Treatment of Human Placental Choriocarcinoma Cells with Formaldehyde and Benzene Induced Growth and Epithelial Mesenchymal Transition via Induction of an Antioxidant Effect



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Chemicals
2.2. Cell Culture and Media
2.3. MTT Assay
2.4. Scratch Assay
2.5. Protein Extraction and Western Blot Assay
2.6. Determination of ROS Production
2.7. Data Analysis
3. Results
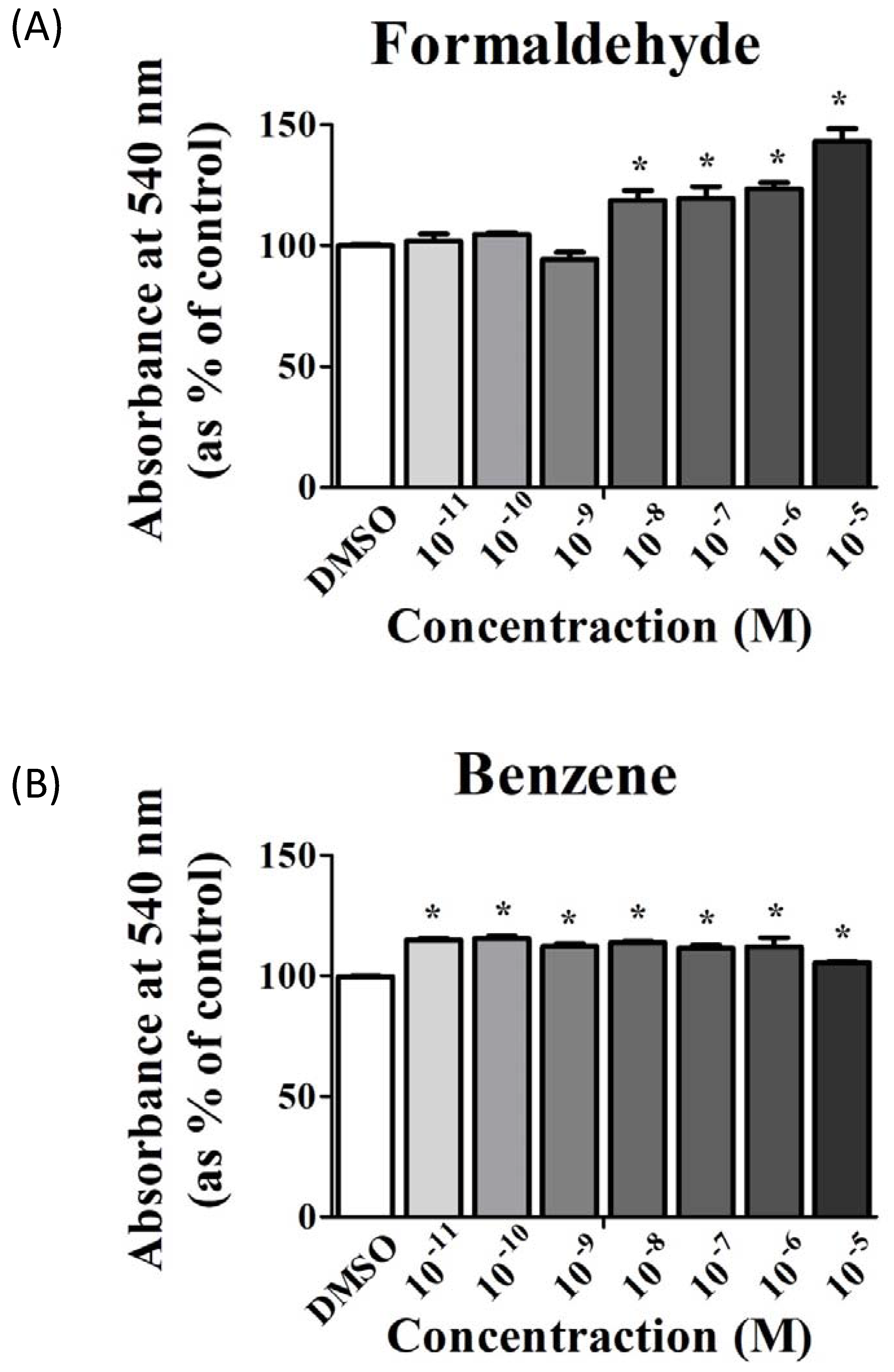
3.1. FA and Bz Induced Increased Cell Proliferation
3.2. Effects of CS Components on Protein Expression of Cell Cycle Regulatory Genes
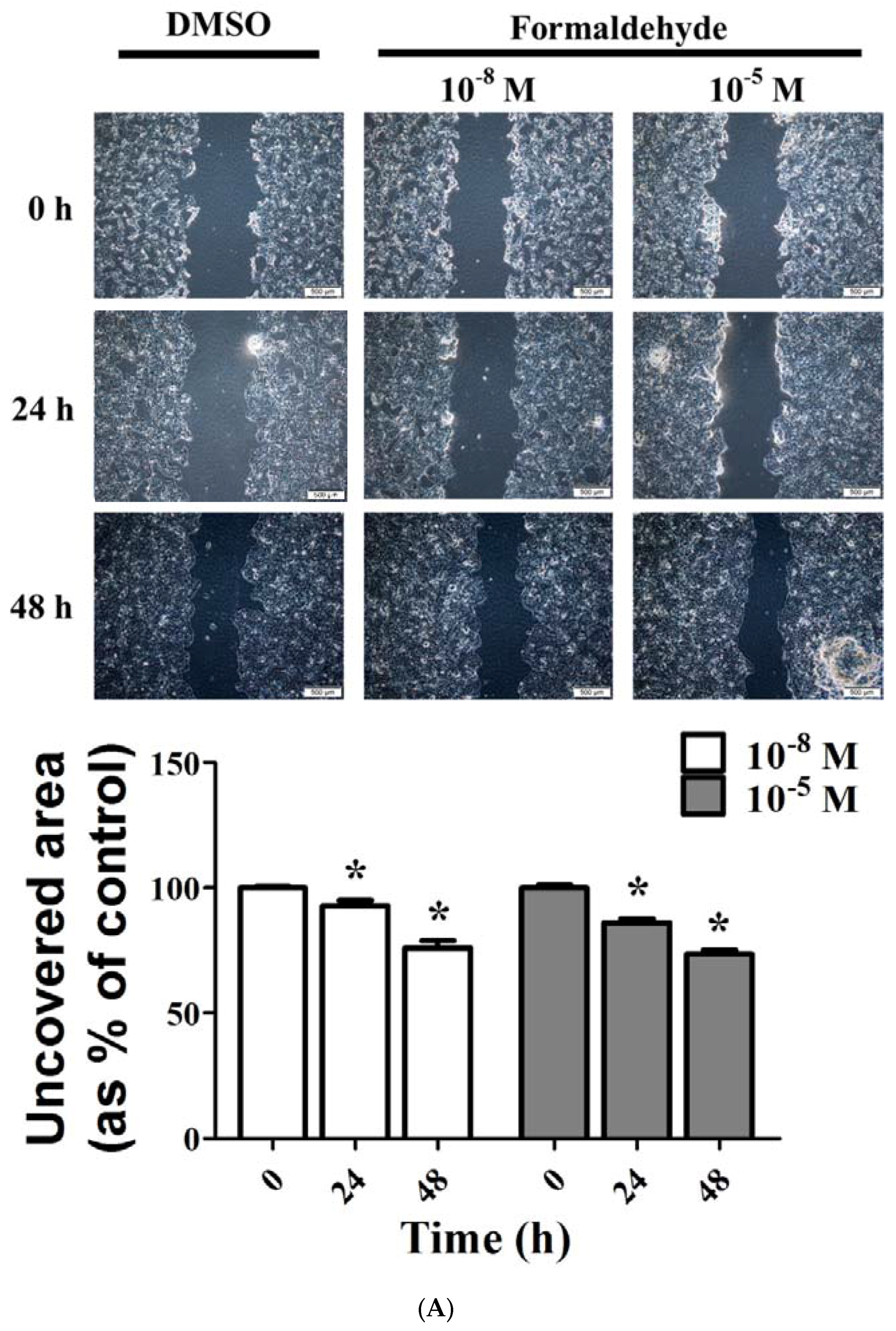
3.3. FA and Bz Induced Activation of Migration in JEG-3
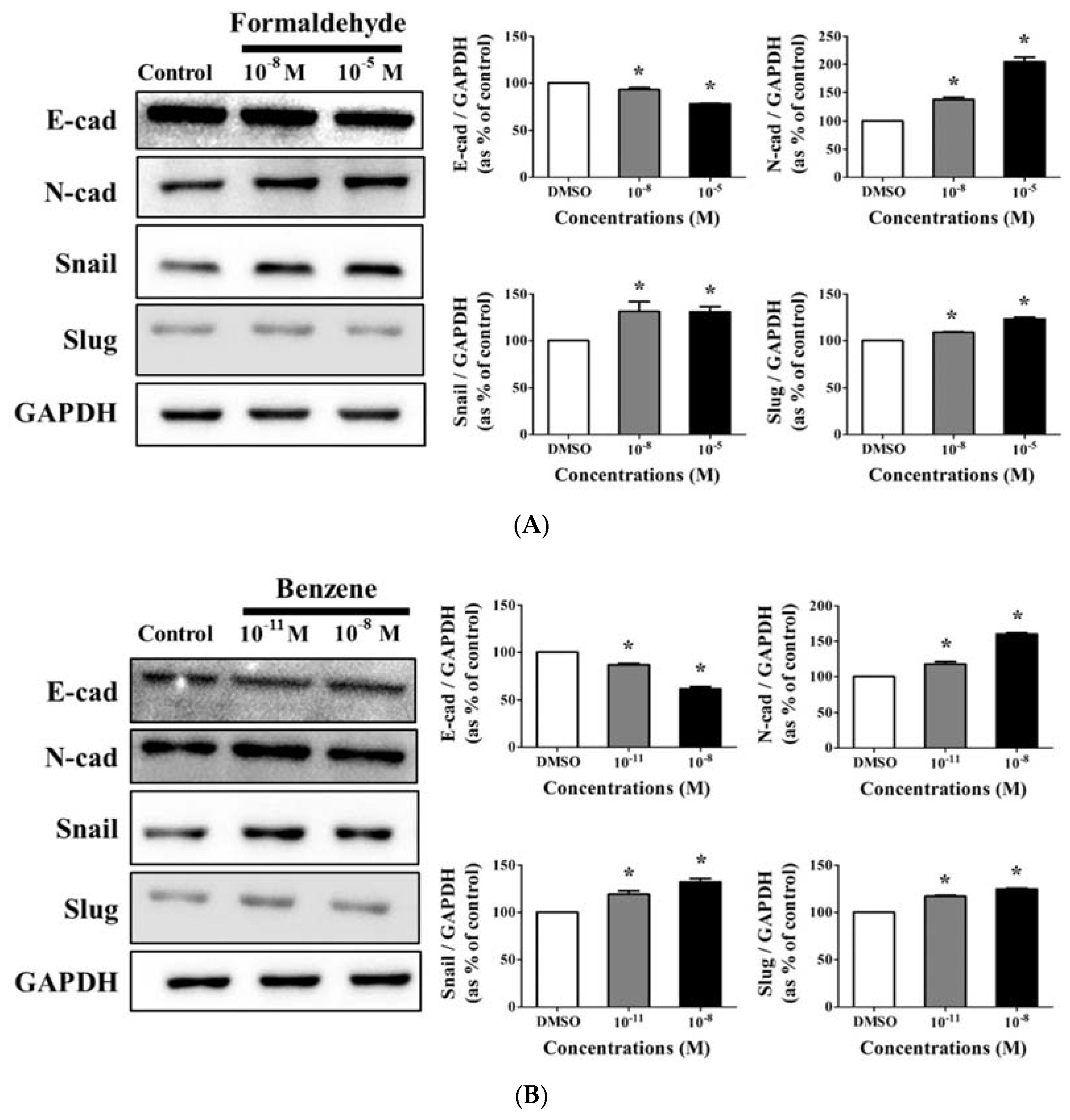
3.4. Effects of CS Components on Protein Expression of EMT Progress Genes
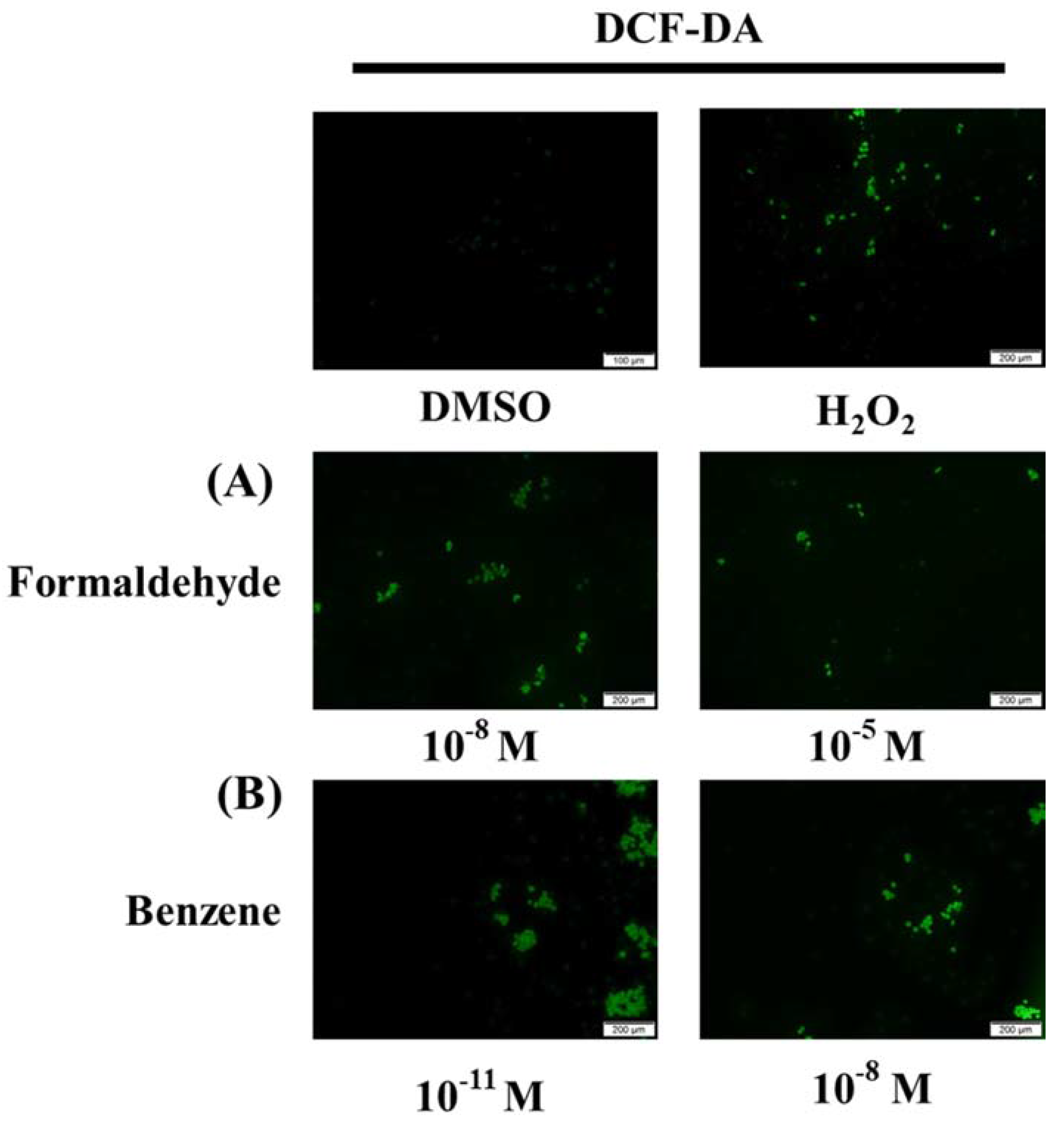
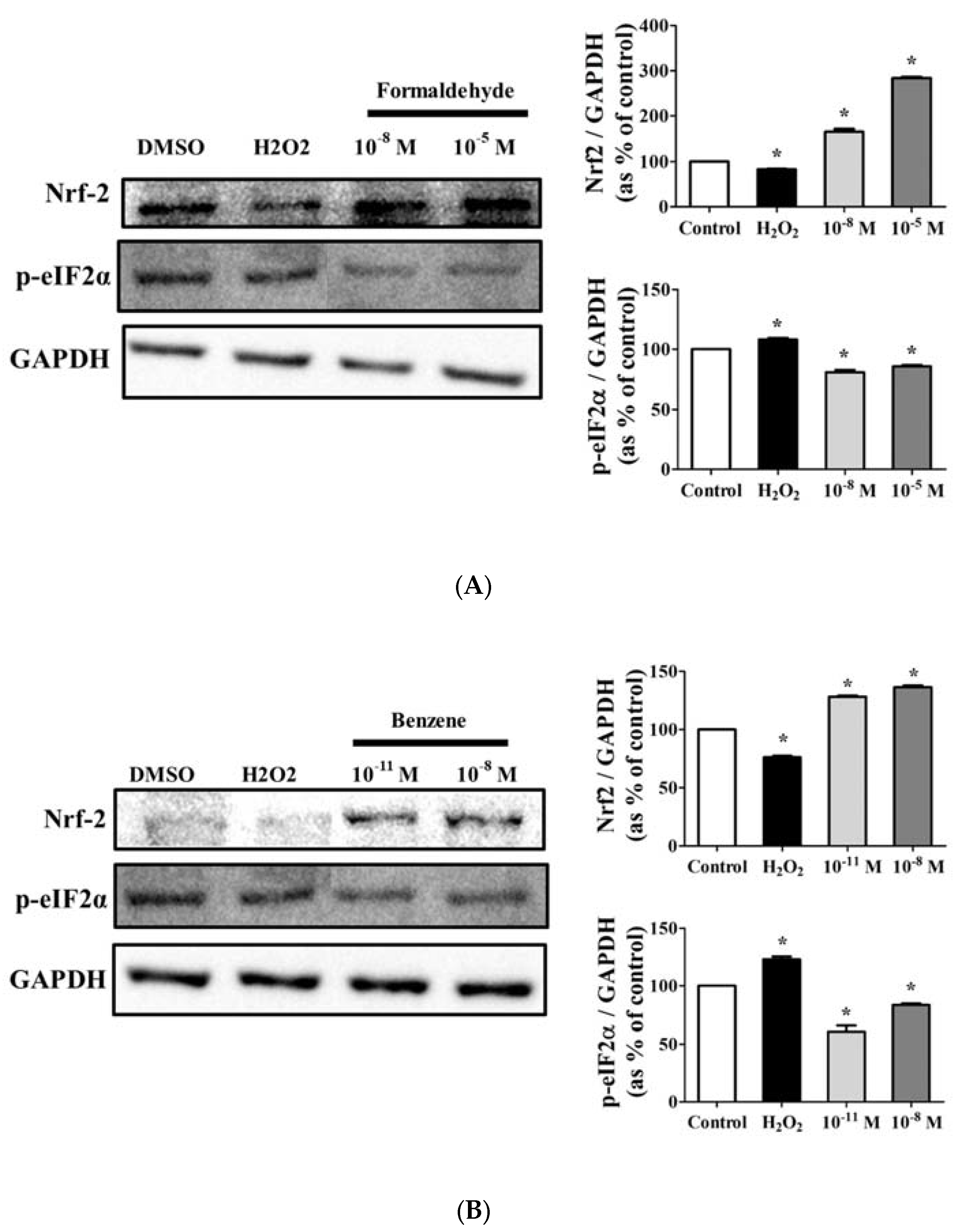
3.5. FA and Bz Activated ROS Synthesis and Increased Antioxidant Factor
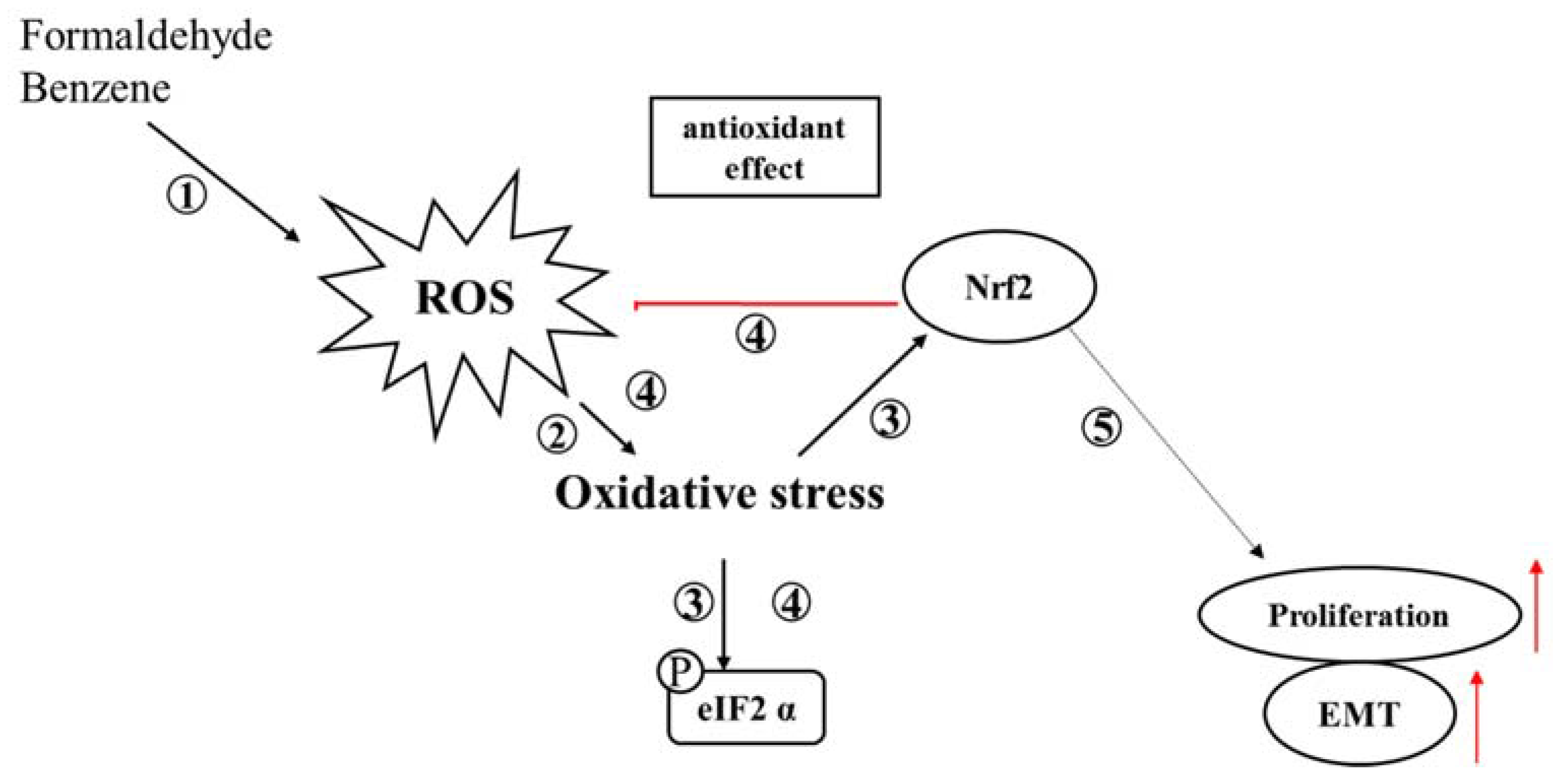
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sobus, S.L.; Warren, G.W. The biologic effects of cigarette smoke on cancer cells. Cancer 2014, 120, 3617–3626. [Google Scholar] [CrossRef] [PubMed]
- Newcomb, P.A.; Carbone, P.P. The health consequences of smoking: Cancer. Med. Clin. N. Am. 1992, 76, 305–331. [Google Scholar] [CrossRef]
- Hanaki, T.; Horikoshi, Y.; Nakaso, K.; Nakasone, M.; Kitagawa, Y.; Amisaki, M.; Arai, Y.; Tokuyasu, N.; Sakamoto, T.; Honjo, S.; et al. Nicotine enhances the malignant potential of human pancreatic cancer cells via activation of atypical protein kinase C. Biochim. Biophys. Acta 2016, 1860 Pt A11, 2404–2415. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.W.; Go, R.E.; Lee, H.M.; Hwang, K.A.; Lee, K.; Kim, B.; Lee, M.Y.; Choi, K.C. Cigarette smoke extracts induced the colon cancer migration via regulating epithelial mesenchymal transition and metastatic genes in human colon cancer cells. Environ. Toxicol. 2016, 32, 690–704. [Google Scholar] [CrossRef] [PubMed]
