Tobacco Smoke Induces and Alters Immune Responses in the Lung Triggering Inflammation, Allergy, Asthma and Other Lung Diseases: A Mechanistic Review




{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
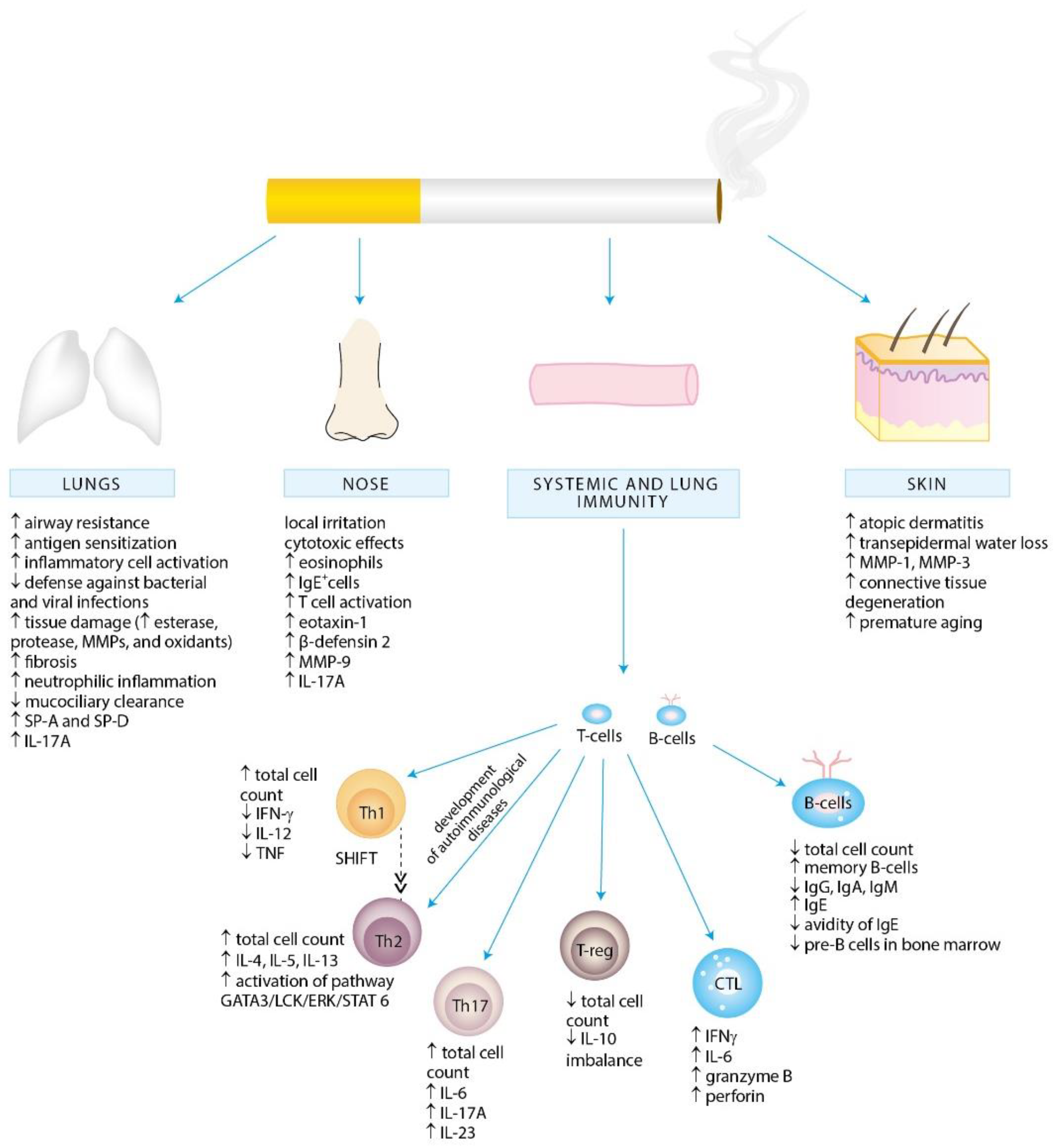
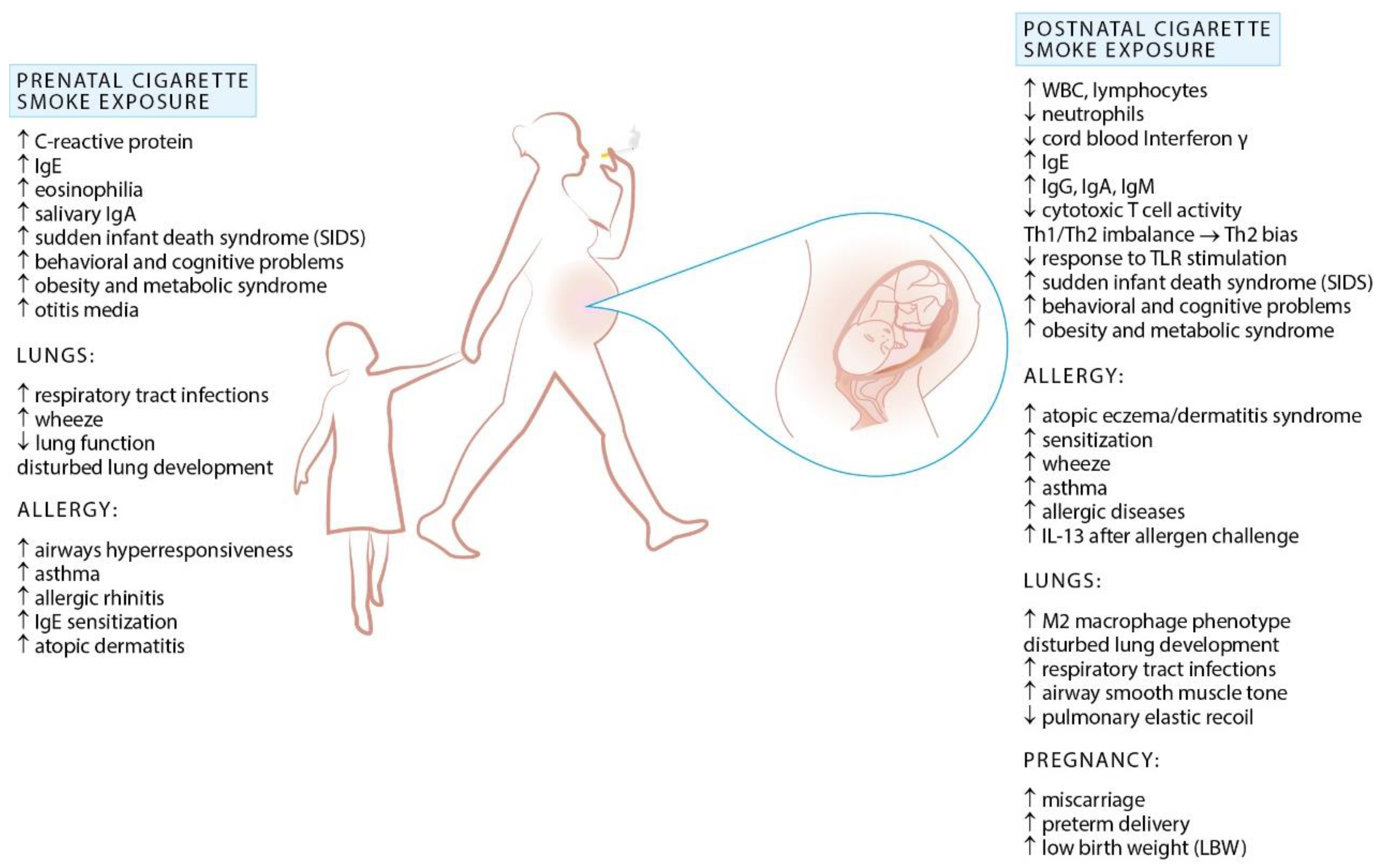
2. Exposure to Cigarette Smoke and Allergy
Prenatal Exposure to Cigarette Smoke and Allergy
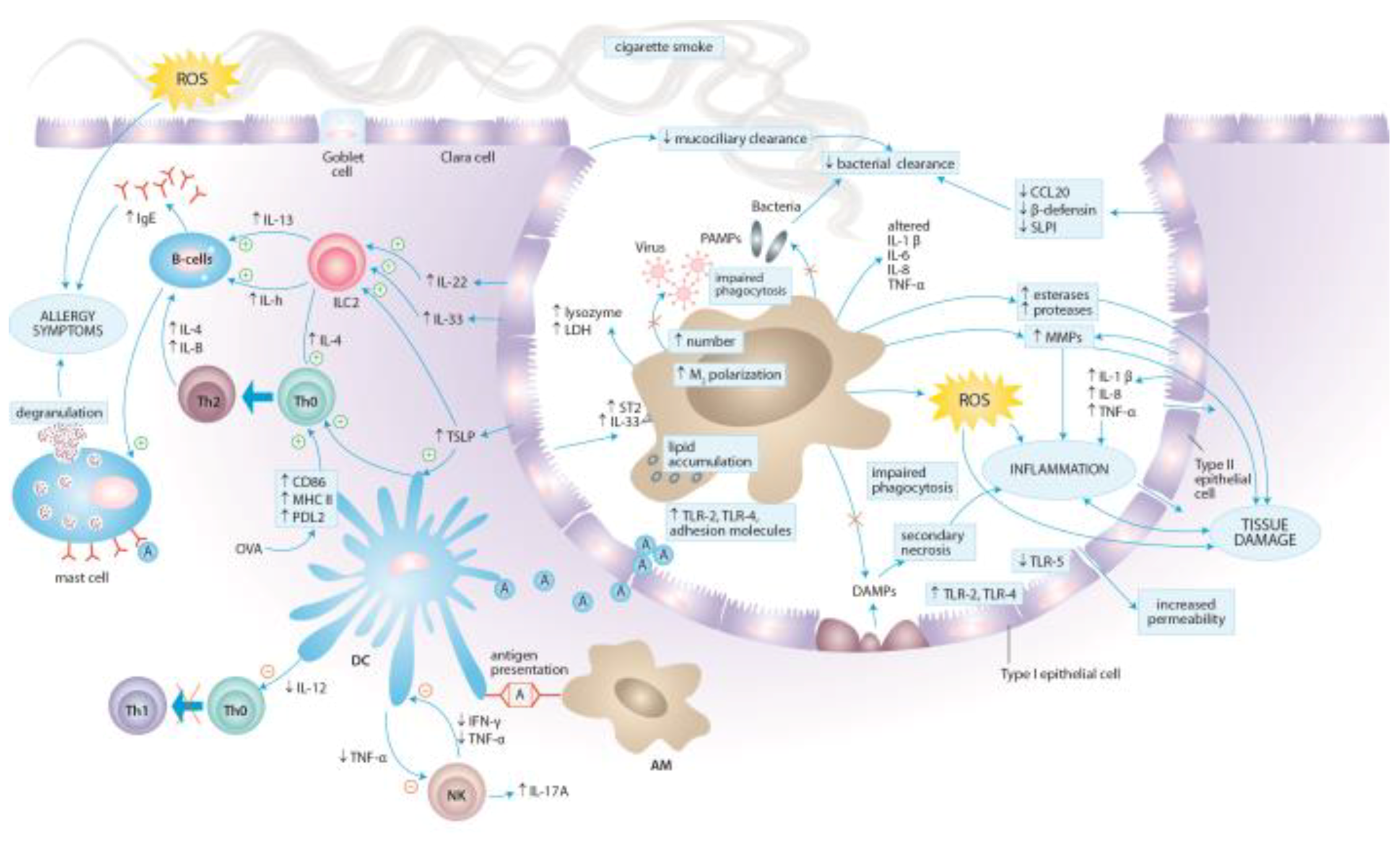
3. Molecular Aspects of Tobacco Smoke Toxicity
4. Effects of Cigarette Smoke Exposure on the Immune System
4.1. Effects of Cigarette Smoke Exposure on Innate Immunity
4.1.1. Epithelial Cells
4.1.2. Alveolar Macrophages
4.1.3. Dendritic Cells
4.1.4. Natural Killer Cells
4.1.5. Neutrophils
4.2. Effects of Cigarette Smoke Exposure on Adaptive Immunity
4.2.1. T Lymphocytes
Th1 and Th2 Cells
Th17 Cells
Treg Cells
4.2.2. B Lymphocytes
5. Conclusions and Further Research
Author Contributions
Acknowledgments
Conflicts of Interest
References
- WHO. WHO Global Report on Trends in Prevalence of Tobacco Smoking; WHO: Geneva, Switzerland, 2015. [Google Scholar]
- Sopori, M. Effects of cigarette smoke on the immune system. Nat. Rev. Immunol. 2002, 2, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Stampfli, M.R.; Anderson, G.P. How cigarette smoke skews immune responses to promote infection, lung disease and cancer. Nat. Rev. Immunol. 2009, 9, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Nuorti, J.P.; Butler, J.C.; Farley, M.M.; Harrison, L.H.; McGeer, A.; Kolczak, M.S.; Breiman, R.F. Cigarette smoking and invasive pneumococcal disease. Active Bacterial Core Surveillance Team. N. Engl. J. Med. 2000, 342, 681–689. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Gender, Women, and the Tobacco Epidemic; Samet, J.M., Yoon, S.-Y., Eds.; World Health Organization: Geneva, Switzerland, 2010; p. 249. [Google Scholar]
- Oberg, M.; Jaakkola, M.S.; Woodward, A.; Peruga, A.; Pruss-Ustun, A. Worldwide burden of disease from exposure to second-hand smoke: A retrospective analysis of data from 192 countries. Lancet 2011, 377, 139–146. [Google Scholar] [CrossRef]
- Wasserman, G.A.; Liu, X.; Pine, D.S.; Graziano, J.H. Contribution of maternal smoking during pregnancy and lead exposure to early child behavior problems. Neurotoxicol. Teratol. 2001, 23, 13–21. [Google Scholar] [CrossRef]
- Prescott, S.L. Effects of early cigarette smoke exposure on early immune development and respiratory disease. Paediatr. Respir. Rev. 2008, 9, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Phaybouth, V.P.; Wang, S.Z.; Hutt, J.A.; McDonald, J.D.; Harrod, K.S.; Barrett, E.G. Cigarette smoke suppresses Th1 cytokine production and increases RSV expression in a neonatal model. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L222–L231. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.A.; Colin, A.A. Infantile respiratory syncytial virus and human rhinovirus infections: Respective role in inception and persistence of wheezing. Eur. Respir. J. 2015, 45, 774–789. [Google Scholar] [CrossRef] [PubMed]
- Cheraghi, M.; Salvi, S. Environmental tobacco smoke (ETS) and respiratory health in children. Eur. J. Pediatr. 2009, 168, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Botelho, F.M.; Gaschler, G.J.; Kianpour, S.; Zavitz, C.C.; Trimble, N.J.; Nikota, J.K.; Bauer, C.M.; Stampfli, M.R. Innate immune processes are sufficient for driving cigarette smoke-induced inflammation in mice. Am. J. Respir. Cell Mol. Biol. 2010, 42, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.A.; Paszkiewicz, G.M.; Hutson, A.D.; Pauly, J.L. Inflammatory response of lung macrophages and epithelial cells to tobacco smoke: A literature review of ex vivo investigations. Immunol. Res. 2010, 46, 94–126. [Google Scholar] [CrossRef] [PubMed]
- Robays, L.J.; Maes, T.; Joos, G.F.; Vermaelen, K.Y. Between a cough and a wheeze: Dendritic cells at the nexus of tobacco smoke-induced allergic airway sensitization. Mucosal Immunol. 2009, 2, 206–219. [Google Scholar] [CrossRef] [PubMed]
- Zavitz, C.C.; Gaschler, G.J.; Robbins, C.S.; Botelho, F.M.; Cox, P.G.; Stampfli, M.R. Impact of cigarette smoke on T and B cell responsiveness. Cell. Immunol. 2008, 253, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Van Hove, C.L.; Moerloose, K.; Maes, T.; Joos, G.F.; Tournoy, K.G. Cigarette smoke enhances Th-2 driven airway inflammation and delays inhalational tolerance. Respir. Res. 2008, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Moerloose, K.B.; Pauwels, R.A.; Joos, G.F. Short-term cigarette smoke exposure enhances allergic airway inflammation in mice. Am. J. Respir. Crit. Care Med. 2005, 172, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Moerloose, K.B.; Robays, L.J.; Maes, T.; Brusselle, G.G.; Tournoy, K.G.; Joos, G.F. Cigarette smoke exposure facilitates allergic sensitization in mice. Respir. Res. 2006, 7, 49. [Google Scholar] [CrossRef] [PubMed]
- Trimble, N.J.; Botelho, F.M.; Bauer, C.M.; Fattouh, R.; Stampfli, M.R. Adjuvant and anti-inflammatory properties of cigarette smoke in murine allergic airway inflammation. Am. J. Respir. Cell Mol. Biol. 2009, 40, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Bozinovski, S.; Seow, H.J.; Chan, S.P.; Anthony, D.; McQualter, J.; Hansen, M.; Jenkins, B.J.; Anderson, G.P.; Vlahos, R. Innate cellular sources of interleukin-17A regulate macrophage accumulation in cigarette- smoke-induced lung inflammation in mice. Clin. Sci. 2015, 129, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Kulig, M.; Luck, W.; Lau, S.; Niggemann, B.; Bergmann, R.; Klettke, U.; Guggenmoos-Holzmann, I.; Wahn, U. Effect of pre- and postnatal tobacco smoke exposure on specific sensitization to food and inhalant allergens during the first 3 years of life. Multicenter Allergy Study Group, Germany. Allergy 1999, 54, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Ronchetti, R.; Macri, F.; Ciofetta, G.; Indinnimeo, L.; Cutrera, R.; Bonci, E.; Antognoni, G.; Martinez, F.D. Increased serum IgE and increased prevalence of eosinophilia in 9-year-old children of smoking parents. J. Allergy Clin. Immunol. 1990, 86, 400–407. [Google Scholar] [CrossRef]
- Lindfors, A.; van Hage-Hamsten, M.; Rietz, H.; Wickman, M.; Nordvall, S.L. Influence of interaction of environmental risk factors and sensitization in young asthmatic children. J. Allergy Clin. Immunol. 1999, 104, 755–762. [Google Scholar] [CrossRef]
- Oryszczyn, M.P.; Annesi-Maesano, I.; Charpin, D.; Paty, E.; Maccario, J.; Kauffmann, F. Relationships of active and passive smoking to total IgE in adults of the Epidemiological Study of the Genetics and Environment of Asthma, Bronchial Hyperresponsiveness, and Atopy (EGEA). Am. J. Respir. Crit. Care Med. 2000, 161, 1241–1246. [Google Scholar] [CrossRef] [PubMed]
- Strachan, D.P.; Cook, D.G. Health effects of passive smoking .5. Parental smoking and allergic sensitisation in children. Thorax 1998, 53, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Feleszko, W.; Ruszczynski, M.; Jaworska, J.; Strzelak, A.; Zalewski, B.M.; Kulus, M. Environmental tobacco smoke exposure and risk of allergic sensitisation in children: A systematic review and meta-analysis. Arch. Dis. Child. 2014, 99, 985–992. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.C.; Chang, S.W.; Hua, M.C.; Liao, S.L.; Tsai, M.H.; Lai, S.H.; Tseng, Y.L.; Yeh, K.W.; Tsai, H.J.; Huang, J.L. Tobacco smoke exposure and multiplexed immunoglobulin E sensitization in children: A population-based study. Allergy 2016, 71, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Goksor, E.; Amark, M.; Alm, B.; Gustafsson, P.M.; Wennergren, G. The impact of pre- and post-natal smoke exposure on future asthma and bronchial hyper-responsiveness. Acta Paediatr. 2007, 96, 1030–1035. [Google Scholar] [CrossRef] [PubMed]
- Young, S.; Le Souef, P.N.; Geelhoed, G.C.; Stick, S.M.; Turner, K.J.; Landau, L.I. The influence of a family history of asthma and parental smoking on airway responsiveness in early infancy. N. Engl. J. Med. 1991, 324, 1168–1173. [Google Scholar] [CrossRef] [PubMed]
- Baena-Cagnani, C.E.; Gomez, R.M.; Baena-Cagnani, R.; Canonica, G.W. Impact of environmental tobacco smoke and active tobacco smoking on the development and outcomes of asthma and rhinitis. Curr. Opin. Allergy Clin. Immunol. 2009, 9, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.F.; Langholz, B.; Salam, M.T.; Gilliland, F.D. Maternal and grandmaternal smoking patterns are associated with early childhood asthma. Chest 2005, 127, 1232–1241. [Google Scholar] [CrossRef]
- Pattenden, S.; Antova, T.; Neuberger, M.; Nikiforov, B.; De, S.M.; Grize, L.; Heinrich, J.; Hruba, F.; Janssen, N.; Luttmann-Gibson, H.; et al. Parental smoking and children’s respiratory health: Independent effects of prenatal and postnatal exposure. Tob. Control 2006, 15, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Thomson, N.C.; Chaudhuri, R.; Livingston, E. Asthma and cigarette smoking. Eur. Respir. J. 2004, 24, 822–833. [Google Scholar] [CrossRef] [PubMed]
- Gilmour, M.I.; Jaakkola, M.S.; London, S.J.; Nel, A.E.; Rogers, C.A. How exposure to environmental tobacco smoke, outdoor air pollutants, and increased pollen burdens influences the incidence of asthma. Environ. Health Persp. 2006, 114, 627–633. [Google Scholar] [CrossRef]
- Chalmers, G.W.; MacLeod, K.J.; Little, S.A.; Thomson, L.J.; McSharry, C.P.; Thomson, N.C. Influence of cigarette smoking on inhaled corticosteroid treatment in mild asthma. Thorax 2002, 57, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, R.; Livingston, E.; McMahon, A.D.; Thomson, L.; Borland, W.; Thomson, N.C. Cigarette smoking impairs the therapeutic response to oral corticosteroids in chronic asthma. Am. J. Respir. Crit. Care Med. 2003, 168, 1308–1311. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, S.C.; Chinchilli, V.M.; Rollings, N.J.; Boushey, H.A.; Cherniack, R.; Craig, T.J.; Deykin, A.; DiMango, E.; Fish, J.E.; Ford, J.G.; et al. Smoking affects response to inhaled corticosteroids or leukotriene receptor antagonists in asthma. Am. J. Respir. Crit. Care Med. 2007, 175, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Van Miert, E.; Sardella, A.; Bernard, A. Biomarkers of early respiratory effects in smoking adolescents. Eur. Respir. J. 2011, 38, 1287–1293. [Google Scholar] [CrossRef] [PubMed]
