Abundances of Clinically Relevant Antibiotic Resistance Genes and Bacterial Community Diversity in the Weihe River, China

Abstract
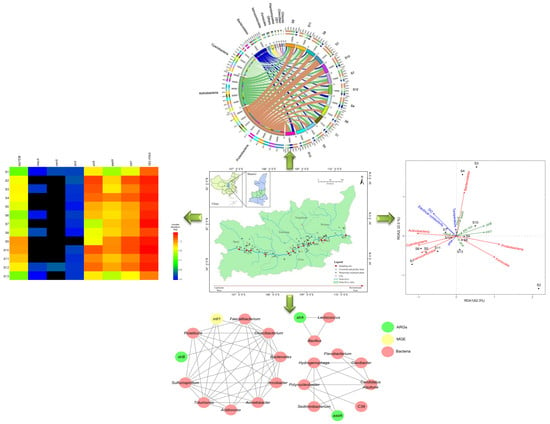
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Sampling Sites and Sample Collection
2.2. Water Sample Processing and Genomic DNA Extraction
2.3. Quantification of Antibiotic Resistance Genes (ARGs) Using Droplet Digital Polymerase Chain Reaction (ddPCR)
2.4. Illumina High-Throughput Sequencing
2.5. Statistical Analysis
3. Results
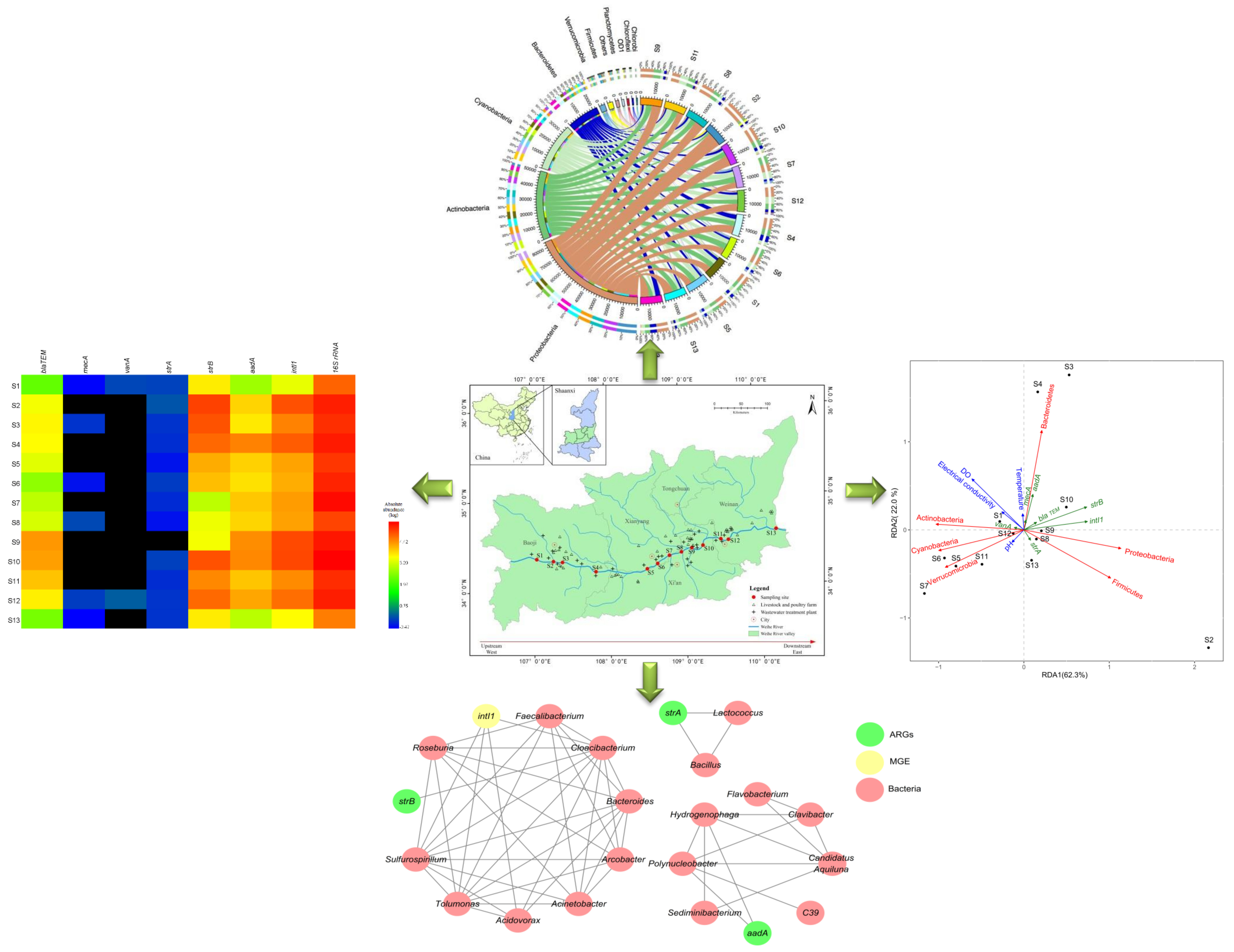
3.1. Distributions of ARGs and the intI1 Gene
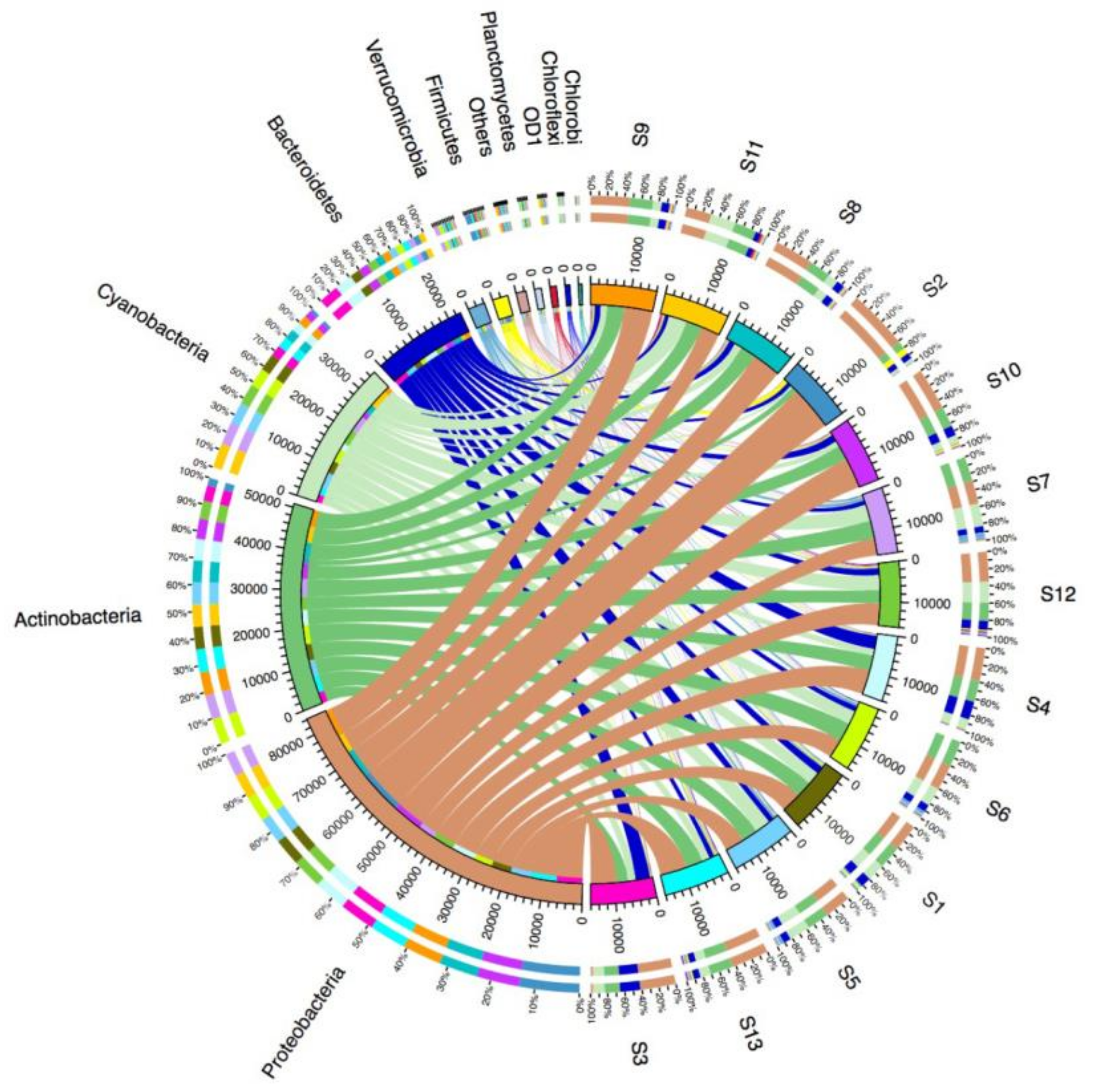
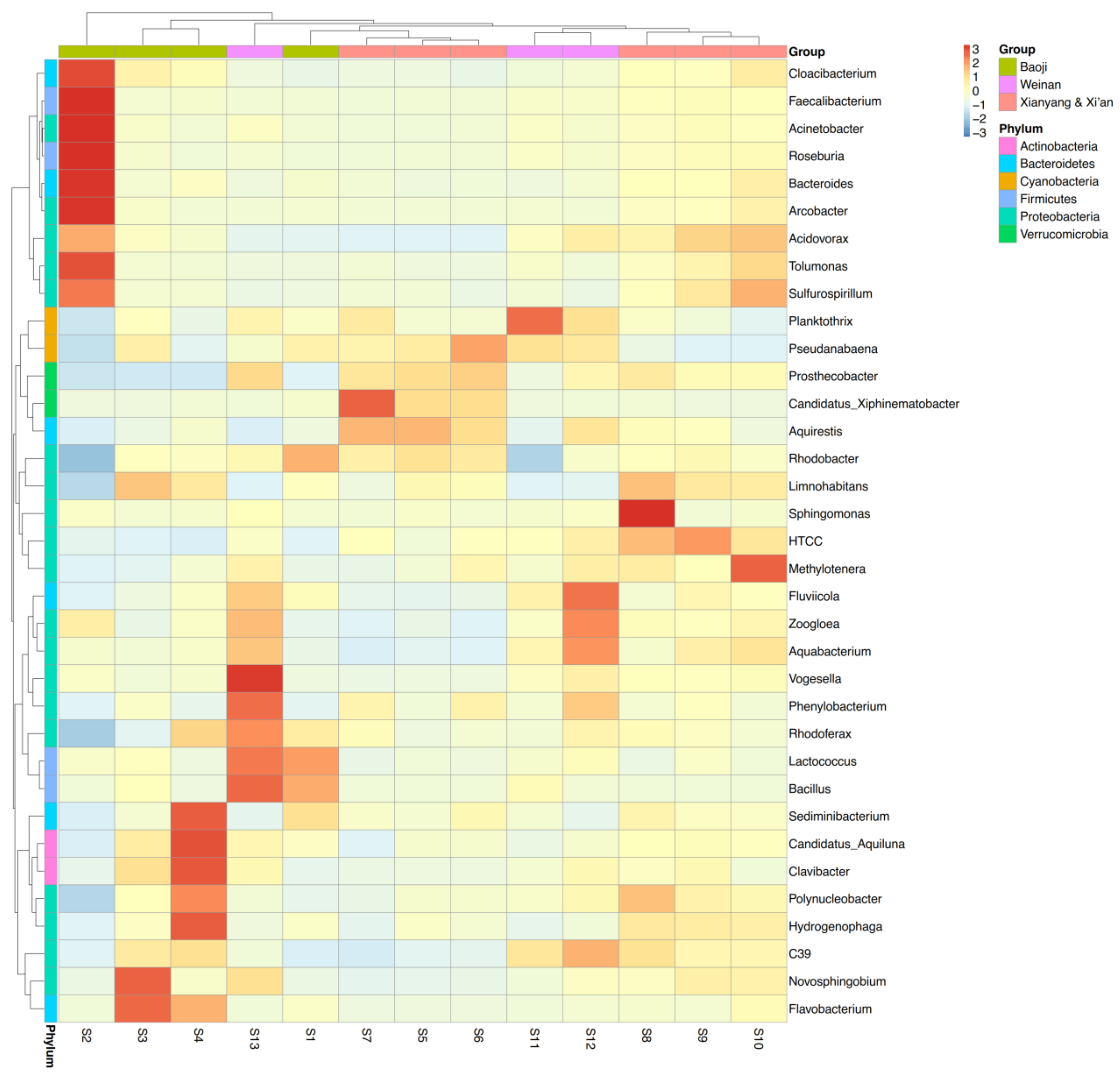
3.2. Microbial Diversity and Community Composition
3.3. Relationships between ARGs, Bacterial Communities, and Environmental Factors
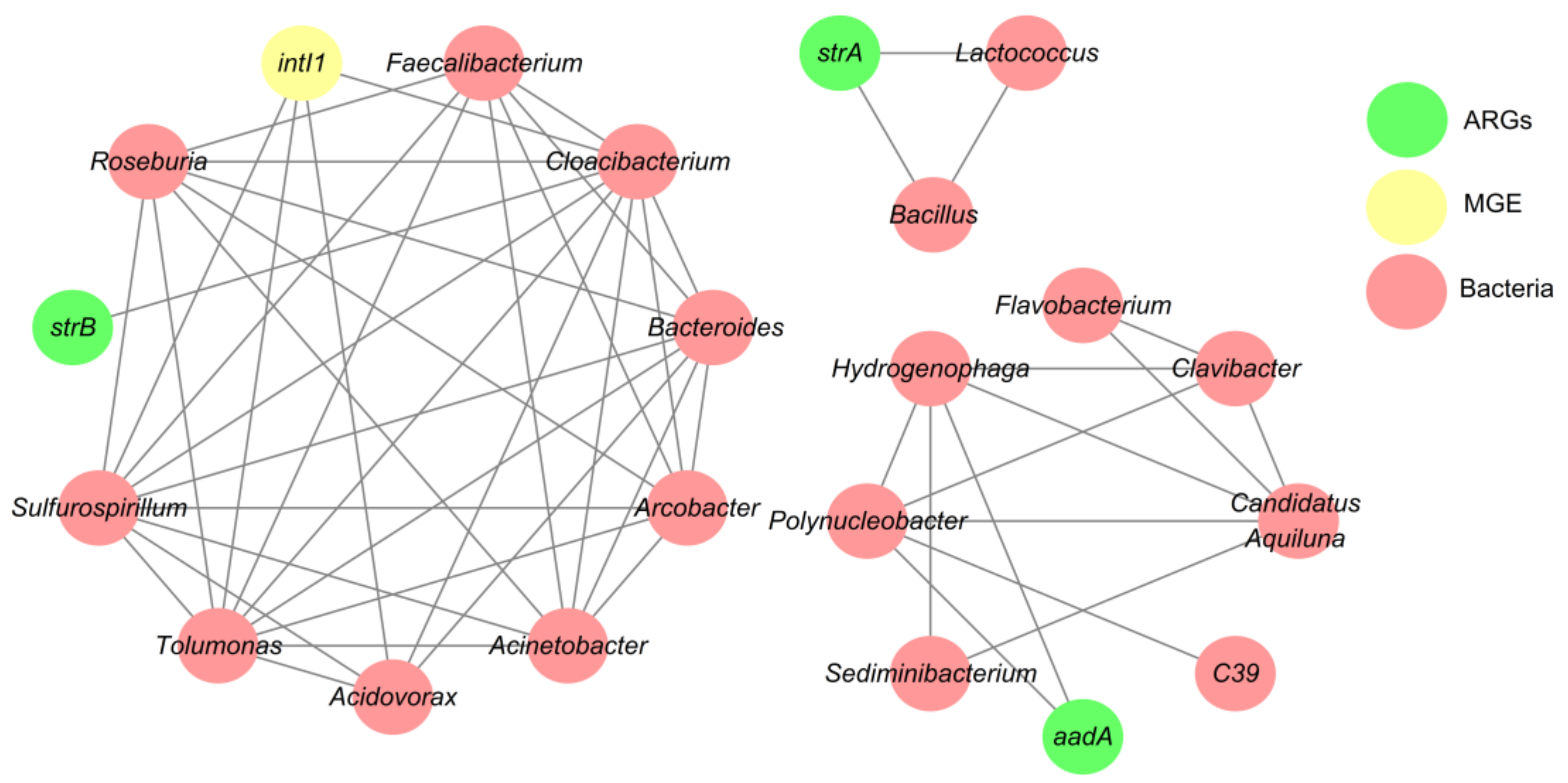
3.4. Network Analysis Based on the intI1 Gene, ARGs, and Bacterial Taxa
4. Discussion
4.1. Prevalence of the intI1 Gene and ARGs in Urban Districts
4.2. Bacterial Community Diversity
4.3. Relationships between ARGs, Bacterial Communities, and Environmental Factors
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pruden, A.; Pei, R.T.; Storteboom, H.; Carlson, K.H. Antibiotic resistance genes as emerging contaminants: Studies in northern Colorado. Environ. Sci. Technol. 2006, 40, 7445–7450. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.-G.; Johnson, T.A.; Su, J.-Q.; Qiao, M.; Guo, G.-X.; Stedtfeld, R.D.; Hashsham, S.A.; Tiedje, J.M. Diverse and abundant antibiotic resistance genes in Chinese swine farms. Proc. Natl. Acad. Sci. USA 2013, 110, 3435–3440. [Google Scholar] [CrossRef] [PubMed]
- Gillings, M.R.; Gaze, W.H.; Pruden, A.; Smalla, K.; Tiedje, J.M.; Zhu, Y.G. Using the class 1 integron-integrase gene as a proxy for anthropogenic pollution. ISME J. 2015, 9, 1269–1279. [Google Scholar] [CrossRef] [PubMed]
