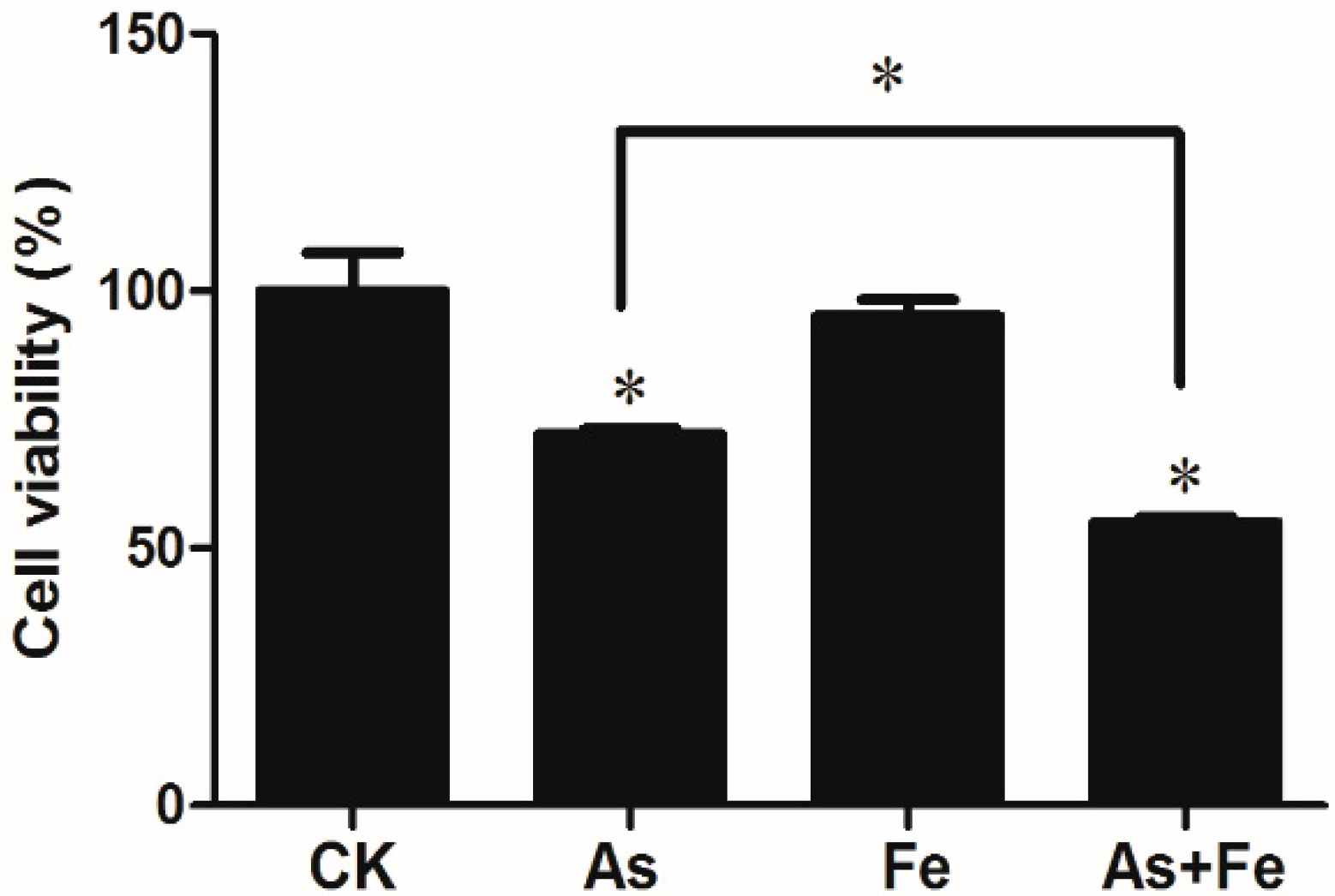
3.1. Cytotoxicity of As and Fe in HepG2
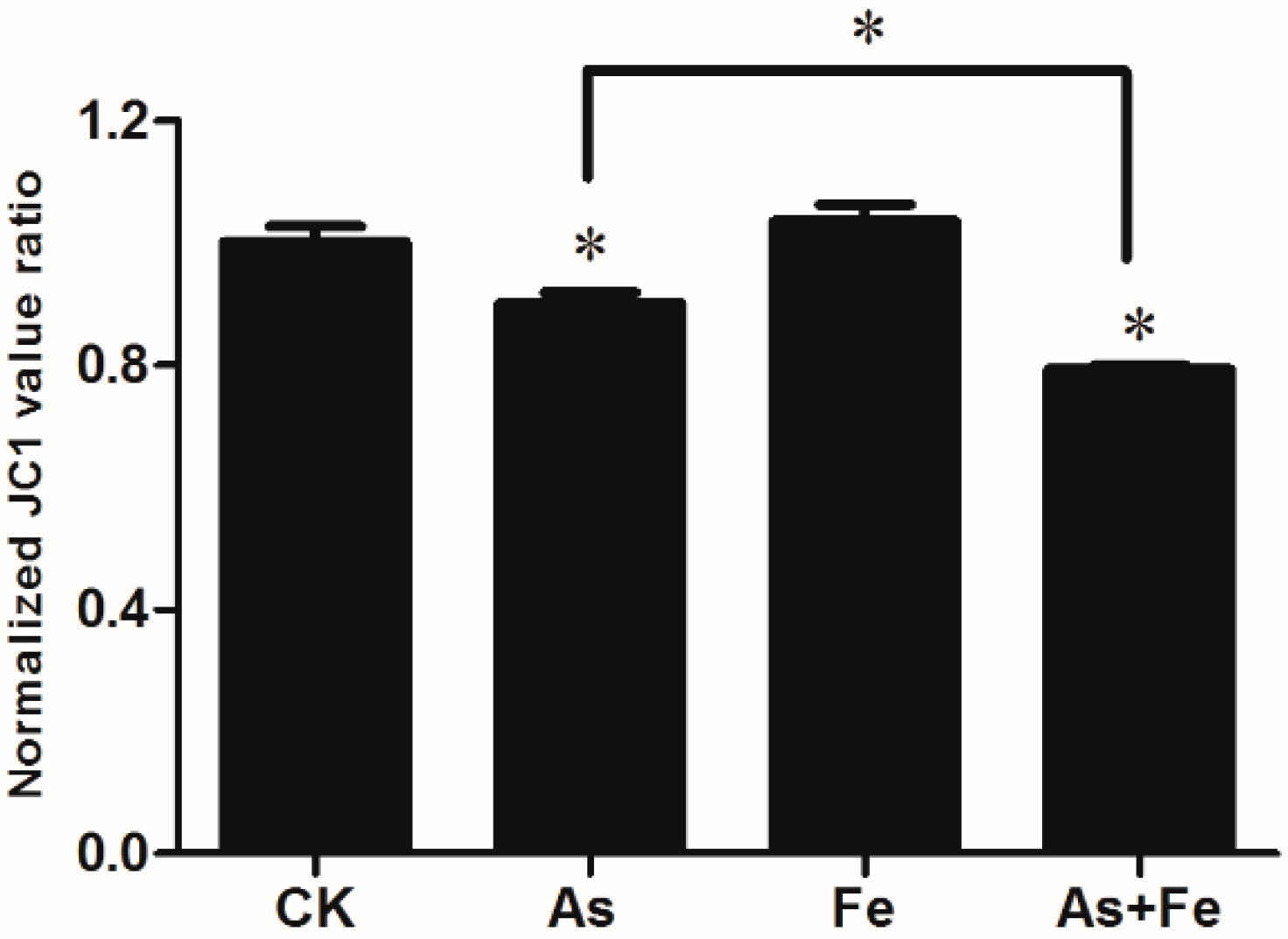
3.2. Changes in Mitochondrial Membrane Potential Induced by As and Fe in HepG2 Cell
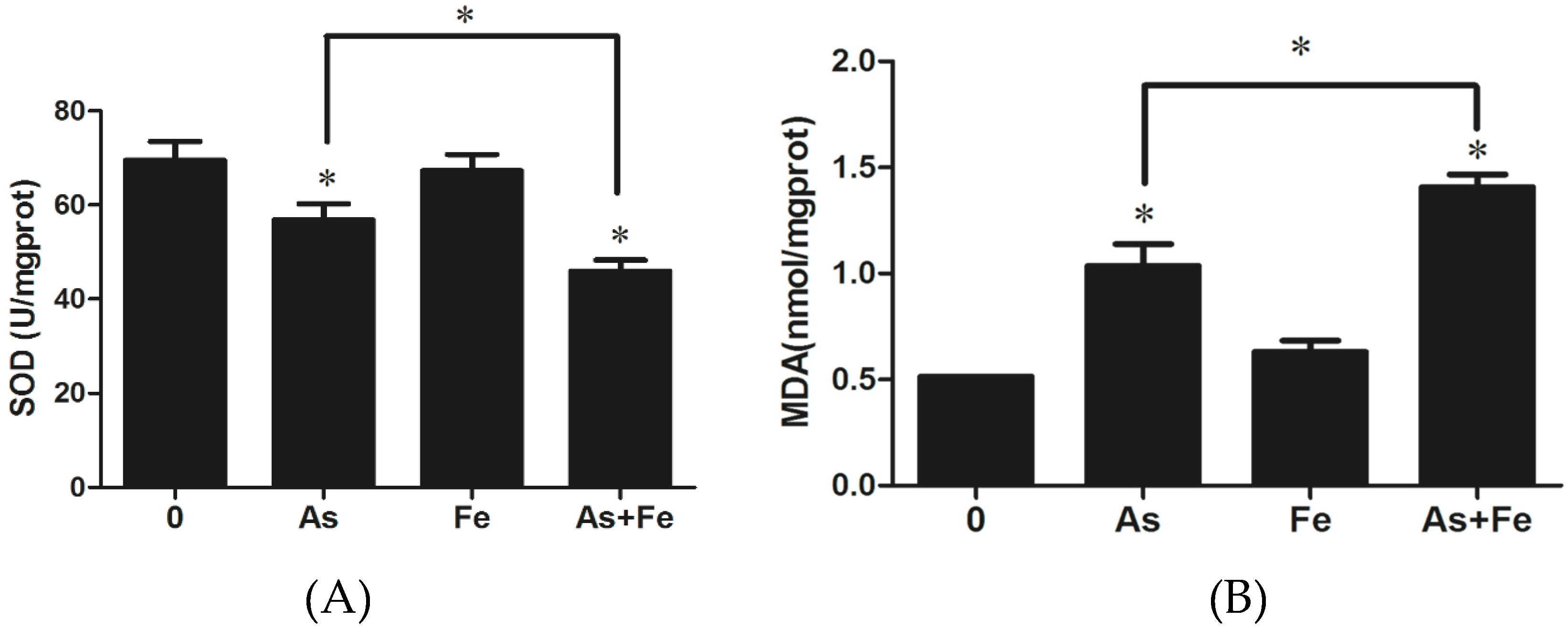
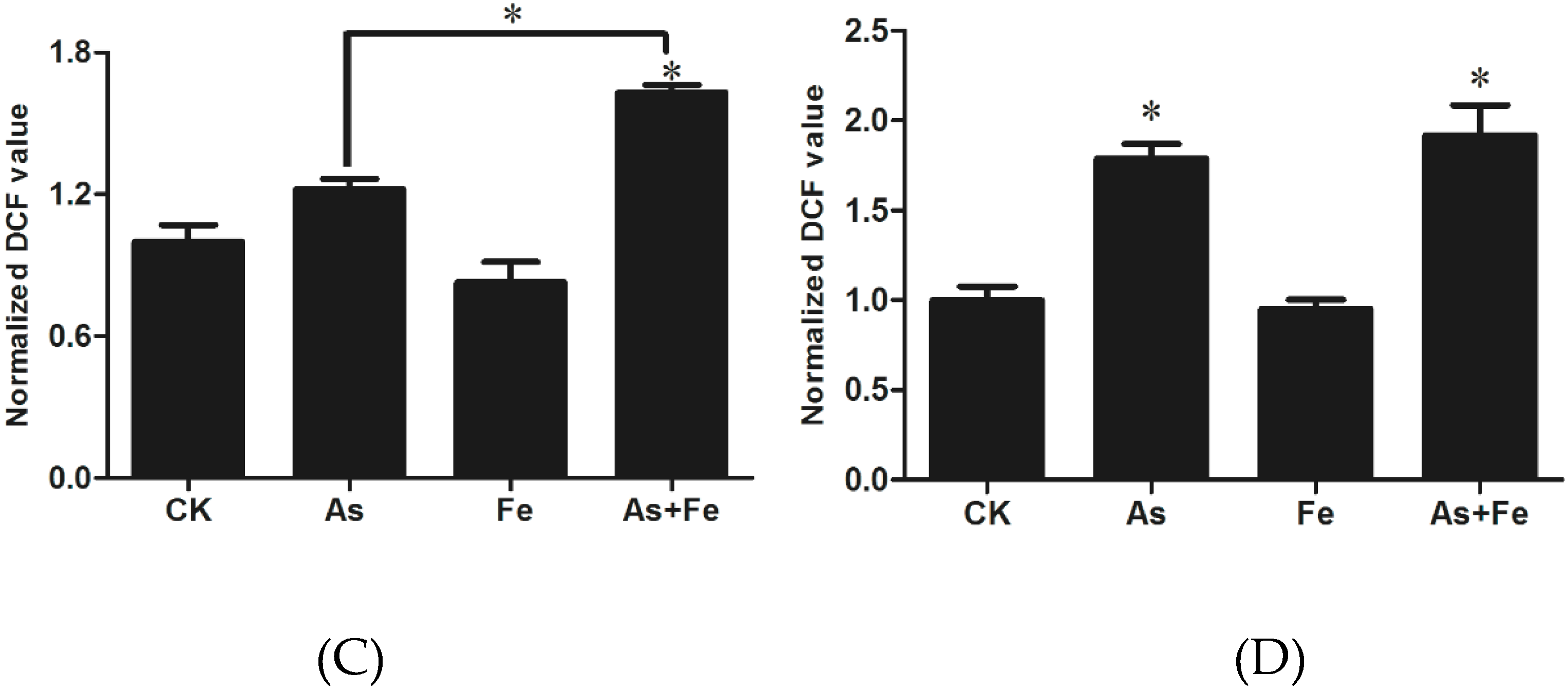
3.3. Determination of Oxidative Stress and Damage in HepG2
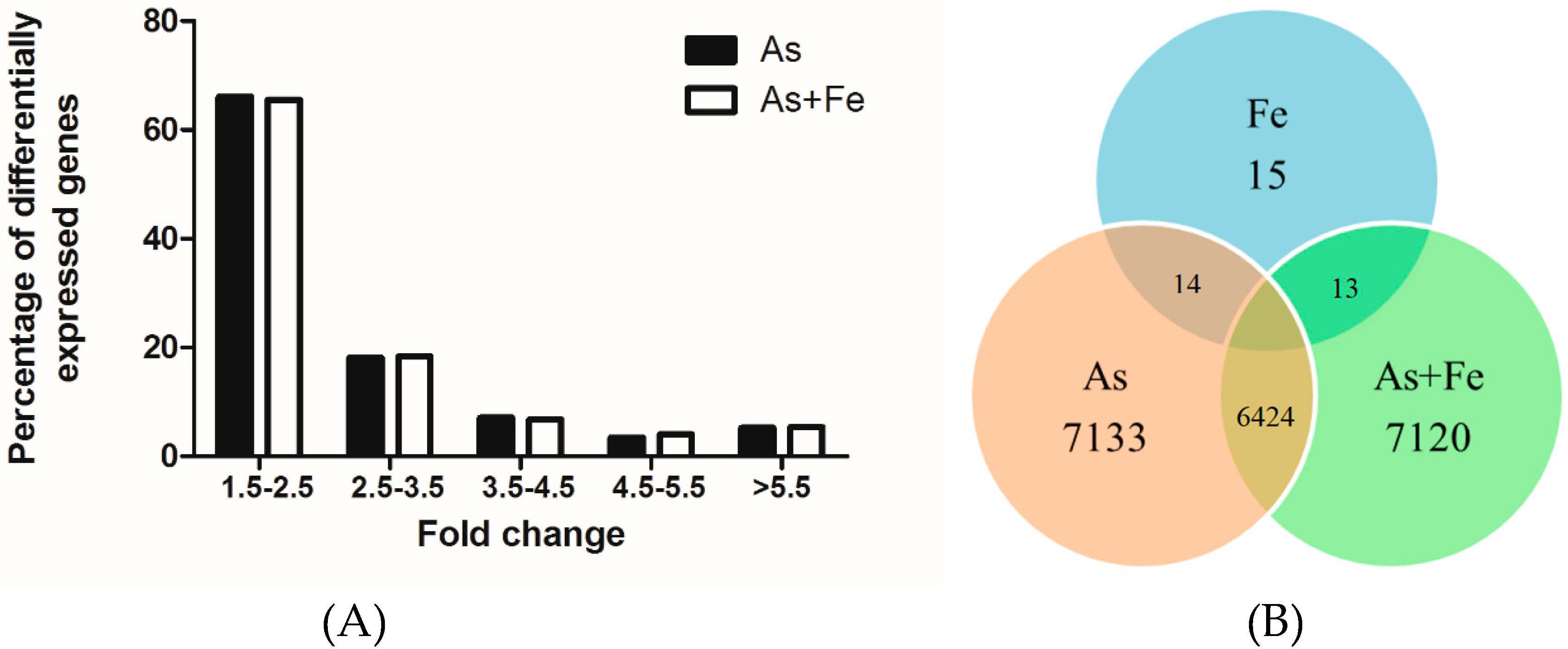
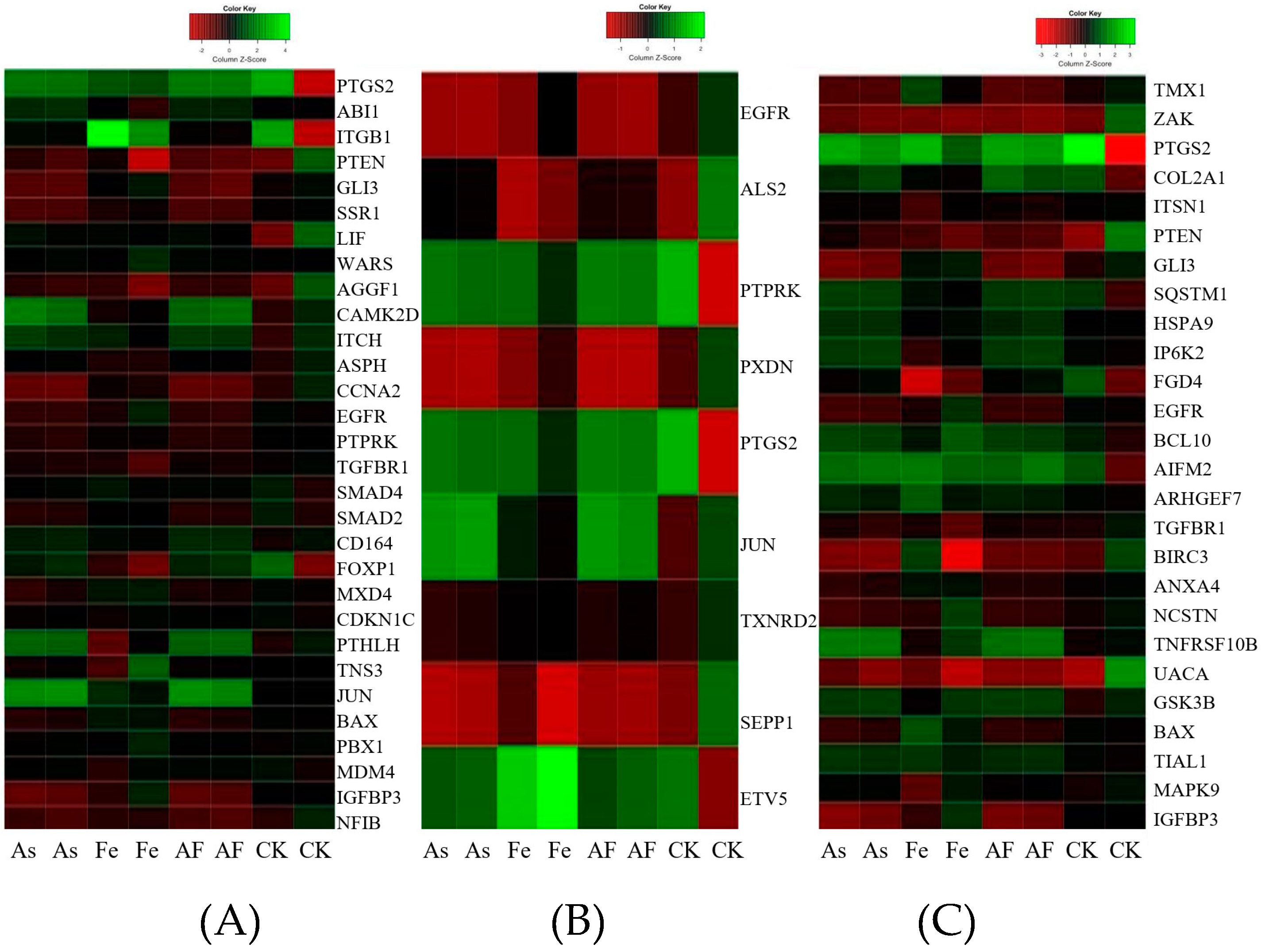
3.4. Gene Expression Profiles in HepG2 Exposed to As and Fe
3.5. Comparative Analysis of Toxicity in HepG2 Exposed to As Alone and Combined As and Fe
3.6. Inhibition of cell Proliferation
3.7. Oxidative Stress and Damage
3.8. Apoptotic singaling Pathway
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Naujokas, M.F.; Anderson, B.; Ahsan, H.; Aposhian, H.V.; Graziano, J.H.; Thompson, C.; Suk, W.A. The broad scope of health effects from chronic arsenic exposure: Update on a worldwide public health problem. Environ. Health Perspect. 2013, 121, 295–302. [Google Scholar] [CrossRef]
- Smith, A.H.; Steinmaus, C.M. Health effects of arsenic and chromium in drinking water: recent human findings. Annu. Rev. Public Health 2009, 30, 107–122. [Google Scholar] [CrossRef] [PubMed]
- Benbrahim-Tallaa, L.; Waalkes, M.P. Inorganic arsenic and human prostate cancer. Environ. Health Perspect. 2008, 116, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Yamauchi, H.; Sun, G.F. Chronic health effects in people exposed to arsenic via the drinking water: Dose-response relationships in review. Toxicol. Appl. Pharmacol. 2004, 198, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, J.S.; Perez, V.; Garry, M.R.; Alexander, D.D. Association of low-level arsenic exposure in drinking water with cardiovascular disease: A systematic review and risk assessment. Toxicology 2014, 323, 78–94. [Google Scholar] [CrossRef] [PubMed]
