Mortality Due to Cystic Fibrosis over a 36-Year Period in Spain: Time Trends and Geographic Variations

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
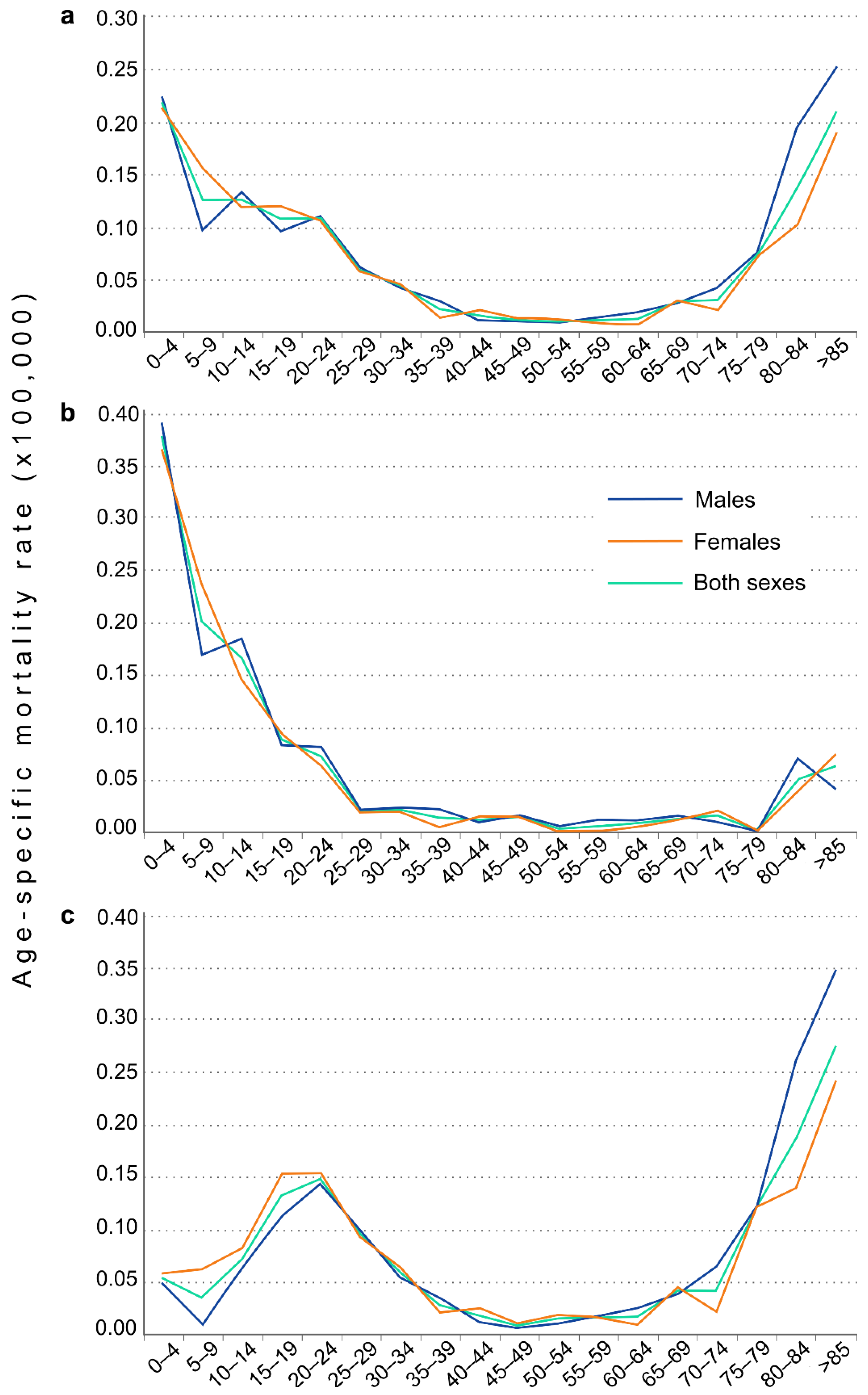
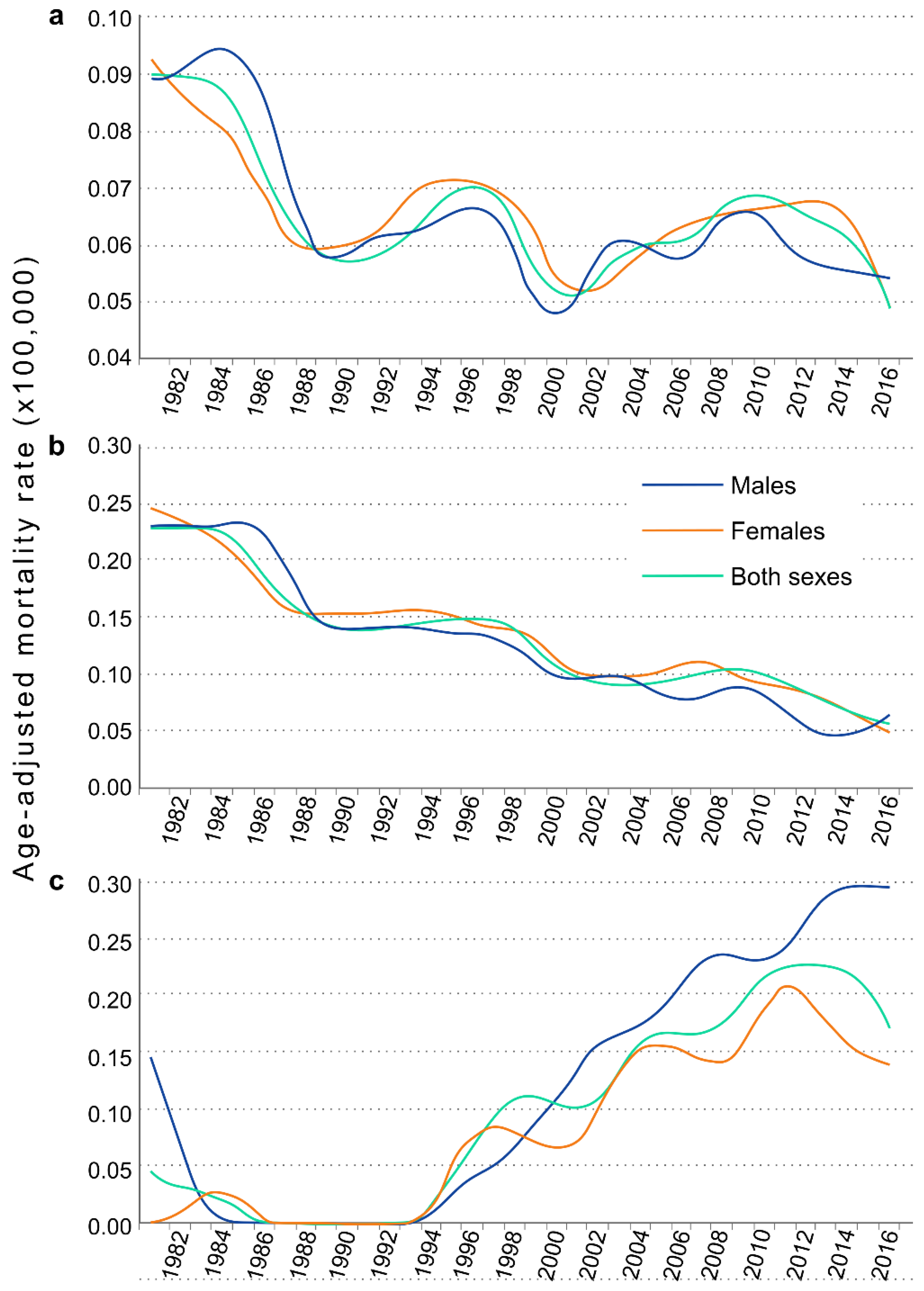
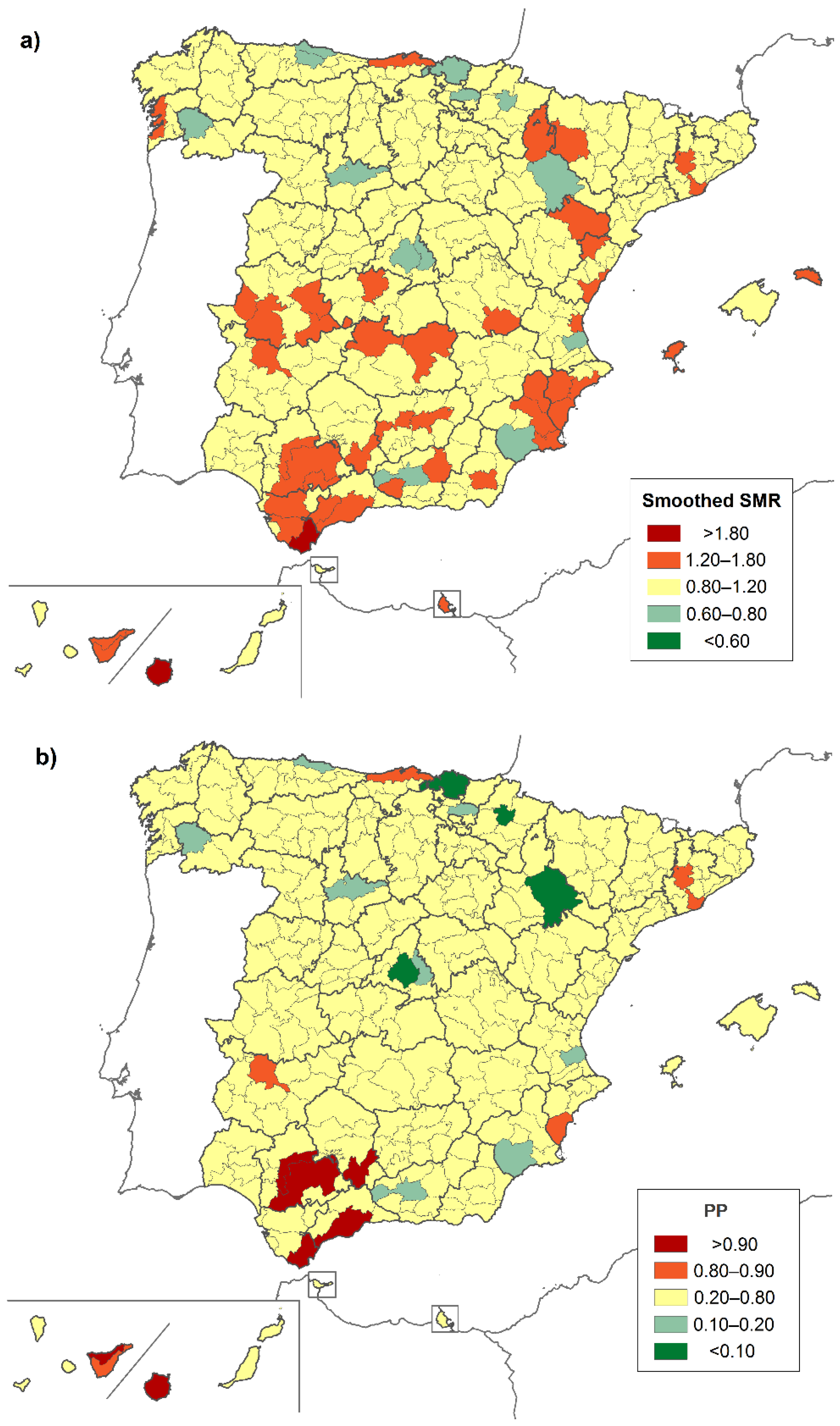
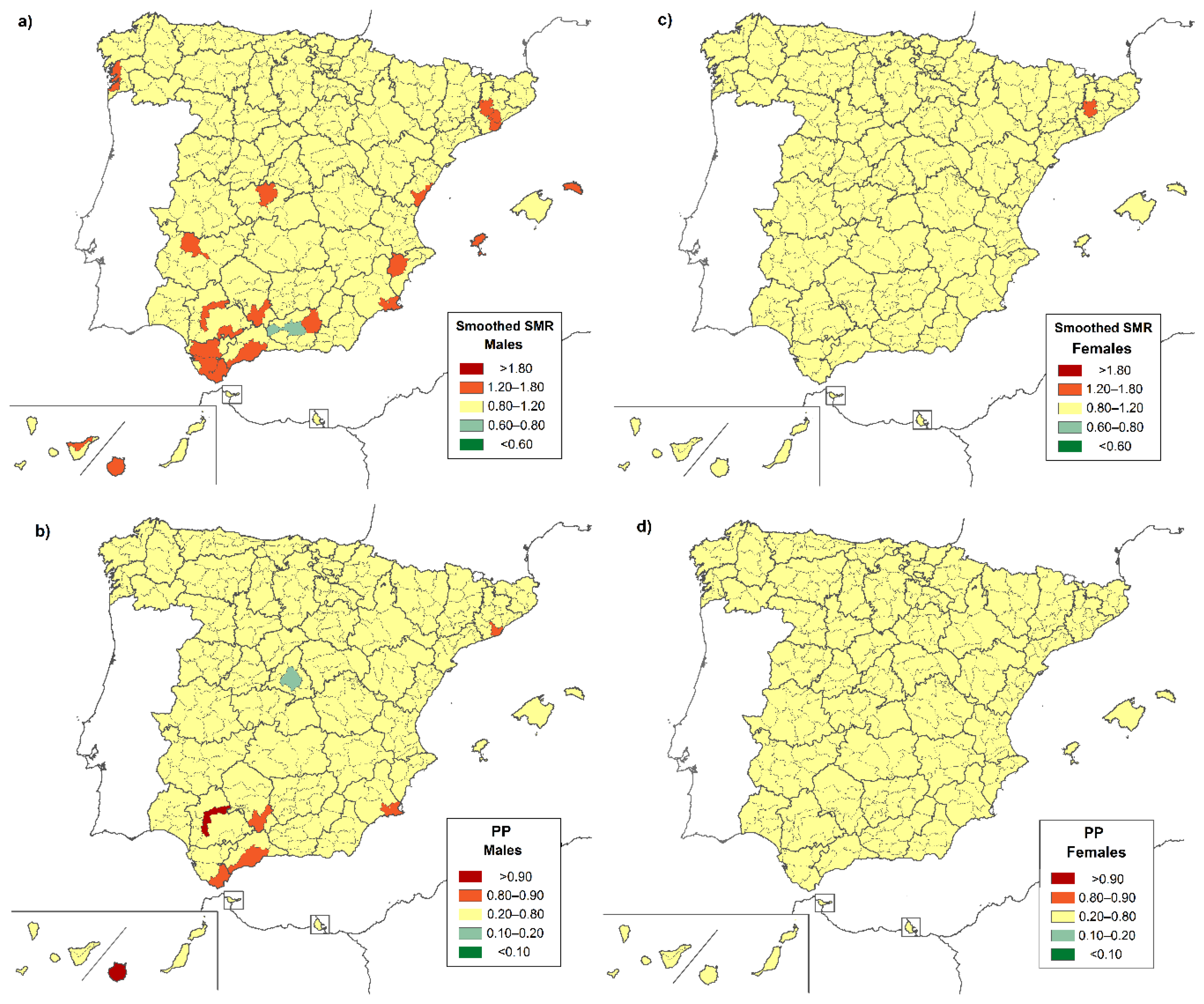
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Research Ethics
References
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef]
- Boyle, M.P.; De Boeck, K. A new era in the treatment of cystic fibrosis: Correction of the underlying CFTR defect. Lancet Respir. Med. 2013, 1, 158–163. [Google Scholar] [CrossRef]
- Rosenfeld, M.; Davis, R.; FitzSimmons, S.; Pepe, M.; Ramsey, B. Gender gap in cystic fibrosis mortality. Am. J. Epidemiol. 1997, 145, 794–803. [Google Scholar] [CrossRef] [PubMed]
- Burgel, P.R.; Bellis, G.; Olesen, H.V.; Viviani, L.; Zolin, A.; Blasi, F.; Elborn, J.S. ERS/ECFS Task Force on Provision of Care for Adults with Cystic Fibrosisin Europe. Future trends in cystic fibrosis demography in 34 