- Stecher, B.; Maier, L.; Hardt, W.-D. ‘Blooming’ in the gut: How dysbiosis might contribute to pathogen evolution. Nat. Rev. Microbiol. 2013, 11, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhang, X.-X.; Ye, L. Plasmid metagenome reveals high levels of antibiotic resistance genes and mobile genetic elements in activated sludge. PLoS ONE 2011, 6, e26041. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.L. Antibiotics and antibiotic resistance genes in natural environments. Science 2008, 321, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Van den Bogaard, A.E.; Stobberingh, E.E. Epidemiology of resistance to antibiotics—Links between animals and humans. Int. J. Antimicrob. Agents 2000, 14, 327–335. [Google Scholar] [CrossRef]
- Venturini, C.; Beatson, S.A.; Djordjevic, S.P.; Walker, M.J. Multiple antibiotic resistance gene recruitment onto the enterohemorrhagic Escherichia coli virulence plasmid. FASEB J. 2010, 24, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Stokes, H.W.; Gillings, M.R. Gene flow, mobile genetic elements and the recruitment of antibiotic resistance genes into Gram-negative pathogens. FEMS Microbiol. Rev. 2011, 35, 790–819. [Google Scholar] [CrossRef] [PubMed]
- Su, H.-C.; Ying, G.-G.; Tao, R.; Zhang, R.-Q.; Zhao, J.-L.; Liu, Y.-S. Class 1 and 2 integrons, sul resistance genes and antibiotic resistance in Escherichia coli isolated from Dongjiang River, South China. Environ. Pollut. 2012, 169, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Yuan, K.; Liang, X.M.; Chen, X.; Zhao, Z.S.; Yang, Y.; Zou, S.C.; Luan, T.G.; Chen, B.W. Occurrences and distribution of sulfonamide and tetracycline resistance genes in the Yangtze River Estuary and nearby coastal area. Mar. Pollut. Bull. 2015, 100, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Li, A.-D.; Yin, X.-L.; Zhang, T. The prevalence of integrons as the carrier of antibiotic resistance genes in natural and man-made environments. Environ. Sci. Technol. 2017, 51, 5721–5728. [Google Scholar] [CrossRef] [PubMed]