- Maull, E.A.; Ahsan, H.; Edwards, J.; Longnecker, M.P.; Navas-Acien, A.; Pi, J.; Silbergeld, E.K.; Styblo, M.; Tseng, C.-H.; Thayer, K.A.; et al. Evaluation of the association between arsenic and diabetes: A national toxicology program workshop review. Environ. Health Perspect. 2012, 120, 1658–1670. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, B.; Xi, S.; Zheng, Q.; Lv, X.; Sun, G. Prolonged environmental exposure of arsenic through drinking water on the risk of hypertension and type 2 diabetes. Environ. Sci. Pollut. Res. 2013, 20, 8151–8161. [Google Scholar] [CrossRef]
- Chen, Y.; Parvez, F.; Gamble, M.; Islam, T.; Ahmed, A.; Argos, M.; Graziano, J.H.; Ahsan, H. Arsenic exposure at low-to-moderate levels and skin lesions, arsenic metabolism, neurological functions, and biomarkers for respiratory and cardiovascular diseases: Review of recent findings from the Health Effects of Arsenic Longitudinal Study (HEALS) in Bangladesh. Toxicol. Appl. Pharmacol. 2009, 239, 184–192. [Google Scholar]
- Herrera, A.; Pineda, J.; Teresa Antonio, M. Toxic effects of perinatal arsenic exposure on the brain of developing rats and the beneficial role of natural antioxidants. Environ. Toxicol. Phar. 2013, 36, 73–79. [Google Scholar] [CrossRef]
- McCarty, K.M.; Houseman, E.A.; Quamruzzaman, Q.; Rahman, M.; Mahiuddin, G.; Smith, T.; Ryan, L.; Christiani, D.C. The impact of diet and betel nut use on skin lesions associated with drinking-water arsenic in Pabna, Bangladesh. Environ. Health Perspect. 2006, 114, 334–340. [Google Scholar] [CrossRef]
- Sinha, D.; Roy, M.; Siddiqi, M.; Bhattacharya, R.K. Arsenic-induced micronuclei formation in mammalian cells and its counteraction by tea. J. Environ. Pathol. Toxicol. Oncol. 2005, 24, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, F. Effects of exogenous glutathione on arsenic distribution and no metabolism in brain of female mice exposed to sodium arsenite through drinking water. J. Environ. Health 2009, 26, 1046–1048. [Google Scholar]