European countries. Eur. Respir. J. 2015, 46, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Corriveau, S.; Sykes, J.; Stephenson, A.L. Cystic fibrosis survival: The changing epidemiology. Curr. Opin. Pulm. Med. 2018, 24, 574–578. [Google Scholar] [CrossRef]
- Mehta, G.; Macek, M., Jr.; Mehta, A.; European Registry Working Group. Cystic fibrosis across Europe: EuroCareCF analysis of demographic data from 35 countries. J. Cyst. Fibros. 2010, 2, S5–S21. [Google Scholar] [CrossRef] [PubMed]
- Farrel, P.M. The prevalence of cystic fibrosis in the European Union. J. Cyst. Fibros. 2008, 7, 450–453. [Google Scholar] [CrossRef]
- Quintana-Gallego, E.; Ruiz-Ramos, M.; Delgado-Pecellin, I.; Calero, C.; Soriano, J.B.; Lopez-Campos, J.L. Mortality from Cystic Fibrosis in Europe: 1994–2010. Pediatric. Pulmonol. 2016, 51, 133–142. [Google Scholar] [CrossRef]
- Dodge, J.A.; Lewis, P.A.; Stanton, M.; Wilsher, J. Cystic fibrosis mortality and survival in the UK: 1947–2003. Eur. Respir. J. 2007, 29, 522–526. [Google Scholar] [CrossRef]