- Pruden, A.; Arabi, M.; Storteboom, H.N. Correlation between upstream human activities and riverine antibiotic resistance genes. Environ. Sci. Technol. 2012, 46, 11541–11549. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Guo, C.; Luo, Y.; Lv, J.; Zhang, Y.; Lin, H.; Wang, L.; Xu, J. Occurrence and distribution of antibiotics, antibiotic resistance genes in the urban rivers in Beijing, China. Environ. Pollut. 2016, 213, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Hu, X.; Xu, T.; Zhang, H.; Sheng, D.; Yin, D. Prevalence of antibiotic resistance genes and their relationship with antibiotics in the Huangpu River and the drinking water sources, Shanghai, China. Sci. Total Environ. 2013, 458, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Niu, Z.G.; Zhang, K.; Zhang, Y. Occurrence and distribution of antibiotic resistance genes in the coastal area of the Bohai Bay, China. Mar. Pollut. Bull. 2016, 107, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Ying, G.-G.; Su, H.-C.; Zhou, H.-W.; Sidhu, J.P.S. Detection of antibiotic resistance and tetracycline resistance genes in Enterobacteriaceae isolated from the Pearl rivers in South China. Environ. Pollut. 2010, 158, 2101–2109. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Shi, J.; Chang, H.; Li, D.; Yang, M.; Kamagata, Y. Phenotyping and genotyping of antibiotic-resistant Escherichia coli isolated from a natural river basin. Environ. Sci. Technol. 2008, 42, 3415–3420. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Mozaz, S.; Chamorro, S.; Marti, E.; Huerta, B.; Gros, M.; Sanchez-Melsio, A.; Borrego, C.M.; Barcelo, D.; Balcazar, J.L. Occurrence of antibiotics and antibiotic resistance genes in hospital and urban wastewaters and their impact on the receiving river. Water Res. 2015, 69, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.F.; He, P.J.; Shao, L.M.; Zhang, H.; Lu, F. Co-occurrence of mobile genetic elements and antibiotic resistance genes in municipal solid waste landfill leachates: A preliminary insight into the role of landfill age. Water Res. 2016, 106, 583–592. [Google Scholar] [CrossRef] [PubMed]