- Vanker, A.; Gie, R.P.; Zar, H.J. The association between environmental tobacco smoke exposure and childhood respiratory disease: A review. Exp. Rev. Respir. Med. 2017, 11, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Burke, H.; Leonardi-Bee, J.; Hashim, A.; Pine-Abata, H.; Chen, Y.; Cook, D.G.; Britton, J.R.; McKeever, T.M. Prenatal and passive smoke exposure and incidence of asthma and wheeze: Systematic review and meta-analysis. Pediatrics 2012, 129, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Hanrahan, J.P.; Tager, I.B.; Segal, M.R.; Tosteson, T.D.; Castile, R.G.; Van, V.H.; Weiss, S.T.; Speizer, F.E. The effect of maternal smoking during pregnancy on early infant lung function. Am. Rev. Respir. Dis. 1992, 145, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, K.; Collaco, J.M.; McGrath-Morrow, S.A. Impact of Tobacco Smoke and Nicotine Exposure on Lung Development. Chest 2016, 149, 552–561. [Google Scholar] [CrossRef] [PubMed]
- De Jong, K.; Vonk, J.M.; Imboden, M.; Lahousse, L.; Hofman, A.; Brusselle, G.G.; Probst-Hensch, N.M.; Postma, D.S.; Boezen, H.M. Genes and pathways underlying susceptibility to impaired lung function in the context of environmental tobacco smoke exposure. Respir. Res. 2017, 18, 142. [Google Scholar] [CrossRef] [PubMed]
- Zuraimi, M.S.; Tham, K.W.; Chew, F.T.; Ooi, P.L.; David, K. Home exposures to environmental tobacco smoke and allergic symptoms among young children in Singapore. Int. Arch. Allergy Immunol. 2008, 146, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Burr, M.L.; Anderson, H.R.; Austin, J.B.; Harkins, L.S.; Kaur, B.; Strachan, D.P.; Warner, J.O. Respiratory symptoms and home environment in children: A national survey. Thorax 1999, 54, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Austin, J.B.; Russell, G. Wheeze, cough, atopy, and indoor environment in the Scottish Highlands. Arch. Dis. Child. 1997, 76, 22–26. [Google Scholar] [CrossRef] [PubMed]
- De, S.; Fenton, J.E.; Jones, A.S.; Clarke, R.W. Passive smoking, allergic rhinitis and nasal obstruction in children. J. Laryngol. Otol. 2005, 119, 955–957. [Google Scholar] [CrossRef] [PubMed]
- Houser, S.M.; Keen, K.J. The role of allergy and smoking in chronic rhinosinusitis and polyposis. Laryngoscope 2008, 118, 1521–1527. [Google Scholar] [CrossRef] [PubMed]
- Comer, D.M.; Elborn, J.S.; Ennis, M. Inflammatory and cytotoxic effects of acrolein, nicotine, acetylaldehyde and cigarette smoke extract on human nasal epithelial cells. BMC Pulm. Med. 2014, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Pace, E.; Ferraro, M.; Di Vincenzo, S.; Gerbino, S.; Bruno, A.; Lanata, L.; Gjomarkaj, M. Oxidative stress and innate immunity responses in cigarette smoke stimulated nasal epithelial cells. Toxicology 2014, 28, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Wang, C.H.; Fu, C.H.; Huang, C.C.; Chang, P.H.; Chen, Y.W.; Wu, C.C.; Wu, P.W.; Lee, T.J. Association between cigarette smoking and interleukin-17A expression in nasal tissues of patients with chronic rhinosinusitis and asthma. Medicine 2016, 95, e5432. [Google Scholar] [CrossRef] [PubMed]
- Montano-Velazquez, B.B.; Flores-Rojas, E.B.; Garcia-Vazquez, F.J.; Jurado-Hernandez, S.; Venancio Hernandez, M.A.; Alanis Flores, A.K.; Jauregui-Renaud, K. Effect of cigarette smoke on counts of immunoreactive cells to eotaxin-1 and eosinophils on the nasal mucosa in young patients with perennial allergic rhinitis. Braz. J. Otorhinolaryngol. 2017, 83, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, J.K.; O’Connell, B.P.; Pasquini, W.; Mulligan, R.M.; Smith, S.; Soler, Z.M.; Atkinson, C.; Schlosser, R.J. Impact of tobacco smoke on upper airway dendritic cell accumulation and regulation by sinonasal epithelial cells. Int. Forum Allergy Rhinol. 2017, 7, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Provoost, S.; Maes, T.; Joos, G.F.; Tournoy, K.G. Monocyte-derived dendritic cell recruitment and allergic T(H)2 responses after exposure to diesel particles are CCR2 dependent. J. Allergy Clin. Immunol. 2012, 129, 483–491. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, B.P.; Schlosser, R.J.; Wentzel, J.L.; Nagel, W.; Mulligan, J.K. Systemic monocyte-derived dendritic cells and associated Th2 skewing in chronic rhinosinusitis. Otolaryngol. Head Neck Surg. 2014, 150, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Jukosky, J.; Gosselin, B.J.; Foley, L.; Dechen, T.; Fiering, S.; Crane-Godreau, M.A. In vivo Cigarette Smoke Exposure Decreases CCL20, SLPI, and BD-1 Secretion by Human Primary Nasal Epithelial Cells. Front. Psychiatry 2015, 6, 185. [Google Scholar] [CrossRef] [PubMed]
- Shen, P.; Morissette, M.C.; Vanderstocken, G.; Gao, Y.; Hassan, M.; Roos, A.; Thayaparan, D.; Merlano, M.; Dorrington, M.G.; Nikota, J.K.; et al. Cigarette Smoke Attenuates the Nasal Host Response to Streptococcus pneumoniae and Predisposes to Invasive Pneumococcal Disease in Mice. Infect. Immun. 2016, 84, 1536–1547. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Sim, S.; Choi, H.G. Atopic dermatitis is associated with active and passive cigarette smoking in adolescents. PLoS ONE 2017, 12, e0187453. [Google Scholar] [CrossRef] [PubMed]
- Kantor, R.; Kim, A.; Thyssen, J.P.; Silverberg, J.I. Association of atopic dermatitis with smoking: A systematic review and meta-analysis. J. Am. Acad. Dermatol. 2016, 75, 1119-1125.e1. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Chuang, H.Y.; Hong, C.H.; Huang, S.K.; Chang, Y.C.; Ko, Y.C.; Yu, H.S. Lifetime exposure to cigarette smoking and the development of adult-onset atopic dermatitis. Br. J. Dermatol. 2011, 164, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Puri, P.; Nandar, S.K.; Kathuria, S.; Ramesh, V. Effects of air pollution on the skin: A review. Indian J. Dermatol. Venereol. Leprol. 2017, 83, 415–423. [Google Scholar] [PubMed]
- Hong, C.H.; Lee, C.H.; Yu, H.S.; Huang, S.K. Benzopyrene, a major polyaromatic hydrocarbon in smoke fume, mobilizes Langerhans cells and polarizes Th2/17 responses in epicutaneous protein sensitization through the aryl hydrocarbon receptor. Int. Immunopharmacol. 2016, 36, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Bonamonte, D.; Vestita, M.; Filoni, A.; Mastrolonardo, M.; Angelini, G.; Foti, C. Tobacco-induced contact dermatitis. Eur. J. Dermatol. 2016, 26, 223–231. [Google Scholar] [PubMed]
- Nielsen, G.D.; Olsen, O.; Larsen, S.T.; Lovik, M.; Poulsen, L.K.; Glue, C.; Brandorff, N.P.; Nielsen, P.J. IgE-mediated sensitisation, rhinitis and asthma from occupational exposures. Smoking as a model for airborne adjuvants? Toxicology 2005, 216, 87–105. [Google Scholar] [CrossRef] [PubMed]
- Tong, V.T.; Dietz, P.M.; Morrow, B.; D’Angelo, D.V.; Farr, S.L.; Rockhill, K.M.; England, L.J. Trends in smoking before, during, and after pregnancy—Pregnancy Risk Assessment Monitoring System, United States, 40 sites, 2000–2010. MMWR Surveill. Summ. 2013, 62, 1–19. [Google Scholar] [PubMed]
- Gilliland, F.D.; Berhane, K.; McConnell, R.; Gauderman, W.J.; Vora, H.; Rappaport, E.B.; Avol, E.; Peters, J.M. Maternal smoking during pregnancy, environmental tobacco smoke exposure and childhood lung function. Thorax 2000, 55, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Moshammer, H.; Hoek, G.; Luttmann-Gibson, H.; Neuberger, M.A.; Antova, T.; Gehring, U.; Hruba, F.; Pattenden, S.; Rudnai, P.; Slachtova, H.; et al. Parental smoking and lung function in children: An international study. Am. J. Respir. Crit. Care Med. 2006, 173, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Gurkan, F.; Kiral, A.; Dagli, E.; Karakoc, F. The effect of passive smoking on the development of respiratory syncytial virus bronchiolitis. Eur. J. Epidemiol. 2000, 16, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Wisborg, K.; Kesmodel, U.; Henriksen, T.B.; Olsen, S.F.; Secher, N.J. A prospective study of smoking during pregnancy and SIDS. Arch. Dis. Child. 2000, 83, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Jaakkola, J.J.; Nafstad, P.; Magnus, P. Environmental tobacco smoke, parental atopy, and childhood asthma. Environ. Health Perspect. 2001, 109, 579–582. [Google Scholar] [CrossRef] [PubMed]
- Saulyte, J.; Regueira, C.; Montes-Martinez, A.; Khudyakov, P.; Takkouche, B. Active or passive exposure to tobacco smoking and allergic rhinitis, allergic dermatitis, and food allergy in adults and children: A systematic review and meta-analysis. PLoS Med. 2014, 11, e1001611. [Google Scholar] [CrossRef] [PubMed]
- Macaubas, C.; de Klerk, N.H.; Holt, B.J.; Wee, C.; Kendall, G.; Firth, M.; Sly, P.D.; Holt, P.G. Association between antenatal cytokine production and the development of atopy and asthma at age 6 years. Lancet 2003, 362, 1192–1197. [Google Scholar] [CrossRef]
- Noakes, P.S.; Holt, P.G.; Prescott, S.L. Maternal smoking in pregnancy alters neonatal cytokine responses. Allergy 2003, 58, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Hinz, D.; Bauer, M.; Roder, S.; Olek, S.; Huehn, J.; Sack, U.; Borte, M.; Simon, J.C.; Lehmann, I.; Herberth, G. Cord blood Tregs with stable FOXP3 expression are influenced by prenatal environment and associated with atopic dermatitis at the age of one year. Allergy 2012, 67, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Almanzar, G.; Eberle, G.; Lassacher, A.; Specht, C.; Koppelstaetter, C.; Heinz-Erian, P.; Trawoger, R.; Bernhard, D.; Prelog, M. Maternal cigarette smoking and its effect on neonatal lymphocyte subpopulations and replication. BMC Pediatr. 2013, 13, 57. [Google Scholar] [CrossRef] [PubMed]