- Li, L.; Qiu, P.; Chen, B.; Lu, Y.; Wu, K.; Thakur, C.; Chang, Q.; Sun, J.; Chen, F. Reactive oxygen species contribute to arsenic-induced EZH2 phosphorylation in human bronchial epithelial cells and lung cancer cells. Toxicol. Appl. Pharm. 2014, 276, 165–170. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tortora, F.; Notariale, R.; Maresca, V.; Good, K.V.; Sorbo, S.; Basile, A.; Piscopo, M.; Manna, C. Phenol-rich Feijoa sellowiana (Pineapple guava) extracts protect human red blood cells from mercury-induced cellular toxicity. Antioxidants 2019, 8, 220. [Google Scholar] [CrossRef] [PubMed]
- Paul, P.C.; Misbahuddin, M.; Ahmed, A.N.N.; Dewan, Z.F.; Mannan, M.A. Accumulation of arsenic in tissues of iron-deficient rats. Toxicol. Lett. 2002, 135, 193–197. [Google Scholar] [CrossRef]
- Rahman, M.M.; Rahman, F.; Sansom, L.; Naidu, R.; Schmidt, O. Arsenic interactions with lipid particles containing iron. Environ. Geochem. Health 2009, 31, 201–206. [Google Scholar] [CrossRef]
- Jomova, K.; Jenisova, Z.; Feszterova, M.; Baros, S.; Liska, J.; Hudecova, D.; Rhodes, C.J.; Valko, M. Arsenic: Toxicity, oxidative stress and human disease. J. Appl. Toxicol. 2011, 31, 95–107. [Google Scholar] [CrossRef]
- Del Razo, L.M.; Quintanilla-Vega, B.; Brambila-Colombres, E.; Calderon-Aranda, E.S.; Manno, M.; Albores, A. Stress proteins induced by arsenic. Toxicol. Appl. Pharm. 2001, 177, 132–148. [Google Scholar] [CrossRef]
- Ahmad, S.; Kitchin, K.T.; Cullen, W.R. Arsenic species that cause release of iron from ferritin and generation of activated oxygen. Arch. Biochem. Biophys. 2000, 382, 195–202. [Google Scholar] [CrossRef]
- Poddar, S.; Mukherjee, P.; Talukder, G.; Sharma, A. Dietary protection by iron against clastogenic effects of short-term exposure to arsenic in mice in vivo. Food Chem. Toxicol. 2000, 38, 735–737. [Google Scholar] [CrossRef]