- LaPara, T.M.; Burch, T.R.; McNamara, P.J.; Tan, D.T.; Yan, M.; Eichmiller, J.J. Tertiary-treated municipal wastewater is a significant point source of antibiotic resistance genes into Duluth-Superior Harbor. Environ. Sci. Technol. 2011, 45, 9543–9549. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.P.; Raith, M.R.; Griffith, J.F. Droplet digital PCR for simultaneous quantification of general and human-associated fecal indicators for water quality assessment. Water Res. 2015, 70, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Cave, L.; Brothier, E.; Abrouk, D.; Bouda, P.S.; Hien, E.; Nazaret, S. Efficiency and sensitivity of the digital droplet PCR for the quantification of antibiotic resistance genes in soils and organic residues. Appl. Microbiol. Biotechnol. 2016, 100, 10597–10608. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Griffith, J.F.; Dorevitch, S.; Weisberg, S.B. Effectiveness of qPCR permutations, internal controls and dilution as means for minimizing the impact of inhibition while measuring Enterococcus in environmental waters. J. Appl. Microbiol. 2012, 113, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Huggett, J.F.; Foy, C.A.; Benes, V.; Emslie, K.; Garson, J.A.; Haynes, R.; Hellemans, J.; Kubista, M.; Nolan, R.; Pfaffl, M.W.; et al. The digital MIQE guidelines: Minimum information for publication of quantitative digital PCR experiments. Clin. Chem. 2013, 59, 892–902. [Google Scholar] [CrossRef] [PubMed]
- Jahn, M.; Vorpahl, C.; Turkowsky, D.; Lindmeyer, M.; Buhler, B.; Harms, H.; Muller, S. Accurate determination of plasmid copy number of flow-sorted cells using droplet digital PCR. Anal. Chem. 2014, 86, 5969–5976. [Google Scholar] [CrossRef] [PubMed]
- Verhaegen, B.; De Reu, K.; De Zutter, L.; Verstraete, K.; Heyndrickx, M.; Van Coillie, E. Comparison of droplet digital PCR and qPCR for the quantification of shiga toxin-producing Escherichia coli in bovine feces. Toxins 2016, 8, 157. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.J.; Lu, A.G.; Dang, S.H.; Chen, F.L. Ammonium nitrogen concentration in the Weihe River, central China during 2005–2015. Environ. Earth Sci. 2016, 75. [Google Scholar] [CrossRef]
- Song, J.; Cheng, D.; Li, Q.; He, X.; Long, Y.; Zhang, B. An evaluation of river health for the Weihe River in Shaanxi Province, China. Adv. Meteorol. 2015, 476020. [Google Scholar] [CrossRef]