- Pesch, B.; Kendzia, B.; Gustavsson, P.; Jockel, K.H.; Johnen, G.; Pohlabeln, H.; Olsson, A.; Ahrens, W.; Gross, I.M.; Bruske, I.; et al. Cigarette smoking and lung cancer-relative risk estimates for the major histological types from a pooled analysis of case-control studies. Int. J. Cancer 2012, 131, 1210–1219. [Google Scholar] [CrossRef] [PubMed]
- Hitchman, S.C.; Fong, G.T. Gender empowerment and female-to-male smoking prevalence ratios. Bull. World Health Organ. 2011, 89, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Chung, S.; Noh, J. Maternal Nicotine Exposure During Late Gestation and Lactation Increases Anxiety-Like and Impulsive Decision-Making Behavior in Adolescent Offspring of Rat. Toxicol. Res. 2016, 32, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Eldridge, A.; Betson, T.R.; Gama, M.V.; McAdam, K. Variation in tobacco and mainstream smoke toxicant yields from selected commercial cigarette products. Regul. Toxicol. Pharmacol. 2015, 71, 409–427. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.D.; O’Mara-Adams, K.J.; Hoffmann, D. Toxic and carcinogenic agents in undiluted mainstream smoke and sidestream smoke of different types of cigarettes. Carcinogenesis 1987, 8, 729–731. [Google Scholar] [CrossRef] [PubMed]
- Adlkofer, F.; Scherer, G.; Conze, C.; Angerer, J.; Lehnert, G. Significance of exposure to benzene and other toxic compounds through environmental tobacco smoke. J. Cancer Res. Clin. Oncol. 1990, 116, 591–598. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Air Quality Guidelines for Europe; WHO: Geneva, Switzerland, 2000; pp. 1–273. [Google Scholar]
- World Health Organization. WHO Guidelines for Indoor Air Quality: Selected Pollutants; WHO: Geneva, Switzerland, 2010. [Google Scholar]