- Devereux, G.; Barker, R.N.; Seaton, A. Antenatal determinants of neonatal immune responses to allergens. Clin. Exp. Allergy 2002, 32, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Murata, Y.; Shimamura, T.; Hamuro, J. The polarization of T(h)1/T(h)2 balance is dependent on the intracellular thiol redox status of macrophages due to the distinctive cytokine production. Int. Immunol. 2002, 14, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.P.; Gundavarapu, S.; Pena-Philippides, J.C.; Rir-Sima-Ah, J.; Mishra, N.C.; Wilder, J.A.; Langley, R.J.; Smith, K.R.; Sopori, M.L. Prenatal secondhand cigarette smoke promotes TH2 polarization and impairs goblet cell differentiation and airway mucus formation. J. Immunol. 2011, 187, 4542–4552. [Google Scholar] [CrossRef] [PubMed]
- Kondo, N.; Kobayashi, Y.; Shinoda, S.; Takenaka, R.; Teramoto, T.; Kaneko, H.; Fukao, T.; Matsui, E.; Kasahara, K.; Yokoyama, Y. Reduced interferon gamma production by antigen-stimulated cord blood mononuclear cells is a risk factor of allergic disorders—6-year follow-up study. Clin. Exp. Allergy 1998, 28, 1340–1344. [Google Scholar] [CrossRef] [PubMed]
- Prescott, S.L.; Macaubas, C.; Smallacombe, T.; Holt, B.J.; Sly, P.D.; Loh, R.; Holt, P.G. Reciprocal age-related patterns of allergen-specific T-cell immunity in normal vs. atopic infants. Clin. Exp. Allergy 1998, 28 (Suppl. 5), 39–44. [Google Scholar] [CrossRef] [PubMed]
- Noakes, P.S.; Hale, J.; Thomas, R.; Lane, C.; Devadason, S.G.; Prescott, S.L. Maternal smoking is associated with impaired neonatal toll-like-receptor-mediated immune responses. Eur. Respir. J. 2006, 28, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Duijts, L.; Jaddoe, V.W.V.; van der Valk, R.J.P.; Henderson, J.A.; Hofman, A.; Raat, H.; Steegers, E.A.P.; Moll, H.A.; de Jongste, J.C. Fetal exposure to maternal and paternal smoking and the risks of wheezing in preschool children: The Generation R Study. Chest 2012, 141, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Kabesch, M.; Hoefler, C.; Carr, D.; Leupold, W.; Weiland, S.K.; von Mutius, E. Glutathione S transferase deficiency and passive smoking increase childhood asthma. Thorax 2004, 59, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Gilliland, F.D.; Li, Y.F.; Dubeau, L.; Berhane, K.; Avol, E.; McConnell, R.; Gauderman, W.J.; Peters, J.M. Effects of glutathione S-transferase M1, maternal smoking during pregnancy, and environmental tobacco smoke on asthma and wheezing in children. Am. J. Respir. Crit. Care Med. 2002, 166, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.L.; Henderson, J.; Northstone, K.; Pembrey, M.; Golding, J. Do grandmaternal smoking patterns influence the etiology of childhood asthma? Chest 2014, 145, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Hatoun, J.; Davis-Plourde, K.; Penti, B.; Cabral, H.; Kazis, L. Tobacco Control Laws and Pediatric Asthma. Pediatrics 2018, 141, S130–S136. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.J.; Hansch, C. The relative toxicity of compounds in mainstream cigarette smoke condensate. Food Chem. Toxicol. 2000, 38, 637–646. [Google Scholar] [CrossRef]
- Proulx, L.I.; Castonguay, A.; Bissonnette, E.Y. Cytokine production by alveolar macrophages is down regulated by the alpha-methylhydroxylation pathway of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK). Carcinogenesis 2004, 25, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Rahman, I.; Macnee, W. Lung glutathione and oxidative stress: Implications in cigarette smoke-induced airway disease. Am. J. Physiol. 1999, 277, L1067–L1088. [Google Scholar] [CrossRef] [PubMed]
- Rahman, I. Oxidative stress, chromatin remodeling and gene transcription in inflammation and chronic lung diseases. J. Biochem. Mol. Biol. 2003, 36, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Rahman, I.; Skwarska, E.; Henry, M.; Davis, M.; O’Connor, C.M.; FitzGerald, M.X.; Greening, A.; Macnee, W. Systemic and pulmonary oxidative stress in idiopathic pulmonary fibrosis. Free Radic. Biol. Med. 1999, 27, 60–68. [Google Scholar] [CrossRef]
- Kelly, F.J.; Mudway, I.; Blomberg, A.; Frew, A.; Sandstrom, T. Altered lung antioxidant status in patients with mild asthma. Lancet 1999, 354, 482–483. [Google Scholar] [CrossRef]
- Macnee, W. Oxidative stress and chronic obstructive pulmonary disease. Manag. Chron. Obstr. Pulm. Dis. 2009, 80, 759–761. [Google Scholar]
- Macnee, W.; Tuder, R.M. New paradigms in the pathogenesis of chronic obstructive pulmonary disease I. Proc. Am. Thorac. Soc. 2009, 6, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Gilmour, P.S.; Donaldson, K.; MacNee, W. Overview of the antioxidant pathways in relation to effects of air pollution. Eur. Respir. Soc. 2002, 21, 241–261. [Google Scholar]
- Moodie, F.M.; Marwick, J.A.; Anderson, C.S.; Szulakowski, P.; Biswas, S.K.; Bauter, M.R.; Kilty, I.; Rahman, I. Oxidative stress and cigarette smoke alter chromatin remodeling but differentially regulate NF-kappaB activation and proinflammatory cytokine release in alveolar epithelial cells. FASEB J. 2004, 18, 1897–1899. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.R.; Chida, A.S.; Bauter, M.R.; Shafiq, N.; Seweryniak, K.; Maggirwar, S.B.; Kilty, I.; Rahman, I. Cigarette smoke induces proinflammatory cytokine release by activation of NF-kappaB and posttranslational modifications of histone deacetylase in macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L46–L57. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.U.; Kim, J.D.; Park, C.S. Gene-Environment Interactions in Asthma: Genetic and Epigenetic Effects. Yonsei Med. J. 2015, 56, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Holt, P.G.; Keast, D. Environmentally induced changes in immunological function: Acute and chronic effects of inhalation of tobacco smoke and other atmospheric contaminants in man and experimental animals. Bacteriol. Rev. 1977, 41, 205–216. [Google Scholar] [PubMed]
- Dvorkin-Gheva, A.; Vanderstocken, G.; Yildirim, A.O.; Brandsma, C.A.; Obeidat, M.; Bosse, Y.; Hassell, J.A.; Stampfli, M.R. Total particulate matter concentration skews cigarette smoke’s gene expression profile. ERJ Open Res. 2016, 2. [Google Scholar] [CrossRef] [PubMed]
- Mio, T.; Romberger, D.J.; Thompson, A.B.; Robbins, R.A.; Heires, A.; Rennard, S.I. Cigarette smoke induces interleukin-8 release from human bronchial epithelial cells. Am. J. Respir. Crit. Care Med. 1997, 155, 1770–1776. [Google Scholar] [CrossRef] [PubMed]
- Hellermann, G.R.; Nagy, S.B.; Kong, X.; Lockey, R.F.; Mohapatra, S.S. Mechanism of cigarette smoke condensate-induced acute inflammatory response in human bronchial epithelial cells. Respir. Res. 2002, 3, 22. [Google Scholar] [CrossRef] [PubMed]
- Churg, A.; Zay, K.; Shay, S.; Xie, C.; Shapiro, S.D.; Hendricks, R.; Wright, J.L. Acute cigarette smoke-induced connective tissue breakdown requires both neutrophils and macrophage metalloelastase in mice. Am. J. Respir. Cell Mol. Biol. 2002, 27, 368–374. [Google Scholar] [CrossRef] [PubMed]
- De Boer, W.I.; Sont, J.K.; van, S.A.; Stolk, J.; van Krieken, J.H.; Hiemstra, P.S. Monocyte chemoattractant protein 1, interleukin 8, and chronic airways inflammation in COPD. J. Pathol. 2000, 190, 619–626. [Google Scholar] [CrossRef]
- Doz, E.; Noulin, N.; Boichot, E.; Guenon, I.; Fick, L.; Le Bert, M.; Lagente, V.; Ryffel, B.; Schnyder, B.; Quesniaux, V.F.; et al. Cigarette smoke-induced pulmonary inflammation is TLR4/MyD88 and IL-1R1/MyD88 signaling dependent. J. Immunol. 2008, 180, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.K.; Iwagaki, H.; Hamano, R.; Yoshino, T.; Tanaka, N.; Nishibori, M. Effect of nicotine on IL-18-initiated immune response in human monocytes. J. Leukoc. Biol. 2006, 80, 1388–1394. [Google Scholar] [CrossRef] [PubMed]
- Nizri, E.; Irony-Tur-Sinai, M.; Lory, O.; Orr-Urtreger, A.; Lavi, E.; Brenner, T. Activation of the cholinergic anti-inflammatory system by nicotine attenuates neuroinflammation via suppression of Th1 and Th17 responses. J. Immunol. 2009, 183, 6681–6688. [Google Scholar] [CrossRef] [PubMed]
- Arnson, Y.; Shoenfeld, Y.; Amital, H. Effects of tobacco smoke on immunity, inflammation and autoimmunity. J. Autoimmun. 2010, 34, J258–J265. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, E.; Spiess, P.C.; Habibovic, A.; Hristova, M.; Bauer, R.A.; Randall, M.J.; Poynter, M.E.; van der Vliet, A. Inhalation of the reactive aldehyde acrolein promotes antigen sensitization to ovalbumin and enhances neutrophilic inflammation. J. Immunotoxicol. 2016, 13, 191–197. [Google Scholar] [CrossRef] [PubMed]
- D’Hulst, A.I.; Maes, T.; Bracke, K.R.; Demedts, I.K.; Tournoy, K.G.; Joos, G.F.; Brusselle, G.G. Cigarette smoke-induced pulmonary emphysema in scid-mice. Is the acquired immune system required? Respir. Res. 2005, 6, 147. [Google Scholar]
- Macnee, W. Pathogenesis of chronic obstructive pulmonary disease. Clin. Chest Med. 2007, 28, 479–513. [Google Scholar] [CrossRef] [PubMed]
- Cosio, M.G.; Saetta, M.; Agusti, A. Immunologic aspects of chronic obstructive pulmonary disease. N. Engl. J. Med. 2009, 360, 2445–2454. [Google Scholar] [CrossRef] [PubMed]
- Honda, Y.; Takahashi, H.; Kuroki, Y.; Akino, T.; Abe, S. Decreased contents of surfactant proteins A and D in BAL fluids of healthy smokers. Chest 1996, 109, 1006–1009. [Google Scholar] [CrossRef] [PubMed]
- Knight, D.A.; Holgate, S.T. The airway epithelium: Structural and functional properties in health and disease. Respirology 2003, 8, 432–446. [Google Scholar] [CrossRef] [PubMed]
- Lambrecht, B.N.; Hammad, H. Allergens and the airway epithelium response: Gateway to allergic sensitization. J. Allergy Clin. Immunol. 2014, 134, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Tam, A.; Wadsworth, S.; Dorscheid, D.; Man, S.F.; Sin, D.D. The airway epithelium: More than just a structural barrier. Ther. Adv. Resp. Dis. 2011, 5, 255–273. [Google Scholar] [CrossRef] [PubMed]