- Li, Q.; Song, J.; Wei, A.; Zhang, B. Changes in major factors affecting the ecosystem health of the Weihe River in Shaanxi Province, China. Front. Environ. Sci. Eng. 2013, 7, 875–885. [Google Scholar] [CrossRef]
- Lachmayr, K.L.; Kerkhof, L.J.; DiRienzo, A.G.; Cavanaugh, C.M.; Ford, T.E. Quantifying nonspecific TEM beta-Lactamase (blaTEM) genes in a wastewater stream. Appl. Environ. Microbiol. 2009, 75, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Volkmann, H.; Schwartz, T.; Bischoff, P.; Kirchen, S.; Obst, U. Detection of clinically relevant antibiotic-resistance genes in municipal wastewater using real-time PCR (TaqMan). J. Microbiol. Methods 2004, 56, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, G.V.; Mellerup, A.; Christiansen, L.E.; Stahl, M.; Olsen, J.E.; Angen, O. Sampling and pooling methods for capturing herd level antibiotic resistance in swine feces using qPCR and CFU approaches. PLoS ONE 2015, 10, e0131672. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Walsh, F.; Ingenfeld, A.; Zampicolli, M.; Hilber-Bodmer, M.; Frey, J.E.; Duffy, B. Real-time PCR methods for quantitative monitoring of streptomycin and tetracycline resistance genes in agricultural ecosystems. J. Microbiol. Methods 2011, 86, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Barraud, O.; Baclet, M.C.; Denis, F.; Ploy, M.C. Quantitative multiplex real-time PCR for detecting class 1, 2 and 3 integrons. J. Antimicrob. Chemother. 2010, 65, 1642–1645. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Gu, J.; Wang, X.; Qian, X.; Tuo, X. Impacts of biochar on the environmental risk of antibiotic resistance genes and mobile genetic elements during anaerobic digestion of cattle farm wastewater. Bioresour. Technol. 2018, 256, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Mao, D.; Rysz, M.; Zhou, Q.; Zhang, H.; Xu, L.; Alvarez, P.J.J. Trends in antibiotic resistance genes occurrence in the Haihe River, China. Environ. Sci. Technol. 2010, 44, 7220–7225. [Google Scholar] [CrossRef] [PubMed]