- Weber-Tschopp, A.; Fischer, T.; Grandjean, E. Physiological and psychological effects of passive smoking (author’s transl). Int. Arch. Occup. Environ. Health 1976, 37, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Red-Horse, K.; Zhou, Y.; Genbacev, O.; Prakobphol, A.; Foulk, R.; McMaster, M.; Fisher, S.J. Trophoblast differentiation during embryo implantation and formation of the maternal-fetal interface. J. Clin. Investig. 2004, 114, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Hod, T.; Cerdeira, A.S.; Karumanchi, S.A. Molecular Mechanisms of Preeclampsia. Cold Spring Harb. Perspect. Med. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.A.; Hwang, K.A.; Choi, K.C. Roles of Dietary Phytoestrogens on the Regulation of Epithelial-Mesenchymal Transition in Diverse Cancer Metastasis. Toxins 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Wolf, K. Tumour-cell invasion and migration: Diversity and escape mechanisms. Nat. Rev. Cancer 2003, 3, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Jeon, S.Y.; Go, R.E.; Heo, J.R.; Kim, C.W.; Hwang, K.A.; Choi, K.C. Effects of cigarette smoke extracts on the progression and metastasis of human ovarian cancer cells via regulating epithelial-mesenchymal transition. Reprod. Toxicol. 2016, 65, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Ding, X.P.; Zhao, Q.N.; Yang, X.J.; An, S.M.; Wang, H.; Xu, L.; Zhu, L.; Chen, H.Z. Role of a7-nicotinic acetylcholine receptor in nicotine-induced invasion and epithelial-to-mesenchymal transition in human non-small cell lung cancer cells. Oncotarget 2016, 7, 59199–59208. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Transcriptional responses to oxidative stress: Pathological and toxicological implications. Pharmacol. Ther. 2010, 125, 376–393. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Saavedra, D.; Sanders, L.; Perez, M.J.; Kosmider, B.; Smith, L.P.; Mitchell, J.D.; Yoshida, T.; Tuder, R.M. RTP801 Amplifies Nox-4-dependent Oxidative Stress Induced by Cigarette Smoke. Am. J. Respir. Cell Mol. Biol. 2016, 56, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Giordano, L.; Deceglie, S.; d’Adamo, P.; Valentino, M.L.; La