- Dye, J.A.; Adler, K.B. Effects of cigarette smoke on epithelial cells of the respiratory tract. Thorax 1994, 49, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Biczysko-Murawa, A.; Stopa, J.; Marszalek, A. Structural changes in tracheal epithelium in environmental smoke exposed rats—Experimental studies. Prz. Lek. 2008, 65, 462–465. [Google Scholar] [PubMed]
- Biczysko-Murawa, A.; Seget, M.; Stopa, J.; Biczysko, W. Changes in tracheal ciliated epithelium of rats exposed to environmental tobacco smoke—Experimental studies. Prz. Lek. 2009, 66, 589–592. [Google Scholar] [PubMed]
- Haswell, L.E.; Hewitt, K.; Thorne, D.; Richter, A.; Gaca, M.D. Cigarette smoke total particulate matter increases mucous secreting cell numbers in vitro: A potential model of goblet cell hyperplasia. Toxicol. In Vitro 2010, 24, 981–987. [Google Scholar] [CrossRef] [PubMed]
- Cohen, N.A.; Zhang, S.; Sharp, D.B.; Tamashiro, E.; Chen, B.; Sorscher, E.J.; Woodworth, B.A. Cigarette smoke condensate inhibits transepithelial chloride transport and ciliary beat frequency. Laryngoscope 2009, 119, 2269–2274. [Google Scholar] [CrossRef] [PubMed]
- Tamashiro, E.; Xiong, G.; Anselmo-Lima, W.T.; Kreindler, J.L.; Palmer, J.N.; Cohen, N.A. Cigarette smoke exposure impairs respiratory epithelial ciliogenesis. Am. J. Rhinol. Allergy 2009, 23, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Biselli, P.J.; Lopes, F.D.; Moriya, H.T.; Rivero, D.H.; Toledo, A.C.; Saldiva, P.H.; Mauad, T.; Martins, M.A. Short-term exposure of mice to cigarette smoke and/or residual oil fly ash produces proximal airspace enlargements and airway epithelium remodeling. Braz. J. Med. Biol. Res. 2011, 44, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wang, X.; Brighton, L.; Hazucha, M.; Jaspers, I.; Carson, J.L. Increased nasal epithelial ciliary beat frequency associated with lifestyle tobacco smoke exposure. Inhal. Toxicol. 2009, 21, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Carson, J.L.; Lu, T.S.; Brighton, L.; Hazucha, M.; Jaspers, I.; Zhou, H. Phenotypic and physiologic variability in nasal epithelium cultured from smokers and nonsmokers exposed to secondhand tobacco smoke. In Vitro Cell. Dev. Biol. Anim. 2010, 46, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Olivera, D.; Knall, C.; Boggs, S.; Seagrave, J. Cytoskeletal modulation and tyrosine phosphorylation of tight junction proteins are associated with mainstream cigarette smoke-induced permeability of airway epithelium. Exp. Toxicol. Pathol. 2010, 62, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Gangl, K.; Reininger, R.; Bernhard, D.; Campana, R.; Pree, I.; Reisinger, J.; Kneidinger, M.; Kundi, M.; Dolznig, H.; Thurnher, D.; et al. Cigarette smoke facilitates allergen penetration across respiratory epithelium. Allergy 2009, 64, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Shaykhiev, R.; Otaki, F.; Bonsu, P.; Dang, D.T.; Teater, M.; Strulovici-Barel, Y.; Salit, J.; Harvey, B.G.; Crystal, R.G. Cigarette smoking reprograms apical junctional complex molecular architecture in the human airway epithelium in vivo. Cell. Mol. Life Sci. 2011, 68, 877–892. [Google Scholar] [CrossRef] [PubMed]
- Kreindler, J.L.; Jackson, A.D.; Kemp, P.A.; Bridges, R.J.; Danahay, H. Inhibition of chloride secretion in human bronchial epithelial cells by cigarette smoke extract. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 288, L894–L902. [Google Scholar] [CrossRef] [PubMed]
- Virgin, F.W.; Azbell, C.; Schuster, D.; Sunde, J.; Zhang, S.; Sorscher, E.J.; Woodworth, B.A. Exposure to cigarette smoke condensate reduces calcium activated chloride channel transport in primary sinonasal epithelial cultures. Laryngoscope 2010, 120, 1465–1469. [Google Scholar] [CrossRef] [PubMed]
- Hessel, J.; Heldrich, J.; Fuller, J.; Staudt, M.R.; Radisch, S.; Hollmann, C.; Harvey, B.G.; Kaner, R.J.; Salit, J.; Yee-Levin, J.; et al. Intraflagellar transport gene expression associated with short cilia in smoking and COPD. PLoS ONE 2014, 9, e85453. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Xu, Z.; Wang, R.; Al-Hijji, M.; Salit, J.; Strulovici-Barel, Y.; Tilley, A.E.; Mezey, J.G.; Crystal, R.G. Genes associated with MUC5AC expression in small airway epithelium of human smokers and nonsmokers. BMC Med. Genom. 2012, 5, 21. [Google Scholar] [CrossRef] [PubMed]
- Kanai, K.; Koarai, A.; Shishikura, Y.; Sugiura, H.; Ichikawa, T.; Kikuchi, T.; Akamatsu, K.; Hirano, T.; Nakanishi, M.; Matsunaga, K.; et al. Cigarette smoke augments MUC5AC production via the TLR3-EGFR pathway in airway epithelial cells. Respir. Investig. 2015, 53, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, T.A.; Heires, A.J.; Sanderson, S.D.; Floreani, A.A. Protein kinase C activation is required for cigarette smoke-enhanced C5a-mediated release of interleukin-8 in human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 1999, 21, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Miller, M.; Cho, J.Y.; Song, D.J.; Karin, M.; Broide, D.H. Inactivation of I kappaB-kinase-beta dependent genes in airway epithelium reduces tobacco smoke induced acute airway inflammation. Int. Immunopharmacol. 2010, 10, 906–912. [Google Scholar] [CrossRef] [PubMed]
- Laan, M.; Bozinovski, S.; Anderson, G.P. Cigarette smoke inhibits lipopolysaccharide-induced production of inflammatory cytokines by suppressing the activation of activator protein-1 in bronchial epithelial cells. J. Immunol. 2004, 173, 4164–4170. [Google Scholar] [CrossRef] [PubMed]
- Pace, E.; Ferraro, M.; Siena, L.; Melis, M.; Montalbano, A.M.; Johnson, M.; Bonsignore, M.R.; Bonsignore, G.; Gjomarkaj, M. Cigarette smoke increases Toll-like receptor 4 and modifies lipopolysaccharide-mediated responses in airway epithelial cells. Immunology 2008, 124, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Damia, A.D.; Gimeno, J.C.; Ferrer, M.J.; Fabregas, M.L.; Folch, P.A.; Paya, J.M. A study of the effect of proinflammatory cytokines on the epithelial cells of smokers, with or without COPD. Arch. Bronconeumol. 2011, 47, 447–453. [Google Scholar] [CrossRef]
- Vlahos, R.; Bozinovski, S.; Jones, J.E.; Powell, J.; Gras, J.; Lilja, A.; Hansen, M.J.; Gualano, R.C.; Irving, L.; Anderson, G.P. Differential protease, innate immunity, and NF-kappaB induction profiles during lung inflammation induced by subchronic cigarette smoke exposure in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L931–L945. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, A.; Caramori, G.; Gnemmi, I.; Contoli, M.; Bristot, L.; Capelli, A.; Ricciardolo, F.L.; Magno, F.; D’Anna, S.E.; Zanini, A.; et al. Association of increased CCL5 and CXCL7 chemokine expression with neutrophil activation in severe stable COPD. Thorax 2009, 64, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Reno, F.; Rocchetti, V.; Migliario, M.; Rizzi, M.; Cannas, M. Chronic exposure to cigarette smoke increases matrix metalloproteinases and Filaggrin mRNA expression in oral keratinocytes: Role of nicotine stimulation. Oral Oncol. 2011, 47, 827–830. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Miyata, M.; Ohba, T.; Ando, T.; Hatsushika, K.; Suenaga, F.; Shimokawa, N.; Ohnuma, Y.; Katoh, R.; Ogawa, H.; et al. Cigarette smoke extract induces thymic stromal lymphopoietin expression, leading to T(H)2-type immune responses and airway inflammation. J. Allergy Clin. Immunol. 2008, 122, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, C.M.; Saglani, S. Epithelial cytokines and pulmonary allergic inflammation. Curr. Opin. Immunol. 2015, 34, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J.; Soumelis, V.; Watanabe, N.; Ito, T.; Wang, Y.H.; Malefyt Rde, W.; Omori, M.; Zhou, B.; Ziegler, S.F. TSLP: An epithelial cell cytokine that regulates T cell differentiation by conditioning dendritic cell maturation. Annu. Rev. Immunol. 2007, 25, 193–219. [Google Scholar] [CrossRef] [PubMed]
- Hodge, S.; Hodge, G.; Holmes, M.; Reynolds, P.N. Increased airway epithelial and T-cell apoptosis in COPD remains despite smoking cessation. Eur. Respir. J. 2005, 25, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Pouwels, S.D.; Zijlstra, G.J.; van der Toorn, M.; Hesse, L.; Gras, R.; Ten Hacken, N.H.; Krysko, D.V.; Vandenabeele, P.; de Vries, M.; van Oosterhout, A.J.; et al. Cigarette smoke-induced necroptosis and DAMP release trigger neutrophilic airway inflammation in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L377–L386. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, P.R.; Kasteler, S.D.; Schmitt, R.E.; Hoidal, J.R. Receptor for advanced glycation end-products signals through Ras during tobacco smoke-induced pulmonary inflammation. Am. J. Respir. Cell Mol. Biol. 2011, 45, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, K.; Radvar, M.; Rezaee, A.; Rafatpanah, H.; Azangoo khiavi, H.; Dadpour, Y.; Radvar, N. Comparison of relative TLR-2 and TLR-4 expression level of disease and healthy gingival tissue of smoking and nonsmoking patients and periodontally healthy control patients. Aust. Dent. J. 2013, 58, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Mortaz, E.; Lazar, Z.; Koenderman, L.; Kraneveld, A.D.; Nijkamp, F.P.; Folkerts, G. Cigarette smoke attenuates the production of cytokines by human plasmacytoid dendritic cells and enhances the release of IL-8 in response to TLR-9 stimulation. Respir. Res. 2009, 10, 47. [Google Scholar] [CrossRef] [PubMed]
- Eltom, S.; Stevenson, C.S.; Rastrick, J.; Dale, N.; Raemdonck, K.; Wong, S.; Catley, M.C.; Belvisi, M.G.; Birrell, M.A. P2X7 receptor and caspase 1 activation are central to airway inflammation observed after exposure to tobacco smoke. PLoS ONE 2011, 6, e24097. [Google Scholar] [CrossRef] [PubMed]