- Chandrasekaran, V.R.M.; Muthaiyan, I.; Huang, P.C.; Liu, M.Y. Using iron precipitants to remove arsenic from water: Is it safe? Water Res. 2010, 44, 5823–5827. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Guo, X.; Zhang, X.; Cui, Y.; Zhang, Y.; Wu, B. Impact of iron precipitant on toxicity of arsenic in water: A combined in vivo and in vitro study. Environ. Sci. Technol. 2013, 47, 3432–3438. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.N.; Niedzwiecki, M.; Liu, X.; Harper, K.N.; Alam, S.; Slavkovich, V.; Ilievski, V.; Levy, D.; Siddique, A.B.; Parvez, F.; et al. Chronic arsenic exposure and blood glutathione and glutathione disulfide concentrations in Bangladeshi adults. Environ. Health Perspect. 2013, 121, 1068–1074. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Zhang, Y.; Zhao, D.; Zhang, X.; Kong, Z.; Cheng, S. Gene expression profiles in liver of mouse after chronic exposure to drinking water. J. Appl. Toxicol. 2009, 29, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Waalkes, M.P. Liver is a target of arsenic carcinogenesis. Toxicol. Sci. 2008, 105, 24–32. [Google Scholar] [CrossRef]
- Chen, C.; Jiang, X.; Ren, Y.; Zhang, Z. Arsenic trioxide co-exposure potentiates benzo(a)pyrene genotoxicity by enhancing the oxidative stress in human lung adenocarcinoma cell. Biol. Trace Elem. Res. 2013, 156, 338–349. [Google Scholar] [CrossRef]
- Kang, Y.H.; Lee, S.J. The role of p38 MAPK and JNK in arsenic trioxide-induced mitochondrial cell death in human cervical cancer cells. J. Cell. Physiol. 2008, 217, 23–33. [Google Scholar] [CrossRef]
- Jiang, X.H.; Wong, B.C.Y.; Yuen, S.T.; Jiang, S.H.; Cho, C.H.; Lai, K.C.; Lin, M.C.M.; Kung, H.F.; Lam, S.K. Arsenic trioxide induces apoptosis in human gastric cancer cells through up-regulation of p53 and activation of caspase-3. Int. J. Cancer 2001, 93, 916. [Google Scholar] [CrossRef][Green Version]