- Vaz-Moreira, I.; Nunes, O.C.; Manaia, C.M. Ubiquitous and persistent Proteobacteria and other Gram-negative bacteria in drinking water. Sci. Total Environ. 2017, 586, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Liang, X.; Nie, X.; Huang, X.; Zou, S.; Li, X. The role of class I integrons in the dissemination of sulfonamide resistance genes in the Pearl River and Pearl River Estuary, South China. J. Hazard. Mater. 2015, 282, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Mroczkowska, J.E.; Barlow, M. Fitness trade-offs in bla(TEM) evolution. Antimicrob. Agents Chemother. 2008, 52, 2340–2345. [Google Scholar] [CrossRef] [PubMed]
- Sundin, G.W.; Bender, C.L. Dissemination of the strA-strB streptomycin-resistance genes among commensal and pathogenic bacteria from humans, animals, and plants. Mol. Ecol. 1996, 5, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Ben Salem, R.; Abbassi, M.S.; García, V.; García-Fierro, R.; Fernández, J.; Kilani, H.; Jaouani, I.; Khayeche, M.; Messadi, L.; Rodicio, M.R. Antimicrobial drug resistance and genetic properties of Salmonella enterica serotype Enteritidis circulating in chicken farms in Tunisia. J. Infect. Public Health 2017, 10, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Huang, S.; Sun, G.; Xu, Z.; Xu, M. Phylogenetic diversity, composition and distribution of bacterioplankton community in the Dongjiang River, China. FEMS Microbiol. Ecol. 2012, 80, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Hu, A.; Yang, X.; Chen, N.; Hou, L.; Ma, Y.; Yu, C.-P. Response of bacterial communities to environmental changes in a mesoscale subtropical watershed, Southeast China. Sci. Total Environ. 2014, 472, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Staley, C.; Gould, T.J.; Wang, P.; Phillips, J.; Cotner, J.B.; Sadowsky, M.J. Species sorting and seasonal dynamics primarily shape bacterial communities in the Upper Mississippi River. Sci. Total Environ. 2015, 505, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Qi, W.; Liang, J.; Qu, J. Using high-throughput sequencing to assess the impacts of treated and untreated wastewater discharge on prokaryotic communities in an urban river. Appl. Microbiol. Biotechnol. 