Morgia, C.; Fracasso, F.; Roberti, M.; Cappellari, M.; Petrosillo, G.; Ciaravolo, S.; et al. Cigarette toxicity triggers Leber’s hereditary optic neuropathy by affecting mtDNA copy number, oxidative phosphorylation and ROS detoxification pathways. Cell Death Dis. 2015, 6, e2021. [Google Scholar] [CrossRef] [PubMed]
- Ashok, I.; Sheeladevi, R.; Wankhar, D. Acute effect of aspartame-induced oxidative stress in Wistar albino rat brain. J. Biomed. Res. 2015, 29, 390–396. [Google Scholar] [PubMed]
- Ryoo, H.D. Long and short (timeframe) of endoplasmic reticulum stress-induced cell death. FEBS J. 2016, 283, 3718–3722. [Google Scholar] [CrossRef] [PubMed]
- Penaranda Fajardo, N.M.; Meijer, C.; Kruyt, F.A. The endoplasmic reticulum stress/unfolded protein response in gliomagenesis, tumor progression and as a therapeutic target in glioblastoma. Biochem. Pharmacol. 2016, 118, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.H.; Lee, J.E.; Kim, H.S.; Jung, Y.J.; Hwang, D.; Lee, J.H.; Yang, S.Y.; Kim, S.C.; Cho, S.K.; An, B.S. Effect of vitamin D3 on production of progesterone in porcine granulosa cells by regulation of steroidogenic enzymes. J. Biomed. Res. 2016, 30, 203–208. [Google Scholar] [PubMed]
- Cullinan, S.B.; Diehl, J.A. Coordination of ER and oxidative stress signaling: The PERK/Nrf2 signaling pathway. Int. J. Biochem. Cell Biol. 2006, 38, 317–332. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Aronowski, J. Nrf2 to pre-condition the brain against injury caused by products of hemolysis after ICH. Transl. Stroke Res. 2013, 4, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Yi, B.R.; Go, R.E.; Hwang, K.A.; Nam, K.H.; Choi, K.C. Methoxychlor and triclosan stimulates ovarian cancer growth by regulating cell cycle- and apoptosis-related genes via an estrogen receptor-dependent pathway. Environ. Toxicol. Pharmacol. 2014, 37, 1264–1274. [Google Scholar] [CrossRef] [PubMed]