- Lucattelli, M.; Cicko, S.; Muller, T.; Lommatzsch, M.; De Cunto, G.; Cardini, S.; Sundas, W.; Grimm, M.; Zeiser, R.; Durk, T.; et al. P2X7 receptor signaling in the pathogenesis of smoke-induced lung inflammation and emphysema. Am. J. Respir. Cell Mol. Biol. 2011, 44, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Ahmed, J.; Wang, G.; Hassan, I.; Strulovici-Barel, Y.; Salit, J.; Mezey, J.G.; Crystal, R.G. Airway epithelial expression of TLR5 is downregulated in healthy smokers and smokers with chronic obstructive pulmonary disease. J. Immunol. 2012, 189, 2217–2225. [Google Scholar] [CrossRef] [PubMed]
- Bauer, C.M.; Dewitte-Orr, S.J.; Hornby, K.R.; Zavitz, C.C.; Lichty, B.D.; Stampfli, M.R.; Mossman, K.L. Cigarette smoke suppresses type I interferon-mediated antiviral immunity in lung fibroblast and epithelial cells. J. Interferon Cytokine Res. 2008, 28, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Eddleston, J.; Lee, R.U.; Doerner, A.M.; Herschbach, J.; Zuraw, B.L. Cigarette smoke decreases innate responses of epithelial cells to rhinovirus infection. Am. J. Respir. Cell Mol. Biol. 2011, 44, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, R.; Rampersaud, R.; Aguilar, J.L.; Randis, T.M.; Kreindler, J.L.; Ratner, A.J. Cigarette smoke inhibits airway epithelial cell innate immune responses to bacteria. Infect. Immun. 2010, 78, 2146–2152. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Case, S.; Bowler, R.P.; Martin, R.J.; Jiang, D.; Chu, H.W. Cigarette smoke modulates PGE2 and host defence against Moraxella catarrhalis infection in human airway epithelial cells. Respirology 2011, 16, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Mahanonda, R.; Sa-Ard-Iam, N.; Eksomtramate, M.; Rerkyen, P.; Phairat, B.; Schaecher, K.E.; Fukuda, M.M.; Pichyangkul, S. Cigarette smoke extract modulates human beta-defensin-2 and interleukin-8 expression in human gingival epithelial cells. J. Periodontal Res. 2009, 44, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Garmendia, J.; Morey, P.; Bengoechea, J.A. Impact of cigarette smoke exposure on host-bacterial pathogen interactions. Eur. Respir. J. 2011, 39, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Drannik, A.G.; Pouladi, M.A.; Robbins, C.S.; Goncharova, S.I.; Kianpour, S.; Stampfli, M.R. Impact of cigarette smoke on clearance and inflammation after Pseudomonas aeruginosa infection. Am. J. Respir. Crit. Care Med. 2004, 170, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Grigg, J.; Walters, H.; Sohal, S.S.; Wood-Baker, R.; Reid, D.W.; Xu, C.B.; Edvinsson, L.; Morissette, M.C.; Stampfli, M.R.; Kirwan, M.; et al. Cigarette smoke and platelet-activating factor receptor dependent adhesion of Streptococcus pneumoniae to lower airway cells. Thorax 2012, 67, 908–913. [Google Scholar] [CrossRef] [PubMed]
- D’Anna, C.; Cigna, D.; Di Sano, C.; Di Vincenzo, S.; Dino, P.; Ferraro, M.; Bini, L.; Bianchi, L.; Di Gaudio, F.; Gjomarkaj, M.; et al. Exposure to cigarette smoke extract and lipopolysaccharide modifies cytoskeleton organization in bronchial epithelial cells. Exp. Lung Res. 2017, 43, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Hudy, M.H.; Traves, S.L.; Wiehler, S.; Proud, D. Cigarette smoke modulates rhinovirus-induced airway epithelial cell chemokine production. Eur. Respir. J. 2010, 35, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Pratt, S.A.; Finley, T.N.; Smith, M.H.; Ladman, A.J. A comparison of alveolar macrophages and pulmonary surfactant(?) obtained from the lungs of human smokers and nonsmokers by endobronchial lavage. Anatom. Rec. 1969, 163, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Domagala-Kulawik, J.; Maskey-Warzechowska, M.; Hermanowicz-Salamon, J.; Chazan, R. Expression of macrophage surface markers in induced sputum of patients with chronic obstructive pulmonary disease. J. Physiol. Pharmacol. 2006, 57 (Suppl. 4), 75–84. [Google Scholar] [PubMed]
- Harris, J.O.; Swenson, E.W.; Johnson, J.E., 3rd. Human alveolar macrophages: Comparison of phagocytic ability, glucose utilization, and ultrastructure in smokers and nonsmokers. J. Clin. Investig. 1970, 49, 2086–2096. [Google Scholar] [CrossRef] [PubMed]
- Morissette, M.C.; Shen, P.; Thayaparan, D.; Stampfli, M.R. Disruption of pulmonary lipid homeostasis drives cigarette smoke-induced lung inflammation in mice. Eur. Respir. J. 2015, 46, 1451–1460. [Google Scholar] [CrossRef] [PubMed]
- Finley, T.N.; Ladman, A.J. Low yield of pulmonary surfactant in cigarette smokers. N. Engl. J. Med. 1972, 286, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Schaberg, T.; Lauer, C.; Lode, H.; Fischer, J.; Haller, H. Increased number of alveolar macrophages expressing adhesion molecules of the leukocyte adhesion molecule family in smoking subjects. Association with cell-binding ability and superoxide anion production. Am. Rev. Respir. Dis. 1992, 146, 1287–1293. [Google Scholar] [CrossRef] [PubMed]
- Skold, C.M.; Lundahl, J.; Hallden, G.; Hallgren, M.; Eklund, A. Chronic smoke exposure alters the phenotype pattern and the metabolic response in human alveolar macrophages. Clin. Exp. Immunol. 1996, 106, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.O.; Olsen, G.N.; Castle, J.R.; Maloney, A.S. Comparison of proteolytic enzyme activity in pulmonary alveolar macrophages and blood leukocytes in smokers and nonsmokers. Am. Rev. Respir. Dis. 1975, 111, 579–586. [Google Scholar] [PubMed]
- Hinman, L.M.; Stevens, C.A.; Matthay, R.A.; Gee, J.B. Elastase and lysozyme activities in human alveolar macrophages. Effects of cigarette smoking. Am. Rev. Respir. Dis. 1980, 121, 263–271. [Google Scholar] [PubMed]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Byrne, A.J.; Mathie, S.A.; Gregory, L.G.; Lloyd, C.M. Pulmonary macrophages: Key players in the innate defence of the airways. Thorax 2015, 70, 1189–1196. [Google Scholar] [CrossRef] [PubMed]
- Varin, A.; Gordon, S. Alternative activation of macrophages: Immune function and cellular biology. Immunobiology 2009, 214, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Shaykhiev, R.; Krause, A.; Salit, J.; Strulovici-Barel, Y.; Harvey, B.G.; O’Connor, T.P.; Crystal, R.G. Smoking-dependent reprogramming of alveolar macrophage polarization: Implication for pathogenesis of chronic obstructive pulmonary disease. J. Immunol. 2009, 183, 2867–2883. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Xie, L.; Lu, J.; Sun, S. Characteristics and potential role of M2 macrophages in COPD. Int. J. Chron. Obstr. Pulm. Dis. 2017, 12, 3029–3039. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Xie, L.; Liu, C.; Zhang, Q.; Sun, S. PTEN/PI3k/AKT Regulates Macrophage Polarization in Emphysematous mice. Scand. J. Immunol. 2017, 85, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, T.; Ning, Q.; Li, F.; Chen, T.; Yao, Y.; Sun, Z. Cigarette smoke extract-treated mast cells promote alveolar macrophage infiltration and polarization in experimental chronic obstructive pulmonary disease. Inhal. Toxicol. 2015, 27, 822–831. [Google Scholar] [CrossRef] [PubMed]
- Oliveira da Silva, C.; Monte-Alto-Costa, A.; Renovato-Martins, M.; Viana Nascimento, F.J.; Dos Santos Valenca, S.; Lagente, V.; Porto, L.C.; Victoni, T. Time Course of the Phenotype of Blood and Bone Marrow Monocytes and Macrophages in the Lung after Cigarette Smoke Exposure In Vivo. Int. J. Mol. Sci. 2017, 18, E1940. [Google Scholar] [CrossRef] [PubMed]
- Geissmann, F.; Jung, S.; Littman, D.R. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 2003, 19, 71–82. [Google Scholar] [CrossRef]
- Eapen, M.S.; Hansbro, P.M.; McAlinden, K.; Kim, R.Y.; Ward, C.; Hackett, T.L.; Walters, E.H.; Sohal, S.S. Abnormal M1/M2 macrophage phenotype profiles in the small airway wall and lumen in smokers and chronic obstructive pulmonary disease (COPD). Sci. Rep. 2017, 7, 13392. [Google Scholar] [CrossRef] [PubMed]
- Draijer, C.; Robbe, P.; Boorsma, C.E.; Hylkema, M.N.; Melgert, B.N. Characterization of macrophage phenotypes in three murine models of house-dust-mite-induced asthma. Med. Inflamm. 2013, 2013, 632049. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, S.D. Elastolytic metalloproteinases produced by human mononuclear phagocytes. Potential roles in destructive lung disease. Am. J. Respir. Crit. Care Med. 1994, 150, S160–S164. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Roche, N.; Oliver, B.G.; Mattos, W.; Barnes, P.J.; Chung, K.F. Balance of matrix metalloprotease-9 and tissue inhibitor of metalloprotease-1 from alveolar macrophages in cigarette smokers. Regulation by interleukin-10. Am. J. Respir. Crit. Care Med. 2000, 162, 1355–1360. [Google Scholar] [CrossRef] [PubMed]
- Babusyte, A.; Stravinskaite, K.; Jeroch, J.; Lotvall, J.; Sakalauskas, R.; Sitkauskiene, B. Patterns of airway inflammation and MMP-12 expression in smokers and ex-smokers with COPD. Respir. Res. 2007, 8, 81. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, P.G.; Koth, L.L.; Yang, Y.H.; Rodriguez, M.W.; Favoreto, S.; Dolganov, G.M.; Paquet, A.C.; Erle, D.J. A distinctive alveolar macrophage activation state induced by cigarette smoking. Am. J. Respir. Crit. Care Med. 2005, 172, 1383–1392. [Google Scholar] [CrossRef] [PubMed]
- Hoidal, J.R.; Fox, R.B.; LeMarbre, P.A.; Takiff, H.E.; Repine, J.E. Oxidative metabolism of alveolar macrophages from young asymptomatic cigarette smokers. Increased superoxide anion release and its potential consequences. Chest 1980, 77, 270–271. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, R.C.; Ogushi, F.; Fells, G.A.; Cantin, A.M.; Jallat, S.; Courtney, M.; Crystal, R.G. Oxidants spontaneously released by alveolar macrophages of cigarette smokers can inactivate the active site of alpha 1-antitrypsin, rendering it ineffective as an inhibitor of neutrophil elastase. J. Clin. Investig. 1987, 80, 1289–1295. [Google Scholar] [CrossRef] [PubMed]