- Shi, H.L.; Shi, X.L.; Liu, K.J. Oxidative mechanism of arsenic toxicity and carcinogenesis. Mol. Cell. Biochem. 2004, 255, 67–78. [Google Scholar] [CrossRef]
- De Vizcaya-Ruiz, A.; Barbier, O.; Ruiz-Ramos, R.; Cebrian, M.E. Biomarkers of oxidative stress and damage in human populations exposed to arsenic. Mutat. Res.-Genent. Toxicol. Environ. Mutagen. 2009, 674, 85–92. [Google Scholar] [CrossRef]
- Marnett, L.J. Lipid peroxidation—DNA damage by malondialdehyde. Mutat. Res./Fundam. Mol. Mech. Mutagen. 1999, 424, 83–95. [Google Scholar] [CrossRef]
- Luster, M.I.; Simeonova, P.P. Arsenic and urinary bladder cell proliferation. Toxicol. Appl. Pharm. 2004, 198, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Levin, J.; Sulpice, E.; Sequeira-Le Grand, A.; Alemany, M.; Caen, J.P.; Han, Z.C. Effect of arsenic trioxide on viability, proliferation, and apoptosis in human megakaryocytic leukemia cell lines. Exp. Hematol. 1999, 27, 845–852. [Google Scholar] [CrossRef]
- Chowdhury, R.; Chatterjee, R.; Giri, A.K.; Mandal, C.; Chaudhuri, K. Arsenic-induced cell proliferation is associated with enhanced ros generation, erk signaling and cyclina expression. Toxicol. Lett. 2010, 198, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Seoane, J.; Le, H.V.; Shen, L.J.; Anderson, S.A.; Massague, J. Integration of Smad and Forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell 2004, 117, 211–223. [Google Scholar] [CrossRef]
- Chen, W.; Martindale, J.L.; Holbrook, N.J.; Liu, Y.S. Tumor promoter arsenite activates extracellular signal-regulated kinase through a signaling pathway mediated by epidermal growth factor receptor and Shc. Mol. Cell. Biol. 1998, 18, 5178–5188. [Google Scholar] [CrossRef]
- Simeonova, P.P.; Wang, S.Y.; Hulderman, T.; Luster, M.I. c-Src-dependent activation of the epidermal growth factor receptor and mitogen-activated protein kinase pathway by arsenic—Role in carcinogenesis. J. Biol. Chem. 2002, 277, 2945–2950. [Google Scholar] [CrossRef]