- Aleskandarany, M.A.; Negm, O.H.; Green, A.R.; Ahmed, M.A.; Nolan, C.C.; Tighe, P.J.; Ellis, I.O.; Rakha, E.A. Epithelial mesenchymal transition in early invasive breast cancer: An immunohistochemical and reverse phase protein array study. Breast Cancer Res. Treat. 2014, 145, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Genbacev, O.; McMaster, M.T.; Lazic, J.; Nedeljkovic, S.; Cvetkovic, M.; Joslin, R.; Fisher, S.J. Concordant in situ and in vitro data show that maternal cigarette smoking negatively regulates placental cytotrophoblast passage through the cell cycle. Reprod. Toxicol. 2000, 14, 495–506. [Google Scholar] [CrossRef]
- Genbacev, O.; McMaster, M.T.; Zdravkovic, T.; Fisher, S.J. Disruption of oxygen-regulated responses underlies pathological changes in the placentas of women who smoke or who are passively exposed to smoke during pregnancy. Reprod. Toxicol. 2003, 17, 509–518. [Google Scholar] [CrossRef]
- Chu, K.M.; Cho, C.H.; Shin, V.Y. Nicotine and gastrointestinal disorders: Its role in ulceration and cancer development. Curr. Pharm. Des. 2013, 19, 5–10. [Google Scholar] [PubMed]
- Castro, L.C.; Allen, R.; Ogunyemi, D.; Roll, K.; Platt, L.D. Cigarette smoking during pregnancy: Acute effects on uterine flow velocity waveforms. Obstet. Gynecol. 1993, 81, 551–555. [Google Scholar] [PubMed]
- Khorram, O.; Han, G.; Magee, T. Cigarette smoke inhibits endometrial epithelial cell proliferation through a nitric oxide-mediated pathway. Fertil. Steril. 2010, 93, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, M.; Khan, K.N.; Fujishita, A.; Masuzaki, H.; Koji, T.; Ishimaru, T. Expression of the arylhydrocarbon receptor in the peri-implantation period of the mouse uterus and the impact of dioxin on mouse implantation. Arch. Histol. Cytol. 2004, 67, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.; Hannon, G.J.; Beach, D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 1993, 366, 704–707. [Google Scholar] [CrossRef] [PubMed]