- Ohta, T.; Yamashita, N.; Maruyama, M.; Sugiyama, E.; Kobayashi, M. Cigarette smoking decreases interleukin-8 secretion by human alveolar macrophages. Respir. Med. 1998, 92, 922–927. [Google Scholar] [CrossRef]
- Soliman, D.M.; Twigg, H.L., 3rd. Cigarette smoking decreases bioactive interleukin-6 secretion by alveolar macrophages. Am. J. Physiol. 1992, 263, L471–L478. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, E.; Itoh, A.; Furuya, K.; Miyamoto, H.; Abe, S.; Kawakami, Y. Release of tumor necrosis factor-alpha from human alveolar macrophages is decreased in smokers. Chest 1993, 103, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Holt, P.G. Immune and inflammatory function in cigarette smokers. Thorax 1987, 42, 241–249. [Google Scholar] [CrossRef] [PubMed]
- McCrea, K.A.; Ensor, J.E.; Nall, K.; Bleecker, E.R.; Hasday, J.D. Altered cytokine regulation in the lungs of cigarette smokers. Am. J. Respir. Crit. Care Med. 1994, 150, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Y.; Virasch, N.; Hao, P.; Aubrey, M.T.; Mukerjee, N.; Bierer, B.E.; Freed, B.M. Suppression of human IL-1beta, IL-2, IFN-gamma, and TNF-alpha production by cigarette smoke extracts. J. Allergy Clin. Immunol. 2000, 106, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Li, X.; Xie, F.; Yang, Z.; Pan, X.; Zhu, M.; Shang, P.; Nie, C.; Liu, H.; Xie, J. Immunomodulatory effects of cigarette smoke condensate in mouse macrophage cell line. Int. J. Immunopathol. Pharmacol. 2017, 30, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Arimilli, S.; Schmidt, E.; Damratoski, B.E.; Prasad, G.L. Role of Oxidative Stress in the Suppression of Immune Responses in Peripheral Blood Mononuclear Cells Exposed to Combustible Tobacco Product Preparation. Inflammation 2017, 40, 1622–1630. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, K.; Klein, T.W.; Friedman, H.; Yamamoto, Y. Involvement of nicotinic acetylcholine receptors in suppression of antimicrobial activity and cytokine responses of alveolar macrophages to Legionella pneumophila infection by nicotine. J. Immunol. 2001, 167, 6518–6524. [Google Scholar] [CrossRef] [PubMed]
- Gaschler, G.J.; Zavitz, C.C.; Bauer, C.M.; Skrtic, M.; Lindahl, M.; Robbins, C.S.; Chen, B.; Stampfli, M.R. Cigarette smoke exposure attenuates cytokine production by mouse alveolar macrophages. Am. J. Respir. Cell Mol. Biol. 2008, 38, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Metcalfe, H.J.; Lea, S.; Hughes, D.; Khalaf, R.; Abbott-Banner, K.; Singh, D. Effects of cigarette smoke on Toll-like receptor (TLR) activation of chronic obstructive pulmonary disease (COPD) macrophages. Clin. Exp. Immunol. 2014, 176, 461–472. [Google Scholar] [CrossRef] [PubMed]
- King, T.E., Jr.; Savici, D.; Campbell, P.A. Phagocytosis and killing of Listeria monocytogenes by alveolar macrophages: Smokers versus nonsmokers. J. Infect. Dis. 1988, 158, 1309–1316. [Google Scholar] [CrossRef] [PubMed]
- Ortega, E.; Barriga, C.; Rodriguez, A.B. Decline in the phagocytic function of alveolar macrophages from mice exposed to cigarette smoke. Comp. Immunol. Microbiol. Infect. Dis. 1994, 17, 77–84. [Google Scholar] [CrossRef]
- Phipps, J.C.; Aronoff, D.M.; Curtis, J.L.; Goel, D.; O’Brien, E.; Mancuso, P. Cigarette smoke exposure impairs pulmonary bacterial clearance and alveolar macrophage complement-mediated phagocytosis of Streptococcus pneumoniae. Infect. Immun. 2010, 78, 1214–1220. [Google Scholar] [CrossRef] [PubMed]
- Harvey, C.J.; Thimmulappa, R.K.; Sethi, S.; Kong, X.; Yarmus, L.; Brown, R.H.; Feller-Kopman, D.; Wise, R.; Biswal, S. Targeting Nrf2 signaling improves bacterial clearance by alveolar macrophages in patients with COPD and in a mouse model. Sci. Transl. Med. 2011, 3, 78ra32. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Jerome, J.A.; Gregory, A.D.; Mallampalli, R.K. Cigarette smoke destabilizes NLRP3 protein by promoting its ubiquitination. Respir. Res. 2017, 18, 2. [Google Scholar] [CrossRef] [PubMed]
- Droemann, D.; Goldmann, T.; Tiedje, T.; Zabel, P.; Dalhoff, K.; Schaaf, B. Toll-like receptor 2 expression is decreased on alveolar macrophages in cigarette smokers and COPD patients. Respir. Res. 2005, 6, 68. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Cowan, M.J.; Hasday, J.D.; Vogel, S.N.; Medvedev, A.E. Tobacco smoking inhibits expression of proinflammatory cytokines and activation of IL-1R-associated kinase, p38, and NF-kappaB in alveolar macrophages stimulated with TLR2 and TLR4 agonists. J. Immunol. 2007, 179, 6097–6106. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Xu, H.; Li, L.; Yuan, W.; Zhang, D.; Huang, W. Susceptibility to Aspergillus Infections in Rats with Chronic Obstructive Pulmonary Disease via Deficiency Function of Alveolar Macrophages and Impaired Activation of TLR2. Inflammation 2016, 39, 1310–1318. [Google Scholar] [CrossRef] [PubMed]
- Lien, E.; Sellati, T.J.; Yoshimura, A.; Flo, T.H.; Rawadi, G.; Finberg, R.W.; Carroll, J.D.; Espevik, T.; Ingalls, R.R.; Radolf, J.D.; et al. Toll-like receptor 2 functions as a pattern recognition receptor for diverse bacterial products. J.Biol. Chem. 1999, 274, 33419–33425. [Google Scholar] [CrossRef] [PubMed]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.Y.; Van Huffel, C.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in Tlr4 gene. Science 1998, 282, 2085–2088. [Google Scholar] [CrossRef] [PubMed]
- Gaschler, G.J.; Skrtic, M.; Zavitz, C.C.; Lindahl, M.; Onnervik, P.O.; Murphy, T.F.; Sethi, S.; Stampfli, M.R. Bacteria challenge in smoke-exposed mice exacerbates inflammation and skews the inflammatory profile. Am. J. Respir. Crit. Care Med. 2009, 179, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Ni, I.; Ji, C.; Vij, N. Second-hand cigarette smoke impairs bacterial phagocytosis in macrophages by modulating CFTR dependent lipid-rafts. PLoS ONE 2015, 10, e0121200. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.O.; Gonzalez-Rothi, R.J. Abnormal phagolysosome fusion in pulmonary alveolar macrophages of rats exposed chronically to cigarette smoke. Am. Rev. Respir. Dis. 1984, 130, 467–471. [Google Scholar] [PubMed]
- Pehote, G.; Bodas, M.; Brucia, K.; Vij, N. Cigarette Smoke Exposure Inhibits Bacterial Killing via TFEB-Mediated Autophagy Impairment and Resulting Phagocytosis Defect. Med. Inflamm. 2017, 2017, 3028082. [Google Scholar] [CrossRef] [PubMed]
- Kammerl, I.E.; Dann, A.; Mossina, A.; Brech, D.; Lukas, C.; Vosyka, O.; Nathan, P.; Conlon, T.M.; Wagner, D.E.; Overkleeft, H.S.; et al. Impairment of Immunoproteasome Function by Cigarette Smoke and in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2016, 193, 1230–1241. [Google Scholar] [CrossRef] [PubMed]
- Hodge, S.; Matthews, G.; Mukaro, V.; Ahern, J.; Shivam, A.; Hodge, G.; Holmes, M.; Jersmann, H.; Reynolds, P.N. Cigarette smoke-induced changes to alveolar macrophage phenotype and function are improved by treatment with procysteine. Am. J. Respir. Cell Mol. Biol. 2011, 44, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Hodge, S.; Hodge, G.; Scicchitano, R.; Reynolds, P.N.; Holmes, M. Alveolar macrophages from subjects with chronic obstructive pulmonary disease are deficient in their ability to phagocytose apoptotic airway epithelial cells. Immunol. Cell Biol. 2003, 81, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Hodge, S.; Hodge, G.; Ahern, J.; Jersmann, H.; Holmes, M.; Reynolds, P.N. Smoking alters alveolar macrophage recognition and phagocytic ability: Implications in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2007, 37, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Huynh, M.L.; Malcolm, K.C.; Kotaru, C.; Tilstra, J.A.; Westcott, J.Y.; Fadok, V.A.; Wenzel, S.E. Defective apoptotic cell phagocytosis attenuates prostaglandin E2 and 15-hydroxyeicosatetraenoic acid in severe asthma alveolar macrophages. Am. J. Respir. Crit. Care Med. 2005, 172, 972–979. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.L.; Gibson, P.G.; Yang, I.A.; Upham, J.; James, A.; Reynolds, P.N.; Hodge, S. Impaired macrophage phagocytosis in non-eosinophilic asthma. Clin. Exp. Allergy 2013, 43, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Hodge, S.; Dean, M.; Hodge, G.; Holmes, M.; Reynolds, P.N. Decreased efferocytosis and mannose binding lectin in the airway in bronchiolitis obliterans syndrome. J. Heart Lung Transplant. 2011, 30, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Hodge, S.; Upham, J.W.; Pizzutto, S.; Petsky, H.L.; Yerkovich, S.; Baines, K.J.; Gibson, P.; Simpson, J.L.; Buntain, H.; Chen, A.C.H.; et al. Is Alveolar Macrophage Phagocytic Dysfunction in Children with Protracted Bacterial Bronchitis a Forerunner to Bronchiectasis? Chest 2016, 149, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.B.; Barnawi, J.; Ween, M.; Hamon, R.; Roscioli, E.; Hodge, G.; Reynolds, P.N.; Pitson, S.M.; Davies, L.T.; Haberberger, R.; et al. Cigarette smoke inhibits efferocytosis via deregulation of sphingosine kinase signaling: Reversal with exogenous S1P and the S1P analogue FTY720. J. Leukoc. Biol. 2016, 100, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.B.; Jersmann, H.; Truong, T.T.; Hamon, R.; Roscioli, E.; Ween, M.; Pitman, M.R.; Pitson, S.M.; Hodge, G.; Reynolds, P.N.; et al. Disrupted epithelial/macrophage crosstalk via Spinster homologue 2-mediated S1P signaling may drive defective macrophage phagocytic function in COPD. PLoS ONE 2017, 12, e0179577. [Google Scholar] [CrossRef] [PubMed]
- Barnawi, J.; Jersmann, H.; Haberberger, R.; Hodge, S.; Meech, R. Reduced DNA methylation of sphingosine-1 phosphate receptor 5 in alveolar macrophages in COPD: A potential link to failed efferocytosis. Respirology 2017, 22, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Barnawi, J.; Tran, H.; Jersmann, H.; Pitson, S.; Roscioli, E.; Hodge, G.; Meech, R.; Haberberger, R.; Hodge, S. Potential Link between the Sphingosine-1-Phosphate (S1P) System and Defective Alveolar Macrophage Phagocytic Function in Chronic Obstructive Pulmonary Disease (COPD). PLoS ONE 2015, 10, e0122771. [Google Scholar] [CrossRef] [PubMed]