- Ten Dijke, P.T.; Goumans, M.J.; Itoh, F.; Itoh, S. Regulation of cell proliferation by Smad proteins. J. Cell. Physiol. 2002, 191, 1–16. [Google Scholar] [CrossRef]
- Sengupta, S.; Jana, S.; Bhattacharyya, A. TGF-beta-Smad2 dependent activation of CDC 25A plays an important role in cell proliferation through NFAT activation in metastatic breast cancer cells. Cell. Signal. 2014, 26, 240–252. [Google Scholar] [CrossRef]
- Lee, Y.S.; Lee, S.J.; Seo, K.W.; Bae, J.U.; Park, S.Y.; Kim, C.D. Homocysteine induces COX-2 expression in macrophages through ROS generated by NMDA receptor-calcium signaling pathways. Free Radic. Res. 2013, 47, 422–431. [Google Scholar] [CrossRef]
- He, J.; Wang, M.; Jiang, Y.; Chen, Q.; Xu, S.; Xu, Q.; Jiang, B.-H.; Liu, L.-Z. Chronic arsenic exposure and angiogenesis in human bronchial epithelial cells via the ROS/miR-199a-5p/HIF-1 alpha/COX-2 pathway. Environ. Health Perspect. 2014, 122, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Edvardsen, H.; Landmark-Hoyvik, H.; Reinertsen, K.V.; Zhao, X.; Grenaker-Alnaes, G.I.; Nebdal, D.; Syvanen, A.-C.; Rodningen, O.; Alsner, J.; Overgaard, J.; et al. SNP in TXNRD2 associated with radiation-induced fibrosis: A study of genetic variation in reactive oxygen species metabolism and signaling. Int. J. Radiat. Oncol. 2013, 86, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Velez, J.M.; Miriyala, S.; Nithipongvanitch, R.; Noel, T.; Plabplueng, C.D.; Oberley, T.; Jungsuwadee, P.; Van Remmen, H.; Vore, M.; Clair, D.K.S. p53 regulates oxidative stress-mediated retrograde signaling: A novel mechanism for chemotherapy-induced cardiac injury. PLoS ONE 2011, 6, e18005. [Google Scholar] [CrossRef] [PubMed]