- Koff, A.; Giordano, A.; Desai, D.; Yamashita, K.; Harper, J.W.; Elledge, S.; Nishimoto, T.; Morgan, D.O.; Franza, B.R.; Roberts, J.M. Formation and activation of a cyclin E-cdk2 complex during the G1 phase of the human cell cycle. Science 1992, 257, 1689–1694. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Hannon, G.J.; Zhang, H.; Casso, D.; Kobayashi, R.; Beach, D. P21 is a universal inhibitor of cyclin kinases. Nature 1993, 366, 701–704. [Google Scholar] [CrossRef] [PubMed]
- Pagano, M.; Tam, S.W.; Theodoras, A.M.; Beer-Romero, P.; Del Sal, G.; Chau, V.; Yew, P.R.; Draetta, G.F.; Rolfe, M. Role of the ubiquitin-proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science 1995, 269, 682–685. [Google Scholar] [CrossRef] [PubMed]
- Heerboth, S.; Housman, G.; Leary, M.; Longacre, M.; Byler, S.; Lapinska, K.; Willbanks, A.; Sarkar, S. EMT and tumor metastasis. Clin. Transl. Med. 2015, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Velnar, T.; Bailey, T.; Smrkolj, V. The wound healing process: An overview of the cellular and molecular mechanisms. J. Int. Med. Res. 2009, 37, 1528–1542. [Google Scholar] [CrossRef] [PubMed]
- Savagner, P.; Yamada, K.M.; Thiery, J.P. The zinc-finger protein slug causes desmosome dissociation, an initial and necessary step for growth factor-induced epithelial-mesenchymal transition. J. Cell Biol. 1997, 137, 1403–1419. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.M.; Kim, C.W.; Hwang, K.A.; Choi, D.W.; Choi, K.C. Three components of cigarette smoke altered the growth and apoptosis of metastatic colon cancer cells via inducing the synthesis of reactive oxygen species and endoplasmic reticulum stress. Environ. Toxicol. Pharmacol. 2016, 45, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Chen, C.; Jiang, X.; Zhang, Z. ROS-mediated endoplasmic reticulum stress and mitochondrial dysfunction underlie apoptosis induced by resveratrol and arsenic trioxide in A549 cells. Chem. Biol. Interact. 