- Donoviel, M.S.; Hait, N.C.; Ramachandran, S.; Maceyka, M.; Takabe, K.; Milstien, S.; Oravecz, T.; Spiegel, S. Spinster 2, a sphingosine-1-phosphate transporter, plays a critical role in inflammatory and autoimmune diseases. FASEB J. 2015, 29, 5018–5028. [Google Scholar] [CrossRef] [PubMed]
- Merad, M.; Sathe, P.; Helft, J.; Miller, J.; Mortha, A. The dendritic cell lineage: Ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu. Rev. Immunol. 2013, 31, 563–604. [Google Scholar] [CrossRef] [PubMed]
- Schultze, J.L.; Aschenbrenner, A.C. Systems immunology allows a new view on human dendritic cells. Semin. Cell Dev. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Joffre, O.; Nolte, M.A.; Sporri, R.; Reis e Sousa, C. Inflammatory signals in dendritic cell activation and the induction of adaptive immunity. Immunol. Rev. 2009, 227, 234–247. [Google Scholar] [CrossRef] [PubMed]
- Soler, P.; Moreau, A.; Basset, F.; Hance, A.J. Cigarette smoking-induced changes in the number and differentiated state of pulmonary dendritic cells/Langerhans cells. Am. Rev. Respir. Dis. 1989, 139, 1112–1117. [Google Scholar] [CrossRef] [PubMed]
- Rogers, A.V.; Adelroth, E.; Hattotuwa, K.; Dewar, A.; Jeffery, P.K. Bronchial mucosal dendritic cells in smokers and ex-smokers with COPD: An electron microscopic study. Thorax 2008, 63, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Tsoumakidou, M.; Elston, W.; Zhu, J.; Wang, Z.; Gamble, E.; Siafakas, N.M.; Barnes, N.C.; Jeffery, P.K. Cigarette smoking alters bronchial mucosal immunity in asthma. Am. J. Respir. Crit. Care Med. 2007, 175, 919–925. [Google Scholar] [CrossRef] [PubMed]
- Lanckacker, E.A.; Tournoy, K.G.; Hammad, H.; Holtappels, G.; Lambrecht, B.N.; Joos, G.F.; Maes, T. Short cigarette smoke exposure facilitates sensitisation and asthma development in mice. Eur. Respir. J. 2013, 41, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Le Rouzic, O.; Koné, B.; Kluza, J.; Marchetti, P.; Hennegrave, F.; Olivier, C.; Kervoaze, G.; Vilain, E.; Mordacq, C.; Just, N.; et al. Cigarette smoke alters the ability of human dendritic cells to promote anti-Streptococcus pneumoniae Th17 response. Respir. Res. 2016, 17, 94. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Du, Y.C.; Xu, J.Y.; Hu, X.Y. Expression and significance of myeloid differentiation factor 88 in marrow dendritic cells in asthmatic rats with cigarette smoke exposure. Chin. Med. J. 2012, 125, 2556–2561. [Google Scholar] [PubMed]
- Robbins, C.S.; Franco, F.; Mouded, M.; Cernadas, M.; Shapiro, S.D. Cigarette smoke exposure impairs dendritic cell maturation and T cell proliferation in thoracic lymph nodes of mice. J. Immunol. 2008, 180, 6623–6628. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.L.; Zhang, H.; Tang, Q.Y.; Bai, J.; He, Z.Y.; Zhang, J.Q.; Li, M.H.; Deng, J.M.; Liu, G.N.; Zhong, X.N. Neutrophil extracellular traps induced by cigarette smoke activate plasmacytoid dendritic cells. Thorax 2017, 72, 1084–1093. [Google Scholar] [CrossRef] [PubMed]
- Castro, S.M.; Chakraborty, K.; Guerrero-Plata, A. Cigarette smoke suppresses TLR-7 stimulation in response to virus infection in plasmacytoid dendritic cells. Toxicol. In Vitro 2011, 25, 1106–1113. [Google Scholar] [CrossRef] [PubMed]
- Nouri-Shirazi, M.; Guinet, E. Evidence for the immunosuppressive role of nicotine on human dendritic cell functions. Immunology 2003, 109, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Kroening, P.R.; Barnes, T.W.; Pease, L.; Limper, A.; Kita, H.; Vassallo, R. Cigarette smoke-induced oxidative stress suppresses generation of dendritic cell IL-12 and IL-23 through ERK-dependent pathways. J. Immunol. 2008, 181, 1536–1547. [Google Scholar] [CrossRef] [PubMed]
- Vassallo, R.; Kroening, P.R.; Parambil, J.; Kita, H. Nicotine and oxidative cigarette smoke constituents induce immune-modulatory and proinflammatory dendritic cell responses. Mol. Immunol. 2008, 45, 3321–3329. [Google Scholar] [CrossRef] [PubMed]
- Vassallo, R.; Tamada, K.; Lau, J.S.; Kroening, P.R.; Chen, L. Cigarette smoke extract suppresses human dendritic cell function leading to preferential induction of Th-2 priming. J. Immunol. 2005, 175, 2684–2691. [Google Scholar] [CrossRef] [PubMed]
- Robays, L.J.; Lanckacker, E.A.; Moerloose, K.B.; Maes, T.; Bracke, K.R.; Brusselle, G.G.; Joos, G.F.; Vermaelen, K.Y. Concomitant inhalation of cigarette smoke and aerosolized protein activates airway dendritic cells and induces allergic airway inflammation in a TLR-independent way. J. Immunol. 2009, 183, 2758–2766. [Google Scholar] [CrossRef] [PubMed]
- Bruggemann, T.R.; Fernandes, P.; Oliveira, L.M.; Sato, M.N.; Martins, M.A.; Arantes-Costa, F.M. Cigarette Smoke Increases CD8alpha(+) Dendritic Cells in an Ovalbumin-Induced Airway Inflammation. Front. Immunol. 2017, 8, 718. [Google Scholar] [CrossRef] [PubMed]
- Kuchroo, V.K.; Das, M.P.; Brown, J.A.; Ranger, A.M.; Zamvil, S.S.; Sobel, R.A.; Weiner, H.L.; Nabavi, N.; Glimcher, L.H. B7-1 and B7-2 costimulatory molecules activate differentially the Th1/Th2 developmental pathways: Application to autoimmune disease therapy. Cell 1995, 80, 707–718. [Google Scholar] [CrossRef]
- Nouri-Shirazi, M.; Guinet, E. A possible mechanism linking cigarette smoke to higher incidence of respiratory infection and asthma. Immunol. Lett. 2006, 103, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Caligiuri, M.A. Human natural killer cells. Blood 2008, 112, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Zamai, L.; Ahmad, M.; Bennett, I.M.; Azzoni, L.; Alnemri, E.S.; Perussia, B. Natural killer (NK) cell-mediated cytotoxicity: Differential use of TRAIL and Fas ligand by immature and mature primary human NK cells. J. Exp. Med. 1998, 188, 2375–2380. [Google Scholar] [CrossRef] [PubMed]
- Ferson, M.; Edwards, A.; Lind, A.; Milton, G.W.; Hersey, P. Low natural killer-cell activity and immunoglobulin levels associated with smoking in human subjects. Int. J. Cancer 1979, 23, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Andersson, A.; Malmhall, C.; Houltz, B.; Tengvall, S.; Sjostrand, M.; Qvarfordt, I.; Linden, A.; Bossios, A. Interleukin-16-producing NK cells and T-cells in the blood of tobacco smokers with and without COPD. Int. J. Chron. Obstr. Pulm. Dis. 2016, 11, 2245–2258. [Google Scholar] [CrossRef] [PubMed]
- Mian, M.F.; Pek, E.A.; Mossman, K.L.; Stampfli, M.R.; Ashkar, A.A. Exposure to cigarette smoke suppresses IL-15 generation and its regulatory NK cell functions in poly I:C-augmented human PBMCs. Mol. Immunol. 2009, 46, 3108–3116. [Google Scholar] [CrossRef] [PubMed]
- Arimilli, S.; Damratoski, B.E.; Prasad, G.L. Combustible and non-combustible tobacco product preparations differentially regulate human peripheral blood mononuclear cell functions. Toxicol. In Vitro 2013, 27, 1992–2004. [Google Scholar] [CrossRef] [PubMed]
- Mian, M.F.; Lauzon, N.M.; Stampfli, M.R.; Mossman, K.L.; Ashkar, A.A. Impairment of human NK cell cytotoxic activity and cytokine release by cigarette smoke. J. Leukoc. Biol. 2008, 83, 774–784. [Google Scholar] [CrossRef] [PubMed]
- Stolberg, V.R.; Martin, B.; Mancuso, P.; Olszewski, M.A.; Freeman, C.M.; Curtis, J.L.; Chensue, S.W. Role of CC chemokine receptor 4 in natural killer cell activation during acute cigarette smoke exposure. Am. J. Pathol. 2014, 184, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Kearley, J.; Silver, J.S.; Sanden, C.; Liu, Z.; Berlin, A.A.; White, N.; Mori, M.; Pham, T.H.; Ward, C.K.; Criner, G.J.; et al. Cigarette smoke silences innate lymphoid cell function and facilitates an exacerbated type I interleukin-33-dependent response to infection. Immunity 2015, 42, 566–579. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Urbanowicz, R.A.; Tighe, P.J.; Todd, I.; Corne, J.M.; Fairclough, L.C. Differential activation of killer cells in the circulation and the lung: A study of current smoking status and chronic obstructive pulmonary disease (COPD). PLoS ONE 2013, 8, e58556. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.S.; Lee, T.H.; Jun, J.A.; Baek, A.R.; Park, J.S.; Koo, S.M.; Kim, Y.K.; Lee, H.S.; Park, C.S. Neutrophilic inflammation in asthma: Mechanisms and therapeutic considerations. Exp. Rev. Respir. Med. 2017, 11, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Conlon, T.M.; Ballester Lopez, C.; Seimetz, M.; Bednorz, M.; Zhou-Suckow, Z.; Weissmann, N.; Eickelberg, O.; Mall, M.A.; Yildirim, A.O. Cigarette smoke causes acute airway disease and exacerbates chronic obstructive lung disease in neonatal mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, l602–1610. [Google Scholar] [CrossRef] [PubMed]
- Moazed, F.; Burnham, E.L.; Vandivier, R.W.; O’Kane, C.M.; Shyamsundar, M.; Hamid, U.; Abbott, J.; Thickett, D.R.; Matthay, M.A.; McAuley, D.F.; et al. Cigarette smokers have exaggerated alveolar barrier disruption in response to lipopolysaccharide inhalation. Thorax 2016, 71, 1130–1136. [Google Scholar] [CrossRef] [PubMed]
- Siew, L.Q.C.; Wu, S.Y.; Ying, S.; Corrigan, C.J. Cigarette smoking increases bronchial mucosal IL-17A expression in asthmatics, which acts in concert with environmental aeroallergens to engender neutrophilic inflammation. Clin. Exp. Allergy 2017, 47, 740–750. [Google Scholar] [CrossRef] [PubMed]
- White, P.C.; Hirschfeld, J.; Milward, M.R.; Cooper, P.R.; Wright, H.J.; Matthews, J.B.; Chapple, I.L.C. Cigarette smoke modifies neutrophil chemotaxis, neutrophil extracellular trap formation and inflammatory response-related gene expression. J. Periodontal Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Chong, M.M.; Littman, D.R. Plasticity of CD4+ T cell lineage differentiation. Immunity 2009, 30, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Raphael, I.; Nalawade, S.; Eagar, T.N.; Forsthuber, T.G. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 2015, 74, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar] [PubMed]