- Sugden, P.H.; Clerk, A. Oxidative stress and growth-regulating intracellular signaling pathways in cardiac myocytes. Antioxid. Redox Signal. 2006, 8, 2111–2124. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Copple, I.M.; Lister, A.; Obeng, A.D.; Kitteringham, N.R.; Jenkins, R.E.; Layfield, R.; Foster, B.J.; Goldring, C.E.; Park, B.K. Physical and Functional Interaction of Sequestosome 1 with Keap1 Regulates the Keap1-Nrf2 Cell Defense Pathway. J. Biol. Chem. 2010, 285, 16782–16788. [Google Scholar] [CrossRef]
- Bartolome, F.; Esteras, N.; Martin-Requero, A.; Boutoleau-Bretonniere, C.; Vercelletto, M.; Gabelle, A.; Le Ber, I.; Honda, T.; Dinkova-Kostova, A.T.; Hardy, J.; et al. Pathogenic p62/SQSTM1 mutations impair energy metabolism through limitation of mitochondrial substrates. Sci. Rep. 2017, 7, 1666. [Google Scholar] [CrossRef]
- Auberger, P. BCR-ABL/p62/SQSTM1: A cannibal embrace. Blood 2012, 120, 3389–3390. [Google Scholar] [CrossRef][Green Version]
- He, P.X.; Zhang, J.; Che, Y.S.; He, Q.J.; Chen, Y.; Ding, J. G226, a new epipolythiodioxopiperazine derivative, triggers DNA damage and apoptosis in human cancer cells in vitro via ROS generation. Acta Pharmacol. Sin. 2014, 35, 1546–1555. [Google Scholar] [CrossRef]
- Simon, H.U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward (5’ to 3’) | Reverse (5’ to 3’) |
|---|---|---|
| β-actin | CGTACCACTGGCATCGTGAT | GTGTTGGCGTACAGGTCTTTG |
| Ptgs2 | GCCCAGCACTTCACGCATCAG | GACCAGGCACCAGACCAAAGACC |
| Tgfbr1 | ACATGATTCAGCCACAGATACC | GCATAGATGTCAGCACGTTTG |
| Sqstm1 | AAATGGGTCCACCAGGAAACTGGA | TCAACTTCAATGCCCAGAGGGCTA |
| Gene | Microarray | qPCR | ||
|---|---|---|---|---|
| As | As + Fe | As | As + Fe | |
| Ptgs2 | 4.01 | 8.57 | 6.26 | 9.65 |
| Tgfbr1 | 0.47 | 0.24 | 0.61 | 0.47 |
| Sqstm1 | 2.48 | 4.82 | 10.86 | 20.73 |
| No. | KEGG Pathway | As | As + Fe |
|---|---|---|---|
| 1 | Pathways in cancer | + | |
| 2 | MAPK (Ras–mitogen-activated protein kinase) signaling pathway | + | + |
| 3 | Cell cycle | + | + |