2016, 245, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.P.; Yang, X.Z.; Cao, G.P. Acetyl-l-carnitine prevents homocysteine-induced suppression of Nrf2/Keap1 mediated antioxidation in human lens epithelial cells. Mol. Med. Rep. 2015, 12, 1145–1150. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.K.; Bisht, B.; Darmawan, D.O.; Chiou, R.; Ha, V.L.; Wallace, W.D.; Chon, A.T.; Hegab, A.E.; Grogan, T.; Elashoff, D.A.; et al. Dynamic changes in intracellular ROS levels regulate airway basal stem cell homeostasis through Nrf2-dependent Notch signaling. Cell Stem Cell 2014, 15, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Zanotto-Filho, A.; Masamsetti, V.P.; Loranc, E.; Tonapi, S.S.; Gorthi, A.; Bernard, X.; Goncalves, R.M.; Moreira, J.C.; Chen, Y.; Bishop, A.J. Alkylating agent induced NRF2 blocks endoplasmic reticulum stress-mediated apoptosis via control of glutathione pools and protein thiol homeostasis. Mol. Cancer Ther. 2016, 15, 3000–3014. [Google Scholar] [CrossRef] [PubMed]
- Yung, H.W.; Alnaes-Katjavivi, P.; Jones, C.J.; El-Bacha, T.; Golic, M.; Staff, A.C.; Burton, G.J. Placental endoplasmic reticulum stress in gestational diabetes: The potential for therapeutic intervention with chemical chaperones and antioxidants. Diabetologia 2016, 59, 2240–2250. [Google Scholar] [CrossRef] [PubMed]
- Ganan-Gomez, I.; Wei, Y.; Yang, H.; Boyano-Adanez, M.C.; Garcia-Manero, G. Oncogenic functions of the transcription factor Nrf2. Free Radic. Biol. Med. 2013, 65, 750–764. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Yang, Y.; Xia, S.; Rao, B.; Zhang, J.; Wang, J. Blockage of Nrf2 suppresses the migration and invasion of esophageal squamous cell carcinoma cells in hypoxic microenvironment. Dis. Esophagus 2014, 27, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, M.; Zhang, L.; Cai, H.; Zhou, S.; Zhang, J.; Wang, Y. Correlation of Nrf2, HO-1, and MRP3 in gallbladder cancer and their relationships to clinicopathologic features and survival. J. Surg. Res. 2010, 164, e99–e105. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, H.J.; Bao, Q.C.; Wang, L.; Guo, T.K.; Chen, W.L.; Xu, L.L.; Zhou, H.S.; Bian, J.L.; Yang, Y.R.; et al. NRF2 promotes breast cancer cell proliferation and metastasis by increasing RhoA/ROCK pathway signal transduction. Oncotarget 2016, 7, 73593–73606. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.S.; Chung, I.; Wong, W.F.; Masamune, A.; Sim, M.S.; Looi, C.Y. Paracrine IL-6 signaling mediates the effects of pancreatic stellate cells on epithelial-mesenchymal transition via Stat3/Nrf2 pathway in pancreatic cancer cells. Biochim. Biophys. Acta 2017, 1861, 296–306. [Google Scholar] [CrossRef] [PubMed]








© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.-M.; Kim, S.-M.; Choi, K.-C. Treatment of Human Placental Choriocarcinoma Cells with Formaldehyde and Benzene Induced Growth and Epithelial Mesenchymal Transition via Induction of an Antioxidant Effect. Int. J. Environ. Res. Public Health 2017, 14, 854. https://doi.org/10.3390/ijerph14080854
Lee H-M, Kim S-M, Choi K-C. Treatment of Human Placental Choriocarcinoma Cells with Formaldehyde and Benzene Induced Growth and Epithelial Mesenchymal Transition via Induction of an Antioxidant Effect. International Journal of Environmental Research and Public Health. 2017; 14(8):854. https://doi.org/10.3390/ijerph14080854
Chicago/Turabian StyleLee, Hae-Miru, Soo-Min Kim, and Kyung-Chul Choi. 2017. "Treatment of Human Placental Choriocarcinoma Cells with Formaldehyde and Benzene Induced Growth and Epithelial Mesenchymal Transition via Induction of an Antioxidant Effect" International Journal of Environmental Research and Public Health 14, no. 8: 854. https://doi.org/10.3390/ijerph14080854
APA StyleLee, H.-M., Kim, S.-M., & Choi, K.-C. (2017). Treatment of Human Placental Choriocarcinoma Cells with Formaldehyde and Benzene Induced Growth and Epithelial Mesenchymal Transition via Induction of an Antioxidant Effect. International Journal of Environmental Research and Public Health, 14(8), 854. https://doi.org/10.3390/ijerph14080854