- Viviani, L.; Bossi, A.; Assael, B.M.; Italian Registry for Cystic Fibrosis Collaborative Group. Absence of a gender gap in survival. An analysis of the Italian registry for cystic fibrosis in the paediatric age. J. Cyst. Fibros. 2011, 10, 313–317. [Google Scholar] [CrossRef]
- Kopp, BT.; Nicholson, L.; Paul, G.; Tobias, J.; Ramanathan, C.; Hayes, D., Jr. Geographic variations in cystic fibrosis: An analysis of the U.S. CF Foundation Registry. Pediatr. Pulmonol. 2015, 50, 754–762. [Google Scholar] [CrossRef]
- Cerda, L.J.; Valdivia, C.G.; Guiraldes, C.E.; Sanchez, D.I. Cystic fibrosis mortality in Chile between 1997 and 2003. Rev. Med. Chil. 2008, 136, 157–162. [Google Scholar]
- Reid, D.W.; Blizzard, C.L.; Shugg, D.M.; Flowers, C.; Cash, C.; Greville, H.M. Changes in cystic fibrosis mortality in Australia, 1979–2005. Med. J. Aust. 2011, 195, 392–395. [Google Scholar] [CrossRef] [PubMed]
- Imaizumi, Y. Incidence and mortality rates of cystic fibrosis in Japan, 1969–1992. Am. J. Med. Genet. 1995, 58, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, R.; Salvatore, D.; Baldo, E.; Forneris, M.P.; Lucidi, V.; Manunza, D.; Marinelli, I.; Messore, B.; Neri, A.S.; Raia, V.; et al. An overview of international literature from cystic fibrosis registries: 1. Mortality and survival studies in cystic fibrosis. J. Cyst. Fibros. 2009, 8, 229–237. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, T.; Gifford, A.H.; Sabadosa, K.A.; Quinton, H.B.; Knapp, E.A.; Goss, C.H.; Marshall, B.C. Longevity of patients with cystic fibrosis in 2000 to 2010 and beyond: Survival analysis of the cystic fibrosis foundation patient registry. Ann. Intern. Med. 2014, 161, 233–241. [Google Scholar] [CrossRef]
- Zolin, A.; Bossi, A.; Cirilli, N.; Kashirskaya, N.; Padoan, R. Cystic Fibrosis Mortality in Childhood. Data from European Cystic Fibrosis Society Patient Registry. Int. J. Environ. Res. Public Health 2018, 15, 2020. [Google Scholar] [CrossRef]
- Ramalle-Gomara, E.; Perucha, M.; González, M.A.; Quiñones, C.; Andrés, J.; Posada, M. Cystic fibrosis mortality trends in Spain among infants and young children: 1981–2004. Eur. J. Epidemiol. 2008, 23, 523–529. [Google Scholar] [CrossRef]
- Psoter, K.J.; De Roos, A.J.; Wakefield, J.; Mayer, J.D.; Bryan, M.; Rosenfeld, M. Association of meteorological and geographical factors and risk of initial Pseudomonas aeruginosa acquisition in young children with cystic fibrosis. Epidemiol. Infect. 2016, 144, 1075–1083. [Google Scholar] [CrossRef]
- Jackson, A.D.; Goss, C.H. Epidemiology of CF: How registries can be used to advance our understanding of the CF population. J. Cyst. Fibros. 2018, 17, 297–305. [Google Scholar] [CrossRef]
- McCormick, J.; Mehta, G.; Olesen, H.V.; Viviani, L.; Macek, M., Jr.; Mehta, A.; European Registry Working Group. Comparative demographics of the European cystic fibrosis population: A cross-sectional database analysis. Lancet 2010, 375, 1007–1013. [Google Scholar] [CrossRef]
- Spanish Cystic Fibrosis Registry Annual Report 2016. Available online: https://fibrosisquistica.org/wp-content/uploads/2018/09/Report2016SpainFinal.pdf (accessed on 15 November 2018).