- Dutton, R.W.; Bradley, L.M.; Swain, S.L. T cell memory. Annu. Rev. Immunol. 1998, 16, 201–223. [Google Scholar] [CrossRef] [PubMed]
- Parkes, G.C.; Whelan, K.; Lindsay, J.O. Smoking in inflammatory bowel disease: Impact on disease course and insights into the aetiology of its effect. J. Crohn Colitis 2014, 8, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Tanigawa, T.; Araki, S.; Nakata, A.; Kitamura, F.; Yasumoto, M.; Sakurai, S.; Kiuchi, T. Increase in memory (CD4+CD29+ and CD4+CD45RO+) T and naive (CD4+CD45RA+) T-cell subpopulations in smokers. Arch. Environ. Health 1998, 53, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Nakata, A.; Takahashi, M.; Irie, M.; Fujioka, Y.; Haratani, T.; Araki, S. Relationship between cumulative effects of smoking and memory CD4+ T lymphocyte subpopulations. Addict. Behav. 2007, 32, 1526–1531. [Google Scholar] [CrossRef] [PubMed]
- Vardavas, C.I.; Plada, M.; Tzatzarakis, M.; Marcos, A.; Warnberg, J.; Gomez-Martinez, S.; Breidenassel, C.; Gonzalez-Gross, M.; Tsatsakis, A.M.; Saris, W.H.; et al. Passive smoking alters circulating naive/memory lymphocyte T-cell subpopulations in children. Pediatr. Allergy Immunol. 2010, 21, 1171–1178. [Google Scholar] [CrossRef] [PubMed]
- Duan, M.C.; Huang, Y.; Zhong, X.N.; Tang, H.J. Th17 cell enhances CD8 T-cell cytotoxicity via IL-21 production in emphysema mice. Med. Inflamm. 2012, 2012, 898053. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.; Gaczkowski, M.; Sturton, G.; Staib, P.; Schinkothe, T.; Klein, E.; Rubbert, A.; Bacon, K.; Wassermann, K.; Erdmann, E. Modification of surface antigens in blood CD8+ T-lymphocytes in COPD: Effects of smoking. Eur. Respir. J. 2007, 29, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Rojas, M.I.; Ramirez-Venegas, A.; Limon-Camacho, L.; Ochoa, L.; Hernandez-Zenteno, R.; Sansores, R.H. Increase of Th17 cells in peripheral blood of patients with chronic obstructive pulmonary disease. Respir. Med. 2011, 105, 1648–1654. [Google Scholar] [CrossRef] [PubMed]
- Forsslund, H.; Mikko, M.; Karimi, R.; Grunewald, J.; Wheelock, A.M.; Wahlstrom, J.; Skold, C.M. Distribution of T-cell subsets in BAL fluid of patients with mild to moderate COPD depends on current smoking status and not airway obstruction. Chest 2014, 145, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Saetta, M.; Baraldo, S.; Corbino, L.; Turato, G.; Braccioni, F.; Rea, F.; Cavallesco, G.; Tropeano, G.; Mapp, C.E.; Maestrelli, P.; et al. CD8+ve cells in the lungs of smokers with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1999, 160, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.Q.; Liu, X.S.; Wang, J.M.; Xu, Y.J. CD8(+) Tc-lymphocytes immunodeviation in peripheral blood and airway from patients of chronic obstructive pulmonary disease and changes after short-term smoking cessation. Chin. Med. J. 2013, 126, 3608–3615. [Google Scholar] [PubMed]
- Harrison, O.J.; Foley, J.; Bolognese, B.J.; Long, E., III; Podolin, P.L.; Walsh, P.T. Airway infiltration of CD4+ CCR6+ Th17 type cells associated with chronic cigarette smoke induced airspace enlargement. Immunol. Lett. 2008, 121, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Romagnani, S. Th1/Th2 cells. Inflamm. Bowel Dis. 1999, 5, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Shaler, C.R.; Horvath, C.N.; McCormick, S.; Jeyanathan, M.; Khera, A.; Zganiacz, A.; Kasinska, J.; Stampfli, M.R.; Xing, Z. Continuous and discontinuous cigarette smoke exposure differentially affects protective Th1 immunity against pulmonary tuberculosis. PLoS ONE 2013, 8, e59185. [Google Scholar] [CrossRef] [PubMed]
- Feleszko, W.; Zawadzka-Krajewska, A.; Matysiak, K.; Lewandowska, D.; Peradzynska, J.; Dinh, Q.T.; Hamelmann, E.; Groneberg, D.A.; Kulus, M. Parental tobacco smoking is associated with augmented IL-13 secretion in children with allergic asthma. J. Allergy Clin. Immunol. 2006, 117, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Bhalla, D.K.; Hirata, F.; Rishi, A.K.; Gairola, C.G. Cigarette smoke, inflammation, and lung injury: A mechanistic perspective. J. Toxicol. Environ. Health Part B Crit. Rev. 2009, 12, 45–64. [Google Scholar] [CrossRef] [PubMed]
- Hartigan-O’Connor, D.J.; Hirao, L.A.; McCune, J.M.; Dandekar, S. Th17 cells and regulatory T cells in elite control over HIV and SIV. Curr. Opin. HIV AIDS 2011, 6, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, W.; Kolls, J.K.; Zheng, Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity 2008, 28, 454–467. [Google Scholar] [CrossRef] [PubMed]
- Sergejeva, S.; Ivanov, S.; Lotvall, J.; Linden, A. Interleukin-17 as a recruitment and survival factor for airway macrophages in allergic airway inflammation. Am. J. Respir. Cell Mol. Biol. 2005, 33, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Molet, S.; Hamid, Q.; Davoine, F.; Nutku, E.; Taha, R.; Page, N.; Olivenstein, R.; Elias, J.; Chakir, J. IL-17 is increased in asthmatic airways and induces human bronchial fibroblasts to produce cytokines. J. Allergy Clin. Immunol. 2001, 108, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Nakae, S.; Komiyama, Y.; Nambu, A.; Sudo, K.; Iwase, M.; Homma, I.; Sekikawa, K.; Asano, M.; Iwakura, Y. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity 2002, 17, 375–387. [Google Scholar] [CrossRef]
- Wang, H.; Peng, W.; Weng, Y.; Ying, H.; Li, H.; Xia, D.; Yu, W. Imbalance of Th17/Treg cells in mice with chronic cigarette smoke exposure. Int. Immunopharmacol. 2012, 14, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.T.; Nakahama, T.; Kishimoto, T. Aryl hydrocarbon receptor and experimental autoimmune arthritis. Semin. Immunopathol. 2013, 35, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Barcelo, B.; Pons, J.; Ferrer, J.M.; Sauleda, J.; Fuster, A.; Agusti, A.G. Phenotypic characterisation of T-lymphocytes in COPD: Abnormal CD4+CD25+ regulatory T-lymphocyte response to tobacco smoking. Eur. Respir. J. 2008, 31, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Roos-Engstrand, E.; Pourazar, J.; Behndig, A.F.; Bucht, A.; Blomberg, A. Expansion of CD4+CD25+ helper T cells without regulatory function in smoking and COPD. Respir. Res. 2011, 12, 74. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Sun, Y.; Hao, Y.; Zhuo, J.; Liu, X.; Bai, P.; Han, J.; Zheng, X.; Zeng, H. Imbalance between subpopulations of regulatory T cells in COPD. Thorax 2013, 68, 1131–1139. [Google Scholar] [CrossRef] [PubMed]
- Brandsma, C.A.; Hylkema, M.N.; Geerlings, M.; van Geffen, W.H.; Postma, D.S.; Timens, W.; Kerstjens, H.A. Increased levels of (class switched) memory B cells in peripheral blood of current smokers. Respir. Res. 2009, 10, 108. [Google Scholar] [CrossRef] [PubMed]
- Brandsma, C.A.; Kerstjens, H.A.; van Geffen, W.H.; Geerlings, M.; Postma, D.S.; Hylkema, M.N.; Timens, W. Differential switching to IgG and IgA in active smoking COPD patients and healthy controls. Eur. Respir. J. 2012, 40, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Hogg, J.C.; Chu, F.; Utokaparch, S.; Woods, R.; Elliott, W.M.; Buzatu, L.; Cherniack, R.M.; Rogers, R.M.; Sciurba, F.C.; Coxson, H.O.; et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N. Engl. J. Med. 2004, 350, 2645–2653. [Google Scholar] [CrossRef] [PubMed]
- Palmer, V.L.; Kassmeier, M.D.; Willcockson, J.; Akhter, M.P.; Cullen, D.M.; Swanson, P.C. N-acetylcysteine increases the frequency of bone marrow pro-B/pre-B cells, but does not reverse cigarette smoking-induced loss of this subset. PLoS ONE 2011, 6, e24804. [Google Scholar] [CrossRef] [PubMed]
- Fusby, J.S.; Kassmeier, M.D.; Palmer, V.L.; Perry, G.A.; Anderson, D.K.; Hackfort, B.T.; Alvarez, G.K.; Cullen, D.M.; Akhter, M.P.; Swanson, P.C. Cigarette smoke-induced effects on bone marrow B-cell subsets and CD4+:CD8+ T-cell ratios are reversed by smoking cessation: Influence of bone mass on immune cell response to and recovery from smoke exposure. Inhal. Toxicol. 2010, 22, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Golpasand Hagh, L.; Zakavi, F.; Ansarifar, S.; Ghasemzadeh, O.; Solgi, G. Association of dental caries and salivary sIgA with tobacco smoking. Aust. Dent. J. 2013, 58, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Bahna, S.L.; Heiner, D.C.; Myhre, B.A. Immunoglobulin E pattern in cigarette smokers. Allergy 1983, 38, 57–64. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strzelak, A.; Ratajczak, A.; Adamiec, A.; Feleszko, W. Tobacco Smoke Induces and Alters Immune Responses in the Lung Triggering Inflammation, Allergy, Asthma and Other Lung Diseases: A Mechanistic Review. Int. J. Environ. Res. Public Health 2018, 15, 1033. https://doi.org/10.3390/ijerph15051033
Strzelak A, Ratajczak A, Adamiec A, Feleszko W. Tobacco Smoke Induces and Alters Immune Responses in the Lung Triggering Inflammation, Allergy, Asthma and Other Lung Diseases: A Mechanistic Review. International Journal of Environmental Research and Public Health. 2018; 15(5):1033. https://doi.org/10.3390/ijerph15051033
Chicago/Turabian StyleStrzelak, Agnieszka, Aleksandra Ratajczak, Aleksander Adamiec, and Wojciech Feleszko. 2018. "Tobacco Smoke Induces and Alters Immune Responses in the Lung Triggering Inflammation, Allergy, Asthma and Other Lung Diseases: A Mechanistic Review" International Journal of Environmental Research and Public Health 15, no. 5: 1033. https://doi.org/10.3390/ijerph15051033
APA StyleStrzelak, A., Ratajczak, A., Adamiec, A., & Feleszko, W. (2018). Tobacco Smoke Induces and Alters Immune Responses in the Lung Triggering Inflammation, Allergy, Asthma and Other Lung Diseases: A Mechanistic Review. International Journal of Environmental Research and Public Health, 15(5), 1033. https://doi.org/10.3390/ijerph15051033