2014, 98, 1841–1851. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Qi, R.; Yang, M.; Zhang, Y.; Yu, T. Bacterial community characteristics under long-term antibiotic selection pressures. Water Res. 2011, 45, 6063–6073. [Google Scholar] [CrossRef] [PubMed]
- Ho, H.T.K.; Lipman, L.J.A.; Gaastra, W. Arcobacter, what is known and unknown about a potential foodborne zoonotic agent! Vet. Microbiol. 2006, 115, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, H.; Maruyama, S.; Morita, Y.; Kubo, M.; Yamamoto, K.; Arai, S.; Izumi, T.; Kobayashi, Y.; Katsube, Y.; Mikami, T. Distribution of Arcobacter species among livestock in Japan. Vet. Microbiol. 2003, 93, 153–158. [Google Scholar] [CrossRef]
- Fera, M.T.; Maugeri, T.L.; Giannone, M.; Gugliandolo, C.; La Camera, E.; Blandino, G.; Carbone, M. In vitro susceptibility of Arcobacter butzleri and Arcobacter cryaerophilus to different antimicrobial agents. Int. J. Antimicrob. Agents 2003, 21, 488–491. [Google Scholar] [CrossRef]
- Zhang, Y.; Marrs, C.F.; Simon, C.; Xi, C. Wastewater treatment contributes to selective increase of antibiotic resistance among Acinetobacter spp. Sci. Total Environ. 2009, 407, 3702–3706. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Mao, D.; Luo, Y.; Wang, L.; Xu, B.; Xu, L. Occurrence of sulfonamide and tetracycline-resistant bacteria and resistance genes in aquaculture environment. Water Res. 2012, 46, 2355–2364. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Li, B.; Li, L.-G.; Zhang, T.; Angelidaki, I. Antibiotic resistance genes and correlations with microbial community and metal resistance genes in full-scale biogas reactors as revealed by metagenomic analysis. Environ. Sci. Technol. 2017, 51, 4069–4080. [Google Scholar] [CrossRef] [PubMed]