- Alonso-Ferreira, V.; Sánchez-Díaz, G.; Villaverde-Hueso, A.; Posada de la Paz, M.; Bermejo-Sánchez, E. A Nationwide Registry-Based Study on Mortality Due to Rare Congenital Anomalies. Int. J. Environ. Res. Public Health 2018, 15, 1715. [Google Scholar] [CrossRef]
- Fitting Segmented Curves Whose Join Points Have to Be Estimated. J. Am. Stat. Assoc. 1966, 61, 1097–1129. Available online: http://amstat.tandfonline.com/doi/abs/10.1080/01621459.1966.10482198 (accessed on 16 November 2018). [CrossRef]
- Velleman, P.F. Definition and comparison of robust nonlinear data smoothing algorithms. J. Am. Stat. Assoc. 1980, 75, 609–615. [Google Scholar] [CrossRef]
- Besag, J.E.; York, J.; Molliè, A. A Bayesian image restoration with two applications in spatial statistics. Ann. Inst. Stat. Math. 1991. [Google Scholar] [CrossRef]
- Kulich, M.; Rosenfeld, M.; Goss, C.H.; Wilmott, R. Improved survival among young patients with cystic fibrosis. J. Pediatr. 2003, 142, 631–636. [Google Scholar] [CrossRef]
- Quintana-Gallego, E.; Delgado-Pecellin, I.; Calero Acuna, C. CFTR protein repair therapy in cystic fibrosis. Arch. Bronconeumol. 2014, 50, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Hamard, C.; Kanaan, R.; Boussaud, V.; Grenet, D.; Abély, M.; Hubert, D.; Munck, A.; Lemonnier, L.; Burgel, P.R. Causes of death in French cystic fibrosis patients: The need for improvement in transplantation referral strategies! J. Cyst. Fibros. 2016, 15, 204–212. [Google Scholar] [CrossRef]
- Barr, H.L.; Britton, J.; Smyth, A.R.; Fogarty, A.W. Association between socioeconomic status, sex, and age at death from cystic fibrosis in England and Wales (1959 to 2008): Cross sectional study. BMJ 2011, 343, d4662. [Google Scholar] [CrossRef]
- Padilla, J.; Calvo, V.; Jordá, C.; Escrivá, J.; Cerón, J.; Peñalver, J.C.; García-Zarza, A.; Pastor, J.; Blasco, E. Lung transplantation in cystic fibrosis: Perioperative mortality. Arch. Bronconeumol. 2005, 41, 489–492. [Google Scholar] [CrossRef]
- Chaparro, C.; Keshavjee, S. Lung transplantation for cystic fibrosis: An update. Expert Rev. Respir. Med. 2016, 10, 1269–1280. [Google Scholar] [CrossRef] [PubMed]
- Buu, M.C.; Sanders, L.M.; Mayo, J.A.; Milla, C.E.; Wise, P.H. Assessing Differences in Mortality Rates and Risk Factors Between Hispanic and Non-Hispanic Patients with Cystic Fibrosis in California. Chest 2016, 149, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Corey, M.; Farewell, V. Determinants of mortality from cystic fibrosis in Canada, 1970–1989. Am. J. Epidemiol. 1996, 143, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Casals, T.; Nunes, V.; Palacio, A.; Giménez, J.; Gaona, A.; Ibáñez, N.; Morral, N.; Estivill, X. Cystic fibrosis in Spain: High frequency of mutation G542X in the Mediterranean coastal area. Hum. Genet. 1993, 91, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Alonso, M.J.; Heine-Suñer, D.; Calvo, M.; Rosell, J.; Giménez, J.; Ramos, M.D.; Telleria, J.J.; Palacio, A.; Estivill, X.; Casals, T. Spectrum of Mutations in the CFTR Gene in Cystic Fibrosis Patients of Spanish Ancestry. Ann. Hum. Genet. 2007, 71, 194–201. [Google Scholar] [CrossRef] [PubMed]
- McPhail, G.L.; Acton, J.D.; Fenchel, M.C.; Amin, R.S.; Seid, M. Improvements in lung function outcomes in children with cystic fibrosis are associated with better nutrition, fewer chronic Pseudomonas aeruginosa infections, and dornase alfa use. J. Pediatr. 2008, 153, 752–757. [Google Scholar] [CrossRef] [PubMed]