| 4 | Purine metabolism | + | + |
| 5 | Pyrimidine metabolism | + | + |
| 6 | p53 signaling pathway | + | + |
| 7 | Colorectal cancer | + | + |
| 8 | DNA replication | + | + |
| 9 | ECM (Extracellular matrix)-receptor interaction | + | |
| 10 | Progesterone-mediated oocyte maturation | + | |
| 11 | ErbB signaling pathway | + | |
| 12 | Base excision repair | + | + |
| 13 | Epithelial cell signaling in Helicobacter pylori infection | + | + |
| 14 | Aminoacyl-tRNA biosynthesis | + | + |
| 15 | Lysine degradation | + | + |
| 16 | Pyruvate metabolism | + | + |
| 17 | Bladder cancer | + | |
| 18 | Notch signaling pathway | + | |
| 19 | Alanine, aspartate and glutamate metabolism | + | + |
| 20 | Porphyrin and chlorophyll metabolism | + | |
| 21 | One carbon pool by folate | + | |
| 22 | Steroid biosynthesis | + | + |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Liu, Y.; Liu, S.; Wu, B. Influence of Iron on Cytotoxicity and Gene Expression Profiles Induced by Arsenic in HepG2 Cells. Int. J. Environ. Res. Public Health 2019, 16, 4484. https://doi.org/10.3390/ijerph16224484
Wang Y, Liu Y, Liu S, Wu B. Influence of Iron on Cytotoxicity and Gene Expression Profiles Induced by Arsenic in HepG2 Cells. International Journal of Environmental Research and Public Health. 2019; 16(22):4484. https://doi.org/10.3390/ijerph16224484
Chicago/Turabian StyleWang, Yonghua, Yuxuan Liu, Su Liu, and Bing Wu. 2019. "Influence of Iron on Cytotoxicity and Gene Expression Profiles Induced by Arsenic in HepG2 Cells" International Journal of Environmental Research and Public Health 16, no. 22: 4484. https://doi.org/10.3390/ijerph16224484
APA StyleWang, Y., Liu, Y., Liu, S., & Wu, B. (2019). Influence of Iron on Cytotoxicity and Gene Expression Profiles Induced by Arsenic in HepG2 Cells. International Journal of Environmental Research and Public Health, 16(22), 4484. https://doi.org/10.3390/ijerph16224484