- Huerta, B.; Marti, E.; Gros, M.; López, P.; Pompêo, M.; Armengol, J.; Barceló, D.; Balcázar, J.L.; Rodríguez-Mozaz, S.; Marcé, R. Exploring the links between antibiotic occurrence, antibiotic resistance, and bacterial communities in water supply reservoirs. Sci. Total Environ. 2013, 456, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Walther, C.; Rossano, A.; Thomann, A.; Perreten, V. Antibiotic resistance in Lactococcus species from bovine milk: Presence of a mutated multidrug transporter mdt(A) gene in susceptible Lactococcus garvieae strains. Vet. Microbiol. 2008, 131, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Han, B.; Gu, J.; Wang, C.; Wang, P.; Ma, Y.; Cao, J.; He, Z. Fate of antibiotic resistant cultivable heterotrophic bacteria and antibiotic resistance genes in wastewater treatment processes. Chemosphere 2015, 135, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Yang, Y.; Ma, L.P.; Ju, F.; Guo, F.; Tiedje, J.M.; Zhang, T. Metagenomic and network analysis reveal wide distribution and co-occurrence of environmental antibiotic resistance genes. ISME J. 2015, 9, 2490–2502. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Gu, J.; Gao, H.; Qian, X.; Li, H. Abundances of Clinically Relevant Antibiotic Resistance Genes and Bacterial Community Diversity in the Weihe River, China. Int. J. Environ. Res. Public Health 2018, 15, 708. https://doi.org/10.3390/ijerph15040708
Wang X, Gu J, Gao H, Qian X, Li H. Abundances of Clinically Relevant Antibiotic Resistance Genes and Bacterial Community Diversity in the Weihe River, China. International Journal of Environmental Research and Public Health. 2018; 15(4):708. https://doi.org/10.3390/ijerph15040708
Chicago/Turabian StyleWang, Xiaojuan, Jie Gu, Hua Gao, Xun Qian, and Haichao Li. 2018. "Abundances of Clinically Relevant Antibiotic Resistance Genes and Bacterial Community Diversity in the Weihe River, China" International Journal of Environmental Research and Public Health 15, no. 4: 708. https://doi.org/10.3390/ijerph15040708
APA StyleWang, X., Gu, J., Gao, H., Qian, X., & Li, H. (2018). Abundances of Clinically Relevant Antibiotic Resistance Genes and Bacterial Community Diversity in the Weihe River, China. International Journal of Environmental Research and Public Health, 15(4), 708. https://doi.org/10.3390/ijerph15040708