- Cantón, R.; Girón, R.; Martínez-Martínez, L.; Oliver, A.; Solé, A.; Valdezate, S.; Máiz, L. Patógenos multirresistentes en la fibrosis quística. Arch. Bronconeumol. 2002, 38, 376–385. [Google Scholar] [CrossRef]
- Lemna, W.K.; Feldman, G.L.; Kerem, B.; Fernbach, S.D.; Zevkovich, E.P.; O’Brien, W.E.; Riordan, J.R.; Collins, F.S.; Tsui, L.C.; Beaudet, A.L. Mutation Analysis for Heterozygote Detection and the Prenatal Diagnosis of Cystic Fibrosis. N. Engl. J. Med. 1990, 322, 291–296. [Google Scholar] [CrossRef]
- Dipple, K.M.; McCabe, E.R. Modifier genes convert “simple” Mendelian disorders to complex traits. Mol. Genet. Metab. 2000, 71, 43–50. [Google Scholar] [CrossRef]
- Sarles, J.; Giorgi, R.; Berthézène, P.; Munck, A.; Cheillan, D.; Dagorn, J.C.; Roussey, M. Neonatal screening for cystic fibrosis: Comparing the performances of IRT/DNA and IRT/ PAP. J. Cyst. Fibros. 2014, 13, 384–390. [Google Scholar] [CrossRef]
- Halliburton, C.S.; Mannino, D.M.; Olney, R.S. Cystic fibrosis deaths in the United States from 1979 through 1991. An analysis using multiple-cause mortality data. Arch. Pediatr. Adolesc. Med. 1996, 150, 1181–1185. [Google Scholar] [CrossRef] [PubMed]
- Panickar, J.R; Dodd, S.R; Smyth, R.L; Couriel, J.M. Trends in deaths from respiratory illness in children in England and Wales from 1968 to 2000. Thorax 2005, 60, 1035–1038. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Both Sexes | Males | Females | ||||
|---|---|---|---|---|---|---|
| Year | Age Adjusted Mortality Rate | 95% CI | Age Adjusted Mortality Rate | 95% CI | Age Adjusted Mortality Rate | 95% CI |
| 1981 | 0.090 | (0.064–0.126) | 0.089 | (0.054–0.154) | 0.092 | (0.055–0.147) |
| 1982 | 0.083 | (0.057–0.117) | 0.076 | (0.043–0.136) | 0.089 | (0.053–0.145) |
| 1983 | 0.093 | (0.066–0.130) | 0.105 | (0.063–0.174) | 0.084 | (0.049–0.138) |
| 1984 | 0.086 | (0.058–0.123) | 0.087 | (0.049–0.150) | 0.085 | (0.047–0.143) |
| 1985 | 0.095 | (0.066–0.132) | 0.111 | (0.069–0.177) | 0.076 | (0.042–0.129) |
| 1986 | 0.078 | (0.051–0.114) | 0.098 | (0.057–0.162) | 0.056 | (0.027–0.107) |
| 1987 | 0.063 | (0.039–0.096) | 0.042 | (0.018–0.091) | 0.084 | (0.047–0.143) |
| 1988 | 0.056 | (0.034–0.089) | 0.060 | (0.028–0.115) | 0.052 | (0.024–0.101) |
| 1989 | 0.085 | (0.057–0.123) | 0.113 | (0.069–0.178) | 0.057 | (0.027–0.109) |
| 1990 | 0.058 | (0.036–0.090) | 0.037 | (0.016–0.079) | 0.079 | (0.042–0.138) |
| 1991 | 0.052 | (0.030–0.085) | 0.049 | (0.022–0.098) | 0.056 | (0.025–0.111) |
| 1992 | 0.062 | (0.039–0.096) | 0.075 | (0.040–0.132) | 0.049 | (0.022–0.099) |
| 1993 | 0.075 | (0.047–0.113) | 0.079 | (0.041–0.139) | 0.071 | (0.034–0.131) |
| 1994 | 0.058 | (0.035–0.093) | 0.040 | (0.017–0.086) | 0.077 | (0.039–0.139) |
| 1995 | 0.060 | (0.038–0.094) | 0.048 | (0.021–0.098) | 0.075 | (0.040–0.132) |
| 1996 | 0.089 | (0.061–0.127) | 0.108 | (0.066–0.171) | 0.067 | (0.035–0.123) |
| 1997 | 0.076 | (0.050–0.114) | 0.083 | (0.045–0.144) | 0.069 | (0.036–0.126) |
| 1998 | 0.057 | (0.034–0.091) | 0.044 | (0.018–0.096) | 0.069 | (0.035–0.128) |
| 1999 | 0.065 | (0.042–0.099) | 0.056 | (0.028–0.107) | 0.074 | (0.040–0.133) |
| 2000 | 0.042 | (0.025–0.070) | 0.045 | (0.022–0.091) | 0.040 | (0.018–0.088) |
| 2001 | 0.050 | (0.030–0.082) | 0.049 | (0.022–0.102) | 0.053 | (0.026–0.105) |
| 2002 | 0.038 | (0.022–0.065) | 0.033 | (0.013–0.077) | 0.041 | (0.019–0.088) |
| 2003 | 0.070 | (0.046–0.105) | 0.079 | (0.043–0.137) | 0.063 | (0.033–0.115) |
| 2004 | 0.090 | (0.062–0.128) | 0.077 | (0.043–0.132) | 0.104 | (0.062–0.168) |
| 2005 | 0.053 | (0.033–0.082) | 0.054 | (0.027–0.100) | 0.052 | (0.026–0.098) |
| 2006 | 0.030 | (0.016–0.052) | 0.029 | (0.011–0.067) | 0.029 | (0.012–0.067) |
| 2007 | 0.064 | (0.041–0.096) | 0.047 | (0.023–0.090) | 0.087 | (0.049–0.146) |
| 2008 | 0.065 | (0.043–0.096) | 0.074 | (0.043–0.123) | 0.058 | (0.029–0.107) |
| 2009 | 0.076 | (0.052–0.109) | 0.068 | (0.037–0.117) | 0.084 | (0.049–0.138) |
| 2010 | 0.072 | (0.048–0.104) | 0.088 | (0.052–0.142) | 0.053 | (0.027–0.098) |
| 2011 | 0.055 | (0.035–0.082) | 0.044 | (0.022–0.082) | 0.067 | (0.037–0.115) |
| 2012 | 0.061 | (0.041–0.090) | 0.056 | (0.030–0.099) | 0.066 | (0.037–0.113) |
| 2013 | 0.079 | (0.054–0.113) | 0.057 | (0.031–0.101) | 0.106 | (0.064–0.167) |
| 2014 | 0.063 | (0.040–0.094) | 0.072 | (0.040–0.124) | 0.053 | (0.026–0.100) |
| 2015 | 0.057 | (0.038–0.085) | 0.050 | (0.027–0.090) | 0.066 | (0.037–0.114) |
| 2016 | 0.049 | (0.030–0.078) | 0.054 | (0.027–0.100) | 0.044 | (0.020–0.090) |
| District | Province | Location | Both Sexes | Males | Females |
|---|---|---|---|---|---|
| Very low risk | |||||
| Área Metropolitana de Madrid | Madrid | C | 0.658 (0.453–0.924) | - | 0.598 (0.341–0.970) |
| High risk | |||||
| Brozas | Cáceres | W | 15.462 (1.736–55.825) | - | 30.304 (3.403–109.413) |
| Alto Maestrazgo | Castellón | E | 12.091 (1.358–43.654) | - | - |
| Logrosán | Cáceres | W | 10.474 (1.176–37.817) | - | - |
| Guadix | Granada | S | 5.468 (1.099–15.978) | - | - |
| Campiña Alta | Córdoba | S | 3.168 (1.269–6.527) | 4.652 (1.499–10.856) | - |
| Campo de Gibraltar | Cádiz | S | 2.892 (1.245–5.698) | 3.783 (1.219–8.829) | - |
| Norte de Tenerife | SC de Tenerife | SW * | 2.473 (1.184–4.549) | - | 2.849 (1.040–6.200) |
| Gran Canaria | Las Palmas | SW * | 2.331 (1.423–3.600) | 2.664 (1.328–4.767) | - |
| La Vega | Sevilla | SW | 2.201 (1.004–4.178) | 2.169 (1.119–3.788) | - |
| La Campiña | Sevilla | SW | 1.934 (1.225–2.902) | - | 2.816 (1.028–6.129) |
| Guadalhorce | Málaga | S | 1.798 (1.126–2.722) | - | - |
| Campo de Cartagena | Murcia | SE | - | 3.395 (1.240–7.390) | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villaverde-Hueso, A.; Sánchez-Díaz, G.; Molina-Cabrero, F.J.; Gallego, E.; Posada de la Paz, M.; Alonso-Ferreira, V. Mortality Due to Cystic Fibrosis over a 36-Year Period in Spain: Time Trends and Geographic Variations. Int. J. Environ. Res. Public Health 2019, 16, 119. https://doi.org/10.3390/ijerph16010119
Villaverde-Hueso A, Sánchez-Díaz G, Molina-Cabrero FJ, Gallego E, Posada de la Paz M, Alonso-Ferreira V. Mortality Due to Cystic Fibrosis over a 36-Year Period in Spain: Time Trends and Geographic Variations. International Journal of Environmental Research and Public Health. 2019; 16(1):119. https://doi.org/10.3390/ijerph16010119
Chicago/Turabian StyleVillaverde-Hueso, Ana, Germán Sánchez-Díaz, Francisco J. Molina-Cabrero, Elisa Gallego, Manuel Posada de la Paz, and Verónica Alonso-Ferreira. 2019. "Mortality Due to Cystic Fibrosis over a 36-Year Period in Spain: Time Trends and Geographic Variations" International Journal of Environmental Research and Public Health 16, no. 1: 119. https://doi.org/10.3390/ijerph16010119
APA StyleVillaverde-Hueso, A., Sánchez-Díaz, G., Molina-Cabrero, F. J., Gallego, E., Posada de la Paz, M., & Alonso-Ferreira, V. (2019). Mortality Due to Cystic Fibrosis over a 36-Year Period in Spain: Time Trends and Geographic Variations. International Journal of Environmental Research and Public Health, 16(1), 119. https://doi.org/10.3390